

Abstract
During the past five years, new high-throughput DNA sequencing technologies have emerged; these technologies are collectively referred to as next generation sequencing (NGS). By virtue of sequencing clonally amplified DNA templates or single DNA molecules in a massively parallel fashion in a flow cell, NGS provides both qualitative and quantitative sequence data. This combination of information has made NGS the technology of choice for complex genetic analyses that were previously either technically infeasible or cost prohibitive. As a result, NGS has had a fundamental and broad impact on many facets of biomedical research. In contrast, the dissemination of NGS into the clinical diagnostic realm is in its early stages. Though NGS is powerful and can be envisioned to have multiple applications in clinical diagnostics, the technology is currently complex. Successful adoption of NGS into the clinical laboratory will require expertise in both molecular biology techniques and bioinformatics. The current report presents principles that underlie NGS including sequencing library preparation, sequencing chemistries, and an introduction to NGS data analysis. These concepts are subsequently further illustrated by showing representative results from a case study using NGS for targeted resequencing of genes implicated in hypertrophic cardiomyopathy.
Next generation sequencing (NGS) refers to high-throughput sequencing technologies that have emerged during the past five years. These technologies share a fundamental process in which clonally amplified DNA templates, or single DNA molecules, are sequenced in a massively parallel fashion in a flow cell.1,2,3 Sequencing is conducted in either a stepwise iterative process or in a continuous real-time manner. By virtue of the highly parallel process, each clonal template or single molecule is “individually” sequenced and can be counted among the total sequences generated. The high-throughput combination of qualitative and quantitative sequence information generated has allowed analyses that were previously either not technically possible or cost prohibitive. This has positioned NGS as the method of choice for large-scale complex genetic analyses including whole genome and transcriptome sequencing, metagenomic characterization of microbial species in environmental and clinical samples, elucidation of DNA binding sites for chromatin and regulatory proteins, and targeted resequencing of regions of the human genome identified by linkage analyses and genome wide association studies.4,5,6,7,8,9,10,11,12 While NGS has experienced wide dissemination throughout biomedical research, its translation into molecular diagnostics is just beginning. This report reviews key process steps of NGS, including library preparation, sequencing, and data analysis. Concepts are subsequently illustrated in the context of a diagnostic application the authors are developing for targeted resequencing of multiple genes whose mutational spectrum lead to the overlapping clinical phenotype of hypertrophic cardiomyopathy.
Next Generation Sequencing
Sample Library Preparation
NGS technologies share general processing steps, as shown in Figure 1, while differing in specific technical details. A major first step in this process is preparation of a “library” comprising DNA fragments ligated to platform-specific oliognucleotide adapters. The input nucleic acid can be genomic DNA, standard or long-range PCR amplicons, or cDNA.
Figure 1.
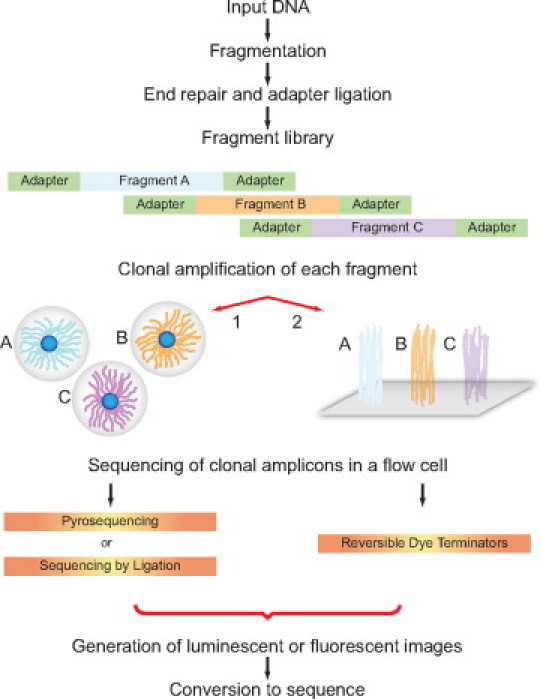
Next generation sequencing process steps for platforms requiring clonally amplified templates (Roche 454, Illumina and Life Technologies). Input DNA is converted to a sequencing library by fragmentation, end repair, and ligation to platform specific oligonucleotide adapters. Individual library fragments are clonally amplified by either (1) water in oil bead–based emulsion PCR (Roche 454 and Life Technologies) or (2) solid surface bridge amplification (Illumina). Flow cell sequencing of clonal templates generates luminescent or fluorescent images that are algorithmically processed into sequence reads.
To achieve fragmentation, the input nucleic acid is subjected to shearing by nebulization, sonication, or enzymatic digestion. The goal is to generate random overlapping fragments typically in the size range of 150–600 bp depending on platform and application requirements. Fragmentation by nebulization uses compressed air flowing through an aqueous solution of nucleic acid for several minutes. This approach is prone to volume loss and potential sample cross-contamination. Further, a broad distribution of fragment sizes is generated, which is disadvantageous when a smaller and more restricted size fragment population is needed. Sonication devices for closed tube fragmentation in the $10-$15,000 range are available, including those manufactured by Diagenode (Sparta, NJ) and Misonix (Farmingdale, NY). However, the premiere instrumentation for fragmentation, in our experience, is manufactured by Covaris (Woburn, MA), which uses acoustic wave energy transmitted into a closed tube containing an aqueous DNA solution. This results in formation and collapse of air bubbles, which generate microscale water jets that cause physical shearing of the nucleic acid. Covaris instruments, which cost $45,000-$125,000 depending on sample throughput capacity, generate the most reproducible and tunable fragment size distributions. In addition, New England Biolabs (Ipswich, MA) has recently introduced a promising enzymatic digestion technology, dsDNA Fragmentase, that uses two enzymes, one that randomly nicks dsDNA and the other that recognizes the nicked site and cuts on the opposite strand to produce dsDNA breaks. Regardless of fragmentation method, optimum conditions must be empirically established based on the size of input nucleic acid and the desired fragment size distribution, with “tighter” distributions generally preferred so as to maximize representation of sequences in the library.
Fragmented nucleic acids have terminal overhangs, which require blunt end repair and phosphorylation. Commonly, fragments are incubated with Klenow (3′ to 5′ exonuclease minus), T4 DNA polymerase (3′ to 5′ exonuclease plus), and polynucleotide kinase in the presence of dNTPs and ATP. T4 DNA polymerase removes 3′ overhangs and the polymerase activity of Klenow and T4 DNA polymerase fill in 5′ overhangs. Phosphorylation of 5′ ends occurs in parallel via T4 polynucleotide kinase activity. Repaired fragments are purified using a spin column or magnetic beads. In some platform protocols, monoadenylation of 3′ ends is subsequently performed using Klenow and dATP. This enhances the efficiency of ligation to platform specific oligonucleotide adapters (with T overhangs). Ligation products are often size separated by gel electrophoresis, and a specific size range is selected compatible with a given platform or application. The adapter modified fragments constitute the “library” of overlapping sequences. For some protocols, the library concentration needs to be increased and this is accomplished by PCR with primers complementary to adapter sequences.
The next major step is to prepare the “library” for massively parallel sequencing. The first wave of NGS platforms manufactured by Roche 454 (Branford, CT), Life Technologies (Carlsbad, CA), and Illumina (San Diego, CA) require their respective libraries to be clonally amplified before sequencing. For the Roche-454 GS-FLX (and GS-Junior) and Life Technologies SOLiD platforms, clonal amplification uses emulsion PCR and requires hybridizing the adapter modified fragment library to beads that display oligonucleotides with sequences complementary to adapter sequences.13,14,15 Hybridization is performed under limiting dilution conditions to achieve hybridization of single library fragments to single beads. This is followed by emulsion PCR, wherein single beads containing single library fragments are segregated into water in oil microdroplets and thermocycled. During cycling, tens of thousands to millions of copies of the starting single fragment sequence are produced on the bead surface (with amplification amount platform specific). Post-PCR, the emulsion is disrupted and beads containing clonally amplified templates are enriched and individually deposited into wells (Roche-454 picotiter plate) or onto a surface-modified glass slide flow cell (Life Technologies SOLiD). The deposition of beads needs to be performed under conditions in which only a single bead is deposited per well or, with the SOLiD technology, beads need to be sufficiently spatially separated on the slide surface. The presence of clonally amplified DNA on the bead surface is required to generate sufficient signal for optical capture from each bead during sequencing. The replicate copies are sequenced “in unison” to yield one sequence read per bead.
For the Illumina Genome Analyzer platform, adapter modified library fragments are automatically dispensed under limiting dilution conditions onto a glass slide flow cell that displays oligonucleotides complementary to Illumina adapter sequences.16 Surface bound individual fragment molecules are clonally amplified using an isothermal bridge amplification method that generates clonal “clusters” of approximately 1000 identical molecules per cluster. By this approach, one fragment is bound to one surface oligonucleotide, undergoes cluster generation, and the replicate copies are sequenced to yield one sequence read.
For the Roche 454, Life Technologies, and Illumina platforms, library preparation is a multistep manual process of pipetting, incubations, and purifications of enzymatic reaction products using spin columns or magnetic beads. For the Illumina technology, a gel purification-based size selection of the library is commonly performed.17 Clonal amplification by emulsion PCR adds considerable complexity to sample processing. However, new front-end automation devices for the emulsion PCR steps have been developed by Roche 454 and Life Technologies. In addition, Epicenter Biotechnologies (Madison, WI) has recently introduced a novel, less manual approach to library preparation wherein fragmentation and adapter addition are accomplished using in vitro transposition and PCR. A Transposome complex comprising free transposon oligonucleotide ends and a transposase is incubated with input DNA resulting in nearly random fragmentation with sizes controlled by enzyme concentration and incubation time. During fragmentation the transposon associated oligonucleotides are covalently attached to the 5′ ends of the target fragment. The fragments are then amplified using tailed PCR primers in which the 5′ primer region contains either Roche 454 or Illumina library specific adapter sequences and the 3′ region is complementary to the transposon oliognucleotides. The resulting library is then ready for either emulsion PCR or bridge amplification.
Important quality control steps are essential during library preparation, notably assuring that fragmentation yielded an appropriate size distribution for the desired application and platform being used. Increasingly, determinations of fragment distributions, size changes due to adapter ligation, and library quantification are performed with high-resolution electrophoresis instruments (eg, Agilent BioAnalyzer, Santa Clara, CA).
Next Generation Sequencing Chemistries
Sequencing of clonally amplified templates on first wave platforms uses nucleotides or their labeled analogs, or oligonucleotide probes, that are incorporated in a stepwise, iterative manner with incorporation events optically monitored and bioinformatically converted to sequence. Chemistries includes pyrosequencing (Roche 454), sequencing by reversible dye terminators (Illumina), and sequencing by sequential ligation of oligonucleotide probes (Life Technologies SOLiD). For pyrosequencing on the Roche 454 GS-FLX and new GS-Junior platforms, dATP, dCTP, dGTP, or dTTP and polymerase are sequentially flowed over the picotitre plate containing bead bound clonally amplified DNA templates. When incorporation of a complementary nucleotide occurs on a growing strand in an individual well, pyrophosphate is released, which drives luciferase-mediated light generation in the well. The luminescent bursts are optically captured with a high-sensitivity CCD camera. Luminescence intensity is directly proportional to the number of nucleotides incorporated, thus homopolymer signals are greater than single base additions and length-dependent. However, accuracy of homopolymer determination decreases with increasing homopolymer length.15 After incorporations events are recorded, residual nucleotides are washed out of the flow cell and the process is repeated.
Illumina sequencing uses a mixture of four fluorescently unique reversible dye terminators that are simultaneously introduced into the flow cell, along with DNA polymerase. Incorporation of complementary bases into individual clusters is recorded by virtue of base specific fluorescent emission spectra. The fluor and termination moieties, linked to the nucleotide base and 3′ deoxyribose sugar position, respectively, are then cleaved and washed away. Successive cycles of dye terminator mixture and DNA polymerase introduction, incorporation, and cleavage yield chain elongation.
Sequencing by ligation on the SOLiD platform involves the iterative introduction of combinations of fluorescently unique oligonucleotide probes containing specific interrogation nucleotides and degenerate nucleotides. Postannealing and ligation, fluorescence is recorded and the labeled section of each probe is cleaved and washed away. Sequencing by ligation of oligonucleotide probes is conceptually different from other NGS technologies in that sequence is inferred from probe hybridization events and includes the process of each base being interrogated twice for each sequence read generated.13
Resulting read lengths vary between chemistries and have been progressively increasing as each technology evolves. Of the first wave technologies, Roche 454 pyrosequencing read lengths are the longest at 400 plus bases. As of this writing, a new platform, requiring clonally amplified templates, has been announced by Ion Torrent (Guilford, CT), whose CEO Jonathan Rothberg earlier founded 454 Life Sciences. The Ion Torrent platform takes clonally amplified DNA templates and individually sequences them on a semiconductor chip consisting of an array of about 1.55 million 3.5-micrometer wells. Underneath the wells is an ion-sensitive layer and one electronic sensor per well. Unmodified nucleotides are sequentially added in the presence of DNA polymerase. With complementary base incorporation, hydrogen ions are released during formation of the 5′ to 3′ phosphodiester bond. The released hydrogen ions decrease the pH of the solution in the well proportional to the number of bases incorporated and the decrease is registered by the sensing system, which is a microscale solid-state pH meter. This approach, which generates read lengths in the 100 base range, represents the first NGS technology that does not depend on light or fluorescent detection.
Single Molecule Sequencing
While the Roche 454, Life Technologies SOLiD, and Illumina platforms currently dominate the field, a second wave of platforms based on single molecule DNA sequencing (SMS) is emerging. Commercially available SMS platforms are those from Helicos BioSciences (Cambridge, MA) and Pacific Biosciences (Menlo Park, CA), with a third platform in development by Life Technologies. Single DNA molecule sequencing inherently reduces the complexity of library preparation in that clonal amplification before sequencing is eliminated. The Helicos BioSciences HeliScope has a library preparative procedure in which DNA fragments are enzymatically polyA 3′ tailed, purified, and hybridized to oligo dT attached to a glass slide flow cell surface. For sequencing on the Heliscope, a variation of dye terminator chemistry comprising one color Cy5 fluorescently labeled, 3′ unblocked reversible dye terminators is used.18,19,20,21 In this approach, sequencing is conducted in a stepwise manner, with each incorporation event monitored at the single molecule level using total internal reflection fluorescence microscopy.
The Pacific BioSciences SMS technology represents the first “real-time” sequencing method based on continuous monitoring of DNA polymerase-mediated incorporation of labeled nucleotide analogues.22,23,24 For library preparation, fragmented DNA is end repaired, monoadenylated, and ligated to adapters that form a stem loop structure on each fragment end. Molecular complexes are then formed in solution comprised of individual DNA library fragments with stem loop adapters that are primed with a sequencing primer complementary to adapter loop sequences, and phi 29 DNA polymerase. Under limiting dilution conditions, individual complexes are deposited into nanoscale wells present in a highly parallel flow cell configuration. Each nanoscale well is optically monitored by a zero mode waveguide. To immobilize the complex, the polymerase is biotinylated and bound to streptavidin on the well floor so as to be optimally oriented in the zero mode waveguide.
To initiate DNA polymerization and hence sequencing, divalent cation and four differently labeled nucleotide analogs are added. A new class of labeled nucleotide analogs was developed for this technology wherein a hexaphosphate moiety is linked to the 5′ position of the deoxyribose sugar. Through a phosphoester bond, nucleotide specific fluors are coupled to the terminal phosphate. As a complementary phospholabeled nucleotide is incorporated into the growing DNA strand, its respective emission fluorescence is observed through excitation in the zero mode waveguide. With incorporation, the phospholinked fluorescent moiety is cleaved and rapidly diffuses away. Successive incorporation events take place at a rate of 2 to 4 bases per second. The ability to distinguish specific incorporation signals from background, due to noncomplementary nucleotide sampling by the polymerase and random nucleotide diffusion, is based on the longer time that a complementary base is “held” by the polymerase. Each incorporation event generates a pulse of nucleotide specific fluorescent emission followed by a return to baseline background fluorescence before the next incorporation event. The phi29 DNA polymerase is a highly processive enzyme with strand displacement activity. When bound to a library fragment with stem loop adapters, the polymerase replicates in a “circular” fashion yielding redundant forward and reverse strand sequence.
A second real-time SMS technology, based on monitoring the incorporation of phospholinked fluorescent nucleotide analogues, is in development by Life Technologies with a planned commercial release in 2011. Although complete details are not yet available, library preparation is anticipated to require DNA fragmentation, end repair, and phosphorylation. The processed fragments are then denatured and ligated, as single strands, to capture oligonucleotides on a glass slide flow cell surface. A primer, complementary to the capture oligonucleotide, is annealed and sequencing is initiated by the addition of four differently labeled nucleotides, a novel DNA polymerase, and divalent cation. The novel DNA polymerase is conjugated to a quantum dot that functions as a donor, whereas the fluorescent moiety on the nucleotide serves as an acceptor in a classic fluorescence resonance energy transfer (FRET) reaction. Excitation of the quantum dot results in a wavelength emission that excites labeled nucleotides that are being incorporated into the growing strand by virtue of their proximity to the DNA polymerase. Nucleotide incorporation is coupled to cleavage and diffusion of the phospholinked fluorescent moiety. In contrast to the use of zero mode waveguides in individual wells, the optical monitoring employs total internal reflection fluorescence microscopy.
Comparative Perspective on Platforms
First wave NGS platforms that sequence clonally amplified templates have continued to improve in terms of accuracy, turnaround times, and throughput. This improvement has been the result of chemistry refinements, instrument modifications to fluidics and optics, higher flow cell template densities, and refined algorithms for signal processing. The maturation of first wave platforms has increased their attractiveness for the clinical diagnostic setting. However, important implementation barriers include complex manual library procedures and expensive reagents costs. Continued automation efforts will reduce the former barrier but run cost reduction is more challenging due to the iterative sequencing process, which consumes reagents with each cycle that need to be replenished.
The transition from clonal template-based NGS to single molecule sequencing offers several potential advantages. Not needing to generate clonal templates as sequencing substrates simplifies library preparation and has the added benefit of reducing representational biases introduced by clonal template amplification. Sequencing run times will be decreased from the current multiple hours (Roche 454) and days (Life Technologies SOLiD and Illumina) to fractions of an hour or two. By not requiring multiple reagent additions and washes, real-time SMS instrument run costs are expected to be on the order of several hundred dollars, as opposed to a few thousand. Read lengths for Pacific Biosciences are now in the 800-1000 plus base range, an improvement that will facilitate alignment and bioinformatic analyses. While SMS technologies are promising, an important concern is higher error rates in individual reads compared with those observed in reads generated from clonal template based sequencing technologies. For further details on chemistries, the reader is referred to recent reviews including one by the authors, which shows diagrams detailing the Roche 454, Illumina, and SOLiD chemistries.3,25,26,27 A summary of key features of next generation sequencing platforms is shown in Table 1.
Table 1.
Characteristics of Next Generation Sequencing Platforms
| Platform | Template preparation | Chemistry | Read length (bases) | Run time (days)* | Gb per run† |
|---|---|---|---|---|---|
| Roche 454 | |||||
| GS FLX Titanium | Clonal-ePCR | Pyrosequencing | 400‡ | 0.42 | 0.40–0.60 |
| GS Junior | Clonal-ePCR | Pyrosequencing | 400‡ | 0.42 | 0.035 |
| Illumina | |||||
| HiSeq 2000 | Clonal Bridge Amplification | Reversible Dye Terminators | 35–100 | 2–4 | 30–100 |
| Genome Analyzer IIX | Clonal Bridge Amplification | Reversible Dye Terminators | 35–100 | 2–4 | 9–25 |
| Genome Analyzer IIE | Clonal Bridge Amplification | Reversible Dye Terminators | 35–100 | 2–5 | 3.5–10 |
| iScanSQ | Clonal Bridge Amplification | Reversible Dye Terminators | 35–75 | 2.5–5 | 4–10 |
| Life Technologies | |||||
| SOLiD 4 | Clonal-ePCR | Oligonucleotide Probe Ligation | 35–50 | 4–7 | 35–50 |
| Helicos Biosciences | |||||
| HeliScope | Single Molecule | Reversible Dye Terminators | 35‡ | 8 | 25 |
| Pacific Biosciences | |||||
| SMRT | Single Molecule | Phospholinked Fluorescent Nucleotides | 800–1000 | 0.02 | Pending |
Run times and gigabase (Gb) output per run for single-end sequencing are denoted by a double asterisk and a single dagger, respectively. Run times and outputs approximately double when performing pair-end sequencing.
Average read lengths for the Roche 454 and Helicos Biosciences platforms.
Sequencing Read Lengths, Accuracy, and Coverage
Each NGS platform generates different read lengths that range from short reads (eg, 35 bases) to greater than 500 bases. For a number of applications, including targeted resequencing, CHiP-Seq, and RNA-Seq, short reads are highly informative and adequate. The advantage of longer reads is evident in applications of de novo genome assembly and when sequencing through areas of repetitive DNA and targets that share regions of high homology (for example, members of a related gene family and functional versus pseudogenes). Most platforms have the important option of being able to sequence both ends of library fragments, termed paired-end or mate-pair sequencing. Different library fragment insert sizes can be used so that the interval between pair end sequence information can be varied. Pair-end sequencing effectively doubles the amount of sequence obtained, facilitates alignment, and improves detection of insertions and deletions occurring between the pair ends.
To address accuracy and coverage, it is important to consider how NGS data are generated. At each iterative cycle (or real-time incorporation), luminescent or fluorescent signals are optically captured and processed. Signals are compared with background and algorithmically converted into nucleotide base information in the form of sequence reads. Each base is assigned a “quality” score that shares conceptual similarity to Phred quality scores used in Sanger sequencing. Direct comparison of quality scores between platforms is not possible as each platform has its own algorithm. With respect to accuracy, NGS chemistries are prone to errors occurring in individual reads at frequencies in the 0.5 to 2% range, depending on platform. These are primarily substitutions and secondarily single base insertions and deletions (with erroneous indels being more pronounced in homopolymer tracts). Therefore, nucleotide variant changes (eg, from a reference sequence) cannot be accurately relied on if present in only a single read. This is addressed by requiring redundancy, that is, the variant must be present in multiple overlapping reads.
The number of times a nucleotide base has been sequenced is referred to as its “coverage.” A not fully-defined parameter is how much coverage is needed for accurate sequencing, if accuracy is defined as ∼99.5 to 99.9%. This varies with platform, but empirical results from the literature have suggested as few as 4 to 5 reads per allele, whereas most groups require 10 to 30 reads per allele.28,29,30 Confidence in variant identification is increased when bidirectional sequencing reads are concordant. NGS is still early in its overall development, and sequencing accuracies will improve with further refinements in chemistry, optics, and processing algorithms.
Redundancy is also inherent to Sanger sequencing in that each base peak of a Sanger electropherogram represents many copies of the same length chain termination product. Based on desired coverage, one can calculate the amount of sequencing required for a given size target(s). However, coverage can vary considerably across target regions. Sources of variability include target enrichment method (see discussion below) and library preparation. Aspects of library preparation that contribute to variability include unequal ligation of adapters to fragment ends and PCR amplification biases. The challenge posed by variable coverage is when coverage is low and the number of reads is less than an empirically derived “cut off” value for accurate variant identification. To offset variability, we devise NGS experiments to yield coverage in the several hundred-fold range to in an effort to achieve 30-fold plus coverage per nucleotide position. While this conservative approach does not take full advantage of NGS throughput capabilities, it minimizes the number of suboptimally covered regions.
Bioinformatics and Variant Identification
Two major computations are performed with NGS reads, those of assembly and alignment. Assemblies are performed primarily when no reference genome exists for the DNA sequenced (for example, a genetically uncharacterized pathogen). Assembly algorithms take sequence reads, align overlapping sections, and generate longer length contigs, which serve as the scaffold for genome assembly and subsequent alignments. For diagnostic applications where NGS is being developed, reference genomes exist (eg, major pathogens such as HIV and the human genome). In this context, the primary computation used is alignment of reads to the reference sequence. For resequencing human gene targets, reads are typically aligned to the relevant, complementary subregions of the human genome (eg, exons or full-length genes) as opposed to aligning against the entire human genome, which is more computationally intensive.
Alignment is the process of determining the best match between sequence reads and reference sequence. To accommodate the large number of reads generated by NGS, a number of new alignment algorithms have been devised. Many share the characteristic that alignment is performed in a multistep or heuristic approach in which the first phase consists of converting either the sequence reads or the reference sequence into an index of shorter length sequences, which are given read identifiers. The index is cross matched (eg, sequence read index against reference sequence or vice versa) using an algorithm whose criteria for alignment is operator-defined, with a key emphasis on the number of nucleotide matches required and, conversely, the number of nucleotide mismatches allowed for the given index sequence length.
A popular method for this type of matching uses traditional computational science-based hash table algorithms. By using indexes of shorter length (sometimes referred to as “seeds”) and permitting mismatches, alignments proceed more rapidly and generate a first pass set of matches. Subsequently, greater stringency is applied to the matched data set, using additional algorithms to yield a more accurately refined final set of alignments that optionally permit gaps for identification of insertions and deletions. These include variants of local sequence alignment approaches such as the traditional Smith-Waterman algorithm and can incorporate criteria based on sequence quality scores. Postalignment, programs generate key information including the number of aligned reads, a list of sequence variants relative to the reference, and the percentage of reads containing the variant. Refinement of variant calling and interpretation is an area of active investigation.
For heterozygous variants, approximately 50% of sequences would be expected to contain the variant (that is, an allelic read percentage of 50), and for a homozygous variant, approximately 100% of the sequences would contain the variant. In practice, variant read percentages for NGS can exhibit a wide range for both heterozygous and homozygous variants. Heterozygous read percentages can range from the low 20 percentile to as high as 80% and homozygous read percentages can range from 60% upwards. These wide ranges complicate variant identification and arise from both technical and bioinformatic sources. For example, during library preparation allelic biases can be introduced by differential PCR amplification. Alignment programs can yield different variant read percentages as a consequence of their unique algorithms and criteria. Cross or misalignment of related sequences to reference genes that share high homology can result in skewing of variant read percentages. Improving variant identification and allelic read percentages will depend on a combination of chemistry and bioinformatic advances including the use of longer read lengths, pair end sequencing (to increase alignment accuracy), algorithm refinements, and sequencing methods that minimize or do not require amplification during library preparation. NGS data analysis has become a burgeoning subdiscipline within bioinformatics. For a further introduction the reader is referred to recent reviews.31,32
Targeted Resequencing
Investigations into the genetic basis of a growing number of inherited disorders have revealed how a clinical phenotype can be due to multiple causative genes with a broad mutational spectrum. Examples include X-linked mental retardation, mitochondrial disorders (secondary to mutations in both the mitochondrial genome and nuclear genes), congenital hearing loss, cardiac arrhythmias, and hypertrophic and dilated cardiomyopathies. A comprehensive diagnostic approach requires the analysis of dozens of genes (upwards of 80 genes for X-linked mental retardation).33,34,35 The magnitude of analysis poses an operational challenge for Sanger sequencing. In the future, when the cost of whole genome sequencing markedly declines, one could envision sequencing the entire genome followed by interpretation of the genes of immediate clinical interest. In the interim, targeted resequencing of the multiple causative genes is an attractive area for NGS diagnostic development. For this approach, a number of target gene enrichment strategies have been tested in conjunction with NGS and are next presented.
Enrichment strategies can be categorized as either amplification-based or oligonucleotide array capture based.36,37 For the former, PCR remains the mainstay approach. Resequencing of exons by targeted PCR enrichment has been demonstrated for a number of genes. Using 96-well formats with either manual or automated reaction setups, amplicons are generated, pooled (preferably at or close to equimolarity), then converted into a NGS library. This approach integrates well with the longer read length technologies (eg, Roche 454). A variation that streamlines library preparation uses amplification primers tailed with platform-specific adapter sequences. An additional variation incorporates identifier or barcode sequences into the adapters. In this scenario, multiple sample libraries are prepared, each with their own barcode, and pooled for sequencing. Post-sequencing, reads are bioinformaticaly assigned to their respective sample of origin.38,39,40 Overlapping long-range PCR with 5- to 10-Kb amplicons can be used to amplify large genes. Postamplification, long-range amplicons are pooled, fragmented, and converted into a library.
Recently, a novel highly parallel PCR microdroplet technology has been introduced by RainDance Technologies (Lexington, MA).41,42 Individual primer pairs for targets are designed (for amplicon lengths 200–600 bp), synthesized, and sequestered in individual stable emulsion microdroplets. Up to a few thousand primer pairs can constitute a microdroplet primer population. For targeted amplification, genomic DNA is first fragmented into a size range of 2000–4000 bp. The fragmented DNA is then randomly distributed into its own microdroplet population. On an automated microfluidic platform, individual microdroplets are merged so that one microdroplet containing fragmented genomic DNA is associated with one microdroplet containing an individual primer pair. The associated droplet pairs are fused while traversing a microfluidic channel across which an electrical potential is applied. Each fused microdroplet contains genomic DNA and a specific primer pair, which are collected into a single tube and thermocycled to achieve highly parallel PCR amplification. Postamplification, the droplets are disrupted to release their contents and the pooled amplification products are converted into a library for sequencing. When using the RainDance technology for enrichment before Roche 454 sequencing, the amplicons are ligated to adapters and converted to clonally amplified templates for sequencing. To streamline, RainDance primers can be synthesized with Roche 454 adapter tails. At present, when using the RainDance technology in conjunction with short read sequencing platforms (eg, Illumina Genome Analyzer or Life Technologies SOLiD), the pooled amplicons are converted by ligation into concatemers, which are then fragmented and processed into the respective sequencing library. While a workable approach, it adds additional technical steps and results in a proportion of junction sequences between unrelated amplicons. The RainDance technology offers PCR specificity and is well suited for resequencing large numbers of exons.
As an additional PCR-based enrichment approach, Fluidigm (South San Francisco, CA) has recently released their Access Array platform. This technology uses a microfluidic chip that contains nanoliter scale reaction chambers separated by valves. In its current configuration, 48 samples can be loaded onto the chip and each sample can be distributed into 48 chambers with unique primer pairs. The loaded chip is placed on a plate thermocycler for PCR amplification. Post amplification, valves are reversed and samples (now comprising up to a 48-amplicon pool) are returned to their original wells. In comparison with RainDance, the Fluidigm technology is non-emulsion based, can accommodate long-range PCR reactions, but, at present, does not have equivalent scalability.
Alternative amplification strategies have been developed using molecular inversion probes (MIPs) and variants thereof.43,44,45 MIPs are single-stranded oligonucleotides that contain a common middle positioned linker sequence flanked by target specific sequences. These can be highly multiplexed and hybridized to denatured genomic DNA. On hybridization, the target specific probe sequences anneal to their genomic complements and the overall probe assumes a circular structure with a gap between the specific annealing sites. The gap can be filled with DNA polymerase followed by ligation. The intact circle, containing the common linker, flanking probe, and intervening genomic sequences, can be amplified by PCR with primers complementary to the common linker sequences. Hundreds to thousands of targets can be captured and amplified in a single tube. MIP technology has not been commercialized, and challenges include the design and synthesis of MIPs and the need to re-design to improve target capture uniformity.
A related technology, termed Selector, is being commercialized by OLINK Genomics (Uppsala, Sweden). In this method, genomic DNA is digested with different combinations of restriction endonucleases. Denatured restriction fragments are hybridized to Selector oligonucleotide probes whose right and left sides are complementary to the fragment ends. Probe hybridization yields a genomic DNA circular structure whose ends are then ligated. For enrichment, ligated circles are amplified using rolling circle amplification. Both MIP and Selector amplification products are subsequently fragmented and converted into next-generation sequencing libraries.
Oligonucleotide array capture methods constitute the second major enrichment strategy.46,47,48,49,50,51,52 With array methods, genomic DNA, or genomic DNA converted into a next-generation sequencing library, is hybridized to oligonucleotides complementary to target regions of interest. Posthybridization, the enriched material is eluted from the array and processed for NGS. The oligonucleotides can be formatted on a solid surface array (eg, Nimblegen, Madison, WI, Agilent, Santa Clara, CA, or Febit, Lexington, MA) or used in solution (Nimblegen and Agilent). In solution or solid surface oligonucleotide array capture experiments require either one or two day hybridization, respectively, and the eluted material can be increased in quantity by PCR before NGS.53 Capture arrays have been designed for both large- and smaller-scale contiguous and noncontiguous target regions. Important technical considerations for array design are the specificity of capture probes and the potential for cocapture and enrichment of nontarget sequences, notably those from closely related genes and pseudogene analogs. When designing an array, it is common to use software such as RepeatMasker (http://www.repeatmasker.org, last accessed on May 12, 2010) that identifies repeat sequences and low complexity regions in target regions. While these will be more often found in introns and other nonexon regions, they can be present in exons, making probe selection challenging.
Despite designing arrays to minimize nonspecific hybridization, cocapture of nontarget regions occurs to a variable degree. Cocapture of highly homologous pseudogene sequences poses an interpretive challenge, which can be mitigated by bioinformatic filtering against pseudogene sequences. The inherent coverage variability in NGS libraries can be compounded by uneven and inadequate capture using arrays, sometimes necessitating array redesign.
Each enrichment methodology has advantages and disadvantages. Array capture is better suited for enrichment of larger target regions up to the scale of the human exome. Amplification-based strategies, particularly those that use PCR, offer increased target enrichment specificity and may ultimately be more appropriate for clinical diagnostic applications where the enhanced specificity will translate to increased diagnostic accuracy.
Case Study: Hypertrophic Cardiomyopathy
In the final section of this report, we present highlights from our initial efforts to develop targeted resequencing of genes implicated in primary hypertrophic cardiomyopathy (HCM). The data selected are representative and illustrate several of the concepts described earlier. HCM has an incidence of approximately 1 in 500 and displays an autosomal dominant pattern of inheritance with variable penetrance.54,55,56,57 HCM manifests primarily in adulthood, although pediatric cases are documented.58 HCM muscle biopsies display disordered cardiac myocyte architecture with interstitial fibrosis. As HCM progresses, left ventricular and septal wall enlargement occurs with development of angina, arrhythmias, and, in its severest form, sudden death. HCM is a disorder of the cardiac sarcomere, the multiprotein contractile unit of the cardiac muscle comprising thick and thin filaments and accessory proteins. More than 450 mutations in 16 genes have been implicated in HCM and the genes with the highest mutation frequency encode for the core sarcomere proteins including myosin heavy and light chains, actin, troponins T and I, and tropomyosin.59,60,61,62 Genetic testing approaches for HCM have included Sanger sequencing of individual genes and a gene panel approach using resequencing arrays.62,63,64 Next generation sequencing now offers a new diagnostic approach for HCM.
In a first phase project to evaluate NGS for HCM diagnostics, we used long-range PCR to amplify 16 genes implicated in HCM in a control DNA (Table 2).62 Primers and reaction conditions were optimized yielding an average amplicon length of 5136 bp with overlaps averaging 550 bases in length. The choice of long-range PCR allowed for the design of fewer amplicons (67 total) and the longer term opportunity to investigate potential deep intronic mutations, which to date have not been extensively studied in HCM. Fourteen of the 16 genes were amplified in full, including exons and introns, while only exons and flanking intronic sequences of PRKAG2 and only one exon of TTN previously reported to contain a HCM associated mutation were amplified. A total of 319,646 nonoverlapping bp of genomic DNA were amplified under uniform reaction conditions using TaKaRa Hot Start DNA polymerase, representing 35,399 bases of exonic DNA.
Table 2.
Major Genes Implicated in Hypertrophic Cardiomyopathy and their Respective Mutation Frequencies
| Protein | Gene | Mutations | Gene size bp |
|---|---|---|---|
| Myosin, heavy chain 7 | MYH7 | 193 | 32,628 |
| Myosin binding protein C | MYBPC3 | 138 | 28,280 |
| Troponin T type 2 | TNNT2 | 33 | 25,673 |
| Troponin I type 3 | TNNI3 | 32 | 12,963 |
| Cysteine and glycine-rich protein 3 | CSRP3 | 12 | 27,024 |
| Tropomyosin 1, α | TPM1 | 11 | 36,274 |
| Myosin, light chain 2 | MYL2 | 10 | 16,758 |
| Actin | ACTC | 7 | 14,631 |
| Myosin, light chain 3 | MYL3 | 5 | 12,617 |
| Protein kinase, AMP-activated, γ 2 | PRKAG2 | 4 | 328,114 |
| Phospholamban | PLN | 2 | 19,112 |
| Troponin C type 1 | TNNC1 | 1 | 9041 |
| Titin | TTN | 2 | 281,434 |
| Myosin, heavy chain 6 | MYH6 | 2 | 32,628 |
| Titin-cap | TCAP | 2 | 9361 |
| Caveolin 3 | CAV3 | 1 | 20,199 |
| Totals | 455 | 906,737 |
For OMIM accession numbers and loci of genes, see Fokstuen et al,62 from which this gene list was derived.
Amplicons were gel purified, and an equimolar amplicon pool was generated, divided, and used to make Roche 454 and Illumina Genome Analyzer libraries, respectively. The two libraries were sequenced on their respective instruments, using either a full picotiter plate (Roche 454 GS-FLX) or a single-flow cell lane (Illumina Genome Analyzer). The average read length for the Roche 454 GS FLX run was 235 bases, whereas the Illumina Genome Analyzer was programmed to generate 36 base length reads. The number and percentage of reads aligning to the HCM reference genes for the Roche 454 GS FLX and Illumina Genome Analyzer were 265,155 (94.1%) and 7,081,505 (89.9%), respectively. Figure 2A shows a coverage plot spanning a portion of the myosin heavy chain 7 (MYH7) gene sequenced from a control individual with no known cardiac abnormality. For the entire gene set, average coverage depths for the GS-FLX and Genome Analyzer were 186 and 782, respectively. The pattern of variable coverage, with peaks and valleys, is typical of NGS data and emphasizes the need to have sufficient sequencing depth.
Figure 2.
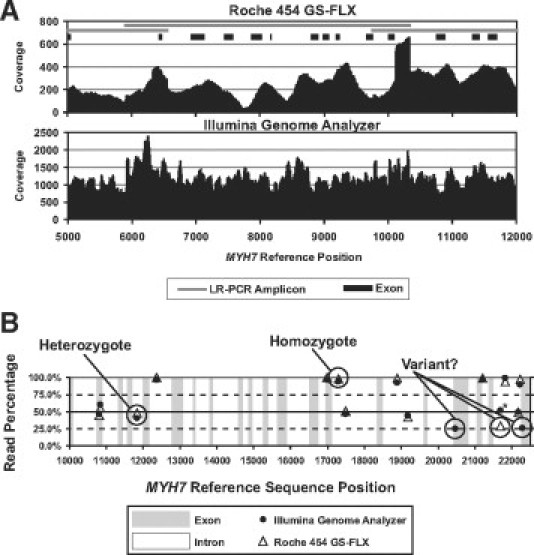
A: Coverage plot of a region of the MYH7 gene with comparison of Roche 454 GS FLX (top panel) and Illumina Genome Analyzer (bottom panel) sequence read data. Coverage is shown on the y axis and MYH7 reference sequence position on the x axis. Overlapping long-range PCR amplicons and exon positions are shown. Note difference in coverage scales between platforms. B: Variant read percentage plot of a region of the MYH7 gene showing variants identified with the Roche 454 GS FLX and Illumina Genome Analyzer platforms. Variant read percentage is shown on the y axis and MYH7 reference sequence position on the x axis. Exons and introns are indicated. Labeled as heterozygote and homozygote are two platform-concordant Sanger-confirmed variants with read percentages consistent with heterozygosity and homozygosity, respectively. Also circled are three variants with read percentages of approximately 25%. Of these three variants, the two identified only with the Illumina Genome Analyzer were determined to be false positives by Sanger sequencing. The third variant, with a read percentage of approximately 25% with the Roche 454 GS FLX, was determined to be a true variant. The complement of this variant is present in the Illumina Genome Analyzer data at a read percentage of approximately 50% (black circle with asterisk).
In Figure 2B, a variant plot for a region of MYH7 is shown. Allelic read percentages are plotted against MYH7 reference sequence position. Circled are two examples of platform concordant variants with allelic read percentages consistent with heterozygosity and homozygosity, respectively. These variants were confirmed by Sanger sequencing. Also shown are three variants with allelic read percentages of approximately 25%, raising the question of whether or not they represent true variants. The two questionable variants, identified in the Illumina Genome Analyzer data only, were determined to be false positives by Sanger sequencing. The other questionable variant, circled for the Roche 454 GS FLX data, was determined to be a true variant, and its complement in the Illumina Genome Analyzer data is present at approximately 50% read percentage.
For variant identification in this control individual sample, we required each variant to have a coverage of 30-fold or greater and an allelic read percentage of 20% or greater. Using this read percentage was a permissive approach in which we expected to encounter false positives. When we applied these criteria to identify exon variants, we delineated 27 platform concordant exon variants. Each of the 27 concordant variants was confirmed by Sanger sequencing and an example of a concordant variant in the actin (ACTC1) gene is shown in Figure 3A. We were able to identify an additional five platform-concordant Sanger-confirmed variants when we reduced our coverage criteria to 5-fold. Of the 32 confirmed exon variants, heterozygous allelic read percentages ranged from 33 to 86% and 21 to 67%, respectively, with the GS FLX and Genome Analyzer platforms, whereas homozygous variant allelic read percentages were either 100% or ranged from 80 to 94%, respectively. Of the 32 Sanger confirmed exon variants, 18 were in coding regions with 16 synonymous and two nonsynonymous. The other 14 variants were located in untranslated regions.
Figure 3.
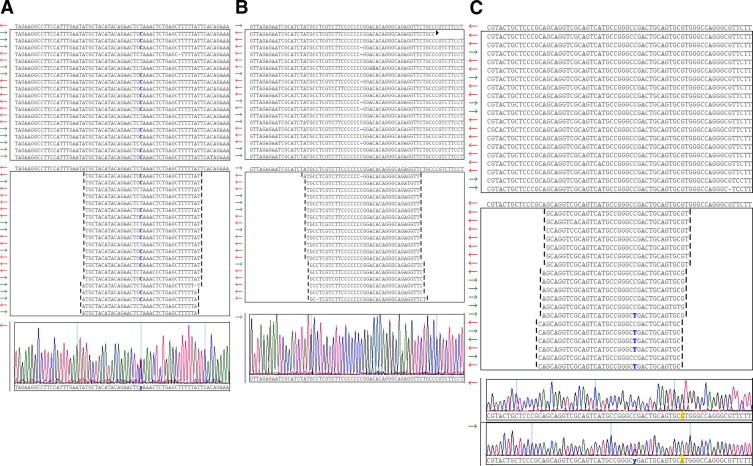
Comparison examples of Roche 454 GS FLX and Illumina Genome Analyzer next generation sequencing results for hypertrophic cardiomyoapathy associated genes. A: Concordant heterozygous variant g.5871488T>C in ACTC1. Roche 454 GS FLX and Illumina Genome Analyzer sequence read data were aligned and viewed in SeqMan NGen version 1.2 software (DNAstar, Madison, WI). Screen shots are shown. Top: GS FLX data showing 19 of 84 reads with the ACTC1 reference sequence shown above the panel box and the variant base designated in blue. Variant read percentage for 84 reads is 50%. Middle: Genome Analyzer data showing 21 of 499 reads and variant base in blue. Variant read percentage for 499 reads is 51.7%. Bottom: Confirmatory Sanger electropherogram. B: Discordant exon variant g.11968221delC in PRKAG2 due to GS FLX homopolymer sequencing error. Roche 454 GS FLX and Illumina Genome Analyzer sequence read data were aligned and viewed in SeqMan NGen version 1.2 software (DNAstar, Madison, WI). Screen shots are shown. Top: GS FLX data showing 19 of 37 reads with the PRKAG2 reference sequence, containing a 7-base polyC tract, shown above the panel box. The g.11968221delC variant in the 7-base poly C tract is present in 32.4% of 37 reads and designated with a dash mark. Middle: Genome Analyzer data showing 19 of 279 reads. The g.11968221delC variant is present in 1.8% of 278 reads. Bottom: Confirmatory Sanger electropherogram. C: Discordant exon variant g.4892783C>T in MYH7 due to misalignment of Genome Analyzer 36-base length reads generated from MYH6 in pooled amplicon library. Roche 454 GS FLX and Illumina Genome Analyzer sequence read data were aligned and viewed in SeqMan NGen version 1.2 software (DNAstar, Madison, WI). Screen shots are shown. Top: GS FLX data showing 19 of 39 reads with the MYH7 reference sequence shown above the panel box. All 39 reads show reference C at position g.4892783. Middle: Genome Analyzer data showing 19 of 293 reads and the variant base designated in blue. Variant read percentage for 293 reads is 21.8%. Bottom: Confirmatory Sanger electropherograms for MYH7 (upper trace) and MYH6 (lower trace) showing variant g.4858071C>T. In this region, MYH7 and MYH6 are 100% identical over 59 bases in the respective reference sequences. Genome Analyzer reads containing the MYH6 variant g.4858071C>T cross aligned to the MHY7 reference sequence in this region resulting in the discordant variant. The longer GS-FLX reads extend beyond this local region of MYH6 and MYH7 homology and avoided misalignment. An additional feature, highlighted in yellow, is a single G/A base difference between MYH6 and MYH7 downstream from the discordant variant. MYH6 Genome Analyzer reads overlying this G/A base difference aligned to MYH6 and not to MYH7.
Discordant exon variant results were observed between the two platforms and segregated into two categories. First, we observed 10 errors in the Roche 454 GS FLX data secondary to homopolymer sequencing errors with an example in the AMP activated protein kinase gene (PRKAG2) shown in Figure 3B. The homopolymer errors were due to either single base deletions or insertions and exhibited allelic read percentages ranging from 20 to 47%. Second, we observed two errors in the Genome Analyzer data that resulted from sequence read misalignments between the closely related MYH7 and MYH6 genes. The high degree of cross homology between these two genes resulted in a skewing of read matching during alignment with an example shown in Figure 3C. Together, these discordants, due to homopolymer and misalignment errors, represent “false positives” in exons and highlight the value of confirmatory Sanger sequencing at this stage of NGS technology evolution. In contrast, no NGS platform false negatives were observed in a total of 23,016 bases that were Sanger sequenced during confirmatory studies. This represents a sampling of 7.2% of the total unique bases that were NGS sequenced.
The highlights of this case study (for full details see Dames et al,65) illustrate the potential of NGS for targeted resequencing of multiple genes implicated in HCM. However, this study also shows important caveats that need to be addressed. Homopolymer errors with the Roche 454 technology have been earlier reported. Roche 454 reports that modifications including metal coating of picotiter well walls to prevent well-to-well cross talk and enhance well specific signals, finer control of pulsed reagent flow and software algorithm adjustments have increased homopolymer accuracy. The misalignment errors we observed with the Genome Analyzer 36 base length reads correlate with the high degree of homology between MYH7 and MYH6. A future approach to mitigate this error type will include longer read lengths (now commercially available) coupled with pair end sequencing. In preliminary experiments with 76 base length Genome Analyzer reads, we have observed improvements in alignments and a reduced range of heterozygous and homozygous allelic read percentages.
Conclusion
Next generation sequencing has fundamentally impacted biomedical research and is poised to begin a transition into the clinical diagnostic space. NGS is still relatively early its evolution, it is fast-paced, and new developments are expected for the foreseeable future. Continued refinement of sequencing chemistries should translate to improved sequencing accuracy, thereby reducing the need for Sanger confirmation. With each year, technical complexity is being reduced through platform improvements and automation. A growing number of enrichment methods and platforms offer options for specific applications from targeted resequencing to whole genome sequencing. Bioinformatic skills are critical for successful analysis and interpretation of NGS data, and this is an area that will present a significant challenge to the diagnostic laboratory. As sequencing throughput increases, the need for more facile approaches to data analysis, variant identification, and correlation with phenotype will become paramount. Balancing the challenges of adopting NGS into the diagnostic laboratory is the potential to use this powerful technology to offer complex genetic diagnostic testing in a more comprehensive manner.
Footnotes
Supported by ARUP Laboratories Institute for Clinical and Experimental Pathology.
S.D. and J.D.D. contributed equally to this study.
This article is partly based on material presented by the authors at the William Beaumont Hospital 18th Annual Symposium on Molecular Pathology: Clinical Applications of Genomic Medicine, which took place September 23–24, 2009, in Troy, MI.
CME Disclosure: None of the authors disclosed any relevant financial relationships.
References
- 1.Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 2.Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 3.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 4.Yeager M, Xiao N, Hayes RB, Bouffard P, Desany B, Burdett L, Orr N, Matthews C, Qi L, Crenshaw A, Markovic Z, Fredrikson KM, Jacobs KB, Amundadottir L, Jarvie TP, Hunter DJ, Hoover R, Thomas G, Harkins TT, Chanock SJ. Comprehensive resequence analysis of a 136 kb region of human chromosome 8q24 associated with prostate and colon cancers. Hum Genet. 2008;124:161–170. doi: 10.1007/s00439-008-0535-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res. 2007;17:1195–1201. doi: 10.1101/gr.6468307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urich T, Lanzen A, Qi J, Huson DH, Schleper C, Schuster SC. Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE. 2008;3:e2527. doi: 10.1371/journal.pone.0002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC, ten Cate JM, Crielaard W. Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res. 2008;87:1016–1020. doi: 10.1177/154405910808701104. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beck S, Rakyan VK. The methylome: approaches for global DNA methylation profiling. Trends Genet. 2008;24:231–237. doi: 10.1016/j.tig.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2008;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKernan KJ, Peckham HE, Costa GL, McLaughlin SF, Fu Y, Tsung EF, Clouser CR, Duncan C, Ichikawa JK, Lee CC, Zhang Z, Ranade SS, Dimalanta ET, Hyland FC, Sokolsky TD, Zhang L, Sheridan A, Fu H, Hendrickson CL, Li B, Kotler L, Stuart JR, Malek JA, Manning JM, Antipova AA, Perez DS, Moore MP, Hayashibara KC, Lyons MR, Beaudoin RE, Coleman BE, Laptewicz MW, Sannicandro AE, Rhodes MD, Gottimukkala RK, Yang S, Bafna V, Bashir A, MacBride A, Alkan C, Kidd JM, Eichler EE, Reese MG, De La Vega FM, Blanchard AP. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, Gomes X, Tartaro K, Niazi F, Turcotte CL, Irzyk GP, Lupski JR, Chinault C, Song XZ, Liu Y, Yuan Y, Nazareth L, Qin X, Muzny DM, Margulies M, Weinstock GM, Gibbs RA, Rothberg JM. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 15.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara ECM, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LJ, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM, Hoschler K, Hurwitz S, Ivanov DV, Johnson MQ, James T, Huw Jones TA, Kang GD, Kerelska TH, Kersey AD, Khrebtukova I, Kindwall AP, Kingsbury Z, Kokko-Gonzales PI, Kumar A, Laurent MA, Lawley CT, Lee SE, Lee X, Liao AK, Loch JA, Lok M, Luo S, Mammen RM, Martin JW, McCauley PG, McNitt P, Mehta P, Moon KW, Mullens JW, Newington T, Ning Z, Ling Ng B, Novo SM, O'Neill MJ, Osborne MA, Osnowski A, Ostadan O, Paraschos LL, Pickering L, Pike AC, Chris Pinkard D, Pliskin DP, Podhasky J, Quijano VJ, Raczy C, Rae VH, Rawlings SR, Chiva Rodriguez A, Roe PM, Rogers J, Rogert Bacigalupo MC, Romanov N, Romieu A, Roth RK, Rourke NJ, Ruediger ST, Rusman E, Sanches-Kuiper RM, Schenker MR, Seoane JM, Shaw RJ, Shiver MK, Short SW, Sizto NL, Sluis JP, Smith MA, Ernest Sohna Sohna J, Spence EJ, Stevens K, Sutton N, Szajkowski L, Tregidgo CL, Turcatti G, Vandevondele S, Verhovsky Y, Virk SM, Wakelin S, Walcott GC, Wang J, Worsley GJ, Yan J, Yau L, Zuerlein M, Mullikin JC, Hurles ME, McCooke NJ, West JS, Oaks FL, Lundberg PL, Klenerman D, Durbin R, Smith AJ. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, Swerdlow H, Turner DJ. A large genome center's improvements to the Illumina sequencing system. Nat Methods. 2008;5:1005–1010. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milos PM. Emergence of single-molecule sequencing and potential for molecular diagnostic applications. Expert Rev Mol Diagn. 2009;9:659–666. doi: 10.1586/erm.09.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ozsolak F, Platt AR, Jones DR, Reifenberger JG, Sass LE, McInerney P, Thompson JF, Bowers J, Jarosz M, Milos PM. Direct RNA sequencing. Nature. 2009;461:814–818. doi: 10.1038/nature08390. [DOI] [PubMed] [Google Scholar]
- 20.Goren A, Ozsolak F, Shoresh N, Ku M, Adli M, Hart C, Gymrek M, Zuk O, Regev A, Milos PM, Bernstein BE. Chromatin profiling by directly sequencing small quantities of immunoprecipitated DNA. Nat Methods. 2010;7:47–49. doi: 10.1038/nmeth.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris TD, Buzby PR, Babcock H, Beer E, Bowers J, Braslavsky I, Causey M, Colonell J, Dimeo J, Efcavitch JW, Giladi E, Gill J, Healy J, Jarosz M, Lapen D, Moulton K, Quake SR, Steinmann K, Thayer E, Tyurina A, Ward R, Weiss H, Xie Z. Single-molecule DNA sequencing of a viral genome. Science. 2008;320:106–109. doi: 10.1126/science.1150427. [DOI] [PubMed] [Google Scholar]
- 22.Korlach J, Bibillo A, Wegener J, Peluso P, Pham TT, Park I, Clark S, Otto GA, Turner SW. Long, processive enzymatic DNA synthesis using 100% dye-labeled terminal phosphate-linked nucleotides. Nucleosides Nucleotides Nucleic Acids. 2008;27:1072–1083. doi: 10.1080/15257770802260741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korlach J, Marks PJ, Cicero RL, Gray JJ, Murphy DL, Roitman DB, Pham TT, Otto GA, Foquet M, Turner SW. Selective aluminum passivation for targeted immobilization of single DNA polymerase molecules in zero-mode waveguide nanostructures. Proc Natl Acad Sci USA. 2008;105:1176–1181. doi: 10.1073/pnas.0710982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S. Real-time DNA sequencing from single polymerase molecules. Science. 2008;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 25.Fuller CW, Middendorf LR, Benner SA, Church GM, Harris T, Huang X, Jovanovich SB, Nelson JR, Schloss JA, Schwartz DC, Vezenov DV. The challenges of sequencing by synthesis. Nature Biotechnol. 2009;27:1013–1023. doi: 10.1038/nbt.1585. [DOI] [PubMed] [Google Scholar]
- 26.Tucker T, Marra M, Friedman JM. Massively parallel sequencing: the next big thing in genetic medicine. Am J Hum Genet. 2009;85:142–154. doi: 10.1016/j.ajhg.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voelkerding KV, Dames SA, Durtschi JD. Next-generation sequencing: from basic research to diagnostics. Clin Chem. 2009;55:641–658. doi: 10.1373/clinchem.2008.112789. [DOI] [PubMed] [Google Scholar]
- 28.Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19:1124–1132. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harismendy O, Ng PC, Strausberg RL, Wang X, Stockwell TB, Beeson KY, Schork NJ, Murray SS, Topol EJ, Levy S, Frazer KA. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith DR, Quinlan AR, Peckham HE, Makowsky K, Tao W, Woolf B, Shen L, Donahue WF, Tusneem N, Stromberg MP, Stewart DA, Zhang L, Ranade SS, Warner JB, Lee CC, Coleman BE, Zhang Z, McLaughlin SF, Malek JA, Sorenson JM, Blanchard AP, Chapman J, Hillman D, Chen F, Rokhsar DS, McKernan KJ, Jeffries TW, Marth GT, Richardson PM. Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res. 2008;18:1638–1642. doi: 10.1101/gr.077776.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flicek P, Birney E. Sense from sequence reads: methods for alignment and assembly. Nat Methods. 2009;6:S6–S12. doi: 10.1038/nmeth.1376. [DOI] [PubMed] [Google Scholar]
- 32.Horner DS, Pavesi G, Castrignano T, De Meo PD, Liuni S, Sammeth M, Picardi E, Pesole G. Bioinformatics approaches for genomics and post genomics applications of next-generation sequencing. Brief Bioinform. 2009;11:181–197. doi: 10.1093/bib/bbp046. [DOI] [PubMed] [Google Scholar]
- 33.Vasta V, Ng SB, Turner EH, Shendure J, Hahn SH. Next generation sequence analysis for mitochondrial disorders. Genome Med. 2009;1:100–110. doi: 10.1186/gm100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen Y, Wu BL, Gusella JF. Large-scale medical resequencing for x-linked mental retardation. Clin Chem. 2010;56:339–341. doi: 10.1373/clinchem.2009.135020. [DOI] [PubMed] [Google Scholar]
- 35.Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, O'Meara S, Latimer C, Dicks E, Menzies A, Stephens P, Blow M, Greenman C, Xue Y, Tyler-Smith C, Thompson D, Gray K, Andrews J, Barthorpe S, Buck G, Cole J, Dunmore R, Jones D, Maddison M, Mironenko T, Turner R, Turrell K, Varian J, West S, Widaa S, Wray P, Teague J, Butler A, Jenkinson A, Jia M, Richardson D, Shepherd R, Wooster R, Tejada MI, Martinez F, Carvill G, Goliath R, de Brouwer AP, van Bokhoven H, Van Esch H, Chelly J, Raynaud M, Ropers HH, Abidi FE, Srivastava AK, Cox J, Luo Y, Mallya U, Moon J, Parnau J, Mohammed S, Tolmie JL, Shoubridge C, Corbett M, Gardner A, Haan E, Rujirabanjerd S, Shaw M, Vandeleur L, Fullston T, Easton DF, Boyle J, Partington M, Hackett A, Field M, Skinner C, Stevenson RE, Bobrow M, Turner G, Schwartz CE, Gecz J, Raymond FL, Futreal PA, Stratton MR. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, Howard E, Shendure J, Turner DJ. Target-enrichment strategies for next-generation sequencing. Nat Methods. 2010;7:111–118. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- 37.Garber K. Fixing the front end. Nature Biotechnol. 2008;26:1101–1104. doi: 10.1038/nbt1008-1101. [DOI] [PubMed] [Google Scholar]
- 38.Binladen J, Gilbert MT, Bollback JP, Panitz F, Bendixen C, Nielsen R, Willerslev E. The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS ONE. 2007;2:e197. doi: 10.1371/journal.pone.0000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Craig DW, Pearson JV, Szelinger S, Sekar A, Redman M, Corneveaux JJ, Pawlowski TL, Laub T, Nunn G, Stephan DA, Homer N, Huentelman MJ. Identification of genetic variants using bar-coded multiplexed sequencing. Nat Methods. 2008;5:887–893. doi: 10.1038/nmeth.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stiller M, Knapp M, Stenzel U, Hofreiter M, Meyer M. Direct multiplex sequencing (DMPS)–a novel method for targeted high-throughput sequencing of ancient and highly degraded DNA. Genome Res. 2009;19:1843–1848. doi: 10.1101/gr.095760.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirkness EF. Targeted sequencing with microfluidics. Nature Biotechnol. 2009;27:998–999. doi: 10.1038/nbt1109-998. [DOI] [PubMed] [Google Scholar]
- 42.Tewhey R, Warner JB, Nakano M, Libby B, Medkova M, David PH, Kotsopoulos SK, Samuels ML, Hutchison JB, Larson JW, Topol EJ, Weiner MP, Harismendy O, Olson J, Link DR, Frazer KA. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nature Biotechnol. 2009;27:1025–1031. doi: 10.1038/nbt.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dahl F, Gullberg M, Stenberg J, Landegren U, Nilsson M. Multiplex amplification enabled by selective circularization of large sets of genomic DNA fragments. Nucleic Acids Res. 2005;33:e71. doi: 10.1093/nar/gni070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li JB, Gao Y, Aach J, Zhang K, Kryukov GV, Xie B, Ahlford A, Yoon JK, Rosenbaum AM, Zaranek AW, LeProust E, Sunyaev SR, Church GM. Multiplex padlock targeted sequencing reveals human hypermutable CpG variations. Genome Res. 2009;19:1606–1615. doi: 10.1101/gr.092213.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dahl F, Stenberg J, Fredriksson S, Welch K, Zhang M, Nilsson M, Bicknell D, Bodmer WF, Davis RW, Ji H. Multigene amplification and massively parallel sequencing for cancer mutation discovery. Proc Natl Acad Sci USA. 2007;104:9387–9392. doi: 10.1073/pnas.0702165104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Summerer D. Enabling technologies of genomic-scale sequence enrichment for targeted high-throughput sequencing. Genomics. 2009;94:363–368. doi: 10.1016/j.ygeno.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 47.Summerer D, Wu H, Haase B, Cheng Y, Schracke N, Stahler CF, Chee MS, Stahler PF, Beier M. Microarray-based multicycle-enrichment of genomic subsets for targeted next-generation sequencing. Genome Res. 2009;19:1616–1621. doi: 10.1101/gr.091942.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bau S, Schracke N, Kranzle M, Wu H, Stahler PF, Hoheisel JD, Beier M, Summerer D. Targeted next-generation sequencing by specific capture of multiple genomic loci using low-volume microfluidic DNA arrays. Anal Bioanal Chem. 2009;393:171–175. doi: 10.1007/s00216-008-2460-7. [DOI] [PubMed] [Google Scholar]
- 49.Hodges E, Xuan Z, Balija V, Kramer M, Molla MN, Smith SW, Middle CM, Rodesch MJ, Albert TJ, Hannon GJ, McCombie WR. Genome-wide in situ exon capture for selective resequencing. Nat Genet. 2007;39:1522–1527. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- 50.Porreca GJ, Zhang K, Li JB, Xie B, Austin D, Vassallo SL, LeProust EM, Peck BJ, Emig CJ, Dahl F, Gao Y, Church GM, Shendure J. Multiplex amplification of large sets of human exons. Nat Methods. 2007;4:931–936. doi: 10.1038/nmeth1110. [DOI] [PubMed] [Google Scholar]
- 51.Albert TJ, Molla MN, Muzny DM, Nazareth L, Wheeler D, Song X, Richmond TA, Middle CM, Rodesch MJ, Packard CJ, Weinstock GM, Gibbs RA. Direct selection of human genomic loci by microarray hybridization. Nat Methods. 2007;4:903–905. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- 52.Okou DT, Steinberg KM, Middle C, Cutler DJ, Albert TJ, Zwick ME. Microarray-based genomic selection for high-throughput resequencing. Nat Methods. 2007;4:907–909. doi: 10.1038/nmeth1109. [DOI] [PubMed] [Google Scholar]
- 53.Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, Gabriel S, Jaffe DB, Lander ES, Nusbaum C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:201–211. doi: 10.1016/j.jacc.2009.02.075. [DOI] [PubMed] [Google Scholar]
- 55.Soor GS, Luk A, Ahn E, Abraham JR, Woo A, Ralph-Edwards A, Butany J. Hypertrophic cardiomyopathy: current understanding and treatment objectives. J Clin Pathol. 2009;62:226–235. doi: 10.1136/jcp.2008.061655. [DOI] [PubMed] [Google Scholar]
- 56.Taylor MR, Carniel E, Mestroni L. Familial hypertrophic cardiomyopathy: clinical features, molecular genetics and molecular genetic testing. Expert Rev Mol Diagn. 2004;4:99–113. doi: 10.1586/14737159.4.1.99. [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez JE, McCudden CR, Willis MS. Familial hypertrophic cardiomyopathy: basic concepts and future molecular diagnostics. Clin Biochem. 2009;42:755–765. doi: 10.1016/j.clinbiochem.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 58.Kaski JP, Syrris P, Esteban MT, Jenkins S, Pantazis A, Deanfield JE, McKenna WJ, Elliott PM. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:436–441. doi: 10.1161/CIRCGENETICS.108.821314. [DOI] [PubMed] [Google Scholar]
- 59.Andersen PS, Havndrup O, Hougs L, Sorensen KM, Jensen M, Larsen LA, Hedley P, Thomsen AR, Moolman-Smook J, Christiansen M, Bundgaard H. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat. 2009;30:363–370. doi: 10.1002/humu.20862. [DOI] [PubMed] [Google Scholar]
- 60.Robin NH, Tabereaux PB, Benza R, Korf BR. Genetic testing in cardiovascular disease. J Am Coll Cardiol. 2007;50:727–737. doi: 10.1016/j.jacc.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 61.Ackerman MJ. Genetic testing for risk stratification in hypertrophic cardiomyopathy and long QT syndrome: fact or fiction? Curr Opin Cardiol. 2005;20:175–181. doi: 10.1097/01.hco.0000163668.44141.89. [DOI] [PubMed] [Google Scholar]
- 62.Fokstuen S, Lyle R, Munoz A, Gehrig C, Lerch R, Perrot A, Osterziel KJ, Geier C, Beghetti M, Mach F, Sztajzel J, Sigwart U, Antonarakis SE, Blouin JL. A DNA resequencing array for pathogenic mutation detection in hypertrophic cardiomyopathy. Hum Mutat. 2008;29:879–885. doi: 10.1002/humu.20749. [DOI] [PubMed] [Google Scholar]
- 63.Waldmuller S, Muller M, Rackebrandt K, Binner P, Poths S, Bonin M, Scheffold T. Array-based resequencing assay for mutations causing hypertrophic cardiomyopathy. Clin Chem. 2008;54:682–687. doi: 10.1373/clinchem.2007.099119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Judge DP. Use of genetics in the clinical evaluation of cardiomyopathy. JAMA. 2009;302:2471–2476. doi: 10.1001/jama.2009.1787. [DOI] [PubMed] [Google Scholar]
- 65.Dames S, Durtschi J, Geiersbach K, Stephens J, Voelkerding KV. J Biomol Tech. 2010;21:73–80. [PMC free article] [PubMed] [Google Scholar]