

Abstract
The majority of colorectal cancer (CRC) cases have chromosomal instability, in which the tumor genome is characterized by gross chromosomal aberrations such as gains in 20q, 13q, 8q, and 7, and losses in 4, 8p, 18q, and 17p. These somatic copy number changes (gains, losses, and somatic uniparental disomies) are crucial to CRC progression as they drive genes toward cancer-promoting (oncogenic or tumor suppressive) states. Numerous studies have shown that the loss of 18q or 8p is associated with poorer clinical outcome in CRCs. Either chromosomal arm may contain a tumor suppressor gene (or genes), whose deactivation by copy loss (loss of wild-type allele, decreased expression) can be crucial to the later stages of cancer progression. Our own integrated genomic analysis (single nucleotide polymorphism array, expression array) of more than 200 CRC tumor and normal samples indicates that the overall down-regulation of genes within the 8p or 18q arm is associated with lower survival rate. Among the often down-regulated, poor prognosis-associated 8p genes is MTUS1, whose gene product (a mitotic spindle-associated protein) was recently demonstrated to have a tumor suppressive property. Within 18q is ATP5A1, which codes for the catalytic a component of mitochondrial H+-ATP synthase. Like SMAD4 (also in 18q), the decreased expression of ATP5A1 appears to be a marker of unfavorable clinical outcome in CRCs.
Colorectal cancer (CRC) is generally associated with more affluent nations. However, recent statistics indicate that the incidence rate of CRC in the developing world from the 1980s until the first few years of this millennium has increased significantly,1 and the disease is turning into a major global health care burden.2 The main culprit may be the growing popularity of Western diet (ie, high in proteins and fats, low in fibers and vegetables) across the globe. In the United States, more than 150,000 people are diagnosed with the disease each year, resulting in over 50,000 deaths.3 In addition to environmental factors (ie, diet, cigarette smoking, and alcohol consumption), genetics can also contribute to CRC incidence. Fewer than 10% of CRC cases may arise from highly-penetrant inherited mutations (eg, mutations in the APC gene for familial adenomatous polyposis or FAP cases and mutations in mismatch repair genes for hereditary nonpolyposis colorectal cancer or HNPCC cases).4 Results from recent genome-wide association studies also indicate that even spontaneous cases (about 70% of CRC cases) may also be influenced by low penetrant predisposition single nucleotide polymorphisms (SNPs).5
CRC cases have two major types of genomic instability. Most CRCs (including FAP cases) are characterized by gross chromosomal aberrations, such as losses or gains of whole or partial chromosomes or chromosomal arms, and are therefore classified as chromosomal instability (CIN) tumors.6 The rest (∼16%7) are classified as microsatellite instability (MIN). MIN tumors are mainly diploid and have a mutator phenotype because of defective mismatch repair. The defective mismatch repair (which may be due to an inherited mutation or promoter hypermethylation of an mismatch repair gene)8,9 results in replication errors within the microsatellite markers. This distinction between MIN and CIN tumors actually has clinical implications since the former is usually associated with better prognosis.10,11
Analytical Tools Used To Detect Somatic Copy Number Aberrations in Cancer (Including CRCs)
The field of cancer cytogenetics may have started when Theodor Boveri and David Hansemann published their seminal works between the late nineteenth and early twentieth centuries.12 Essentially, these two pioneers hypothesized a link between carcinogenesis and abnormal constitution of the chromosomes. The discovery of correct number of human chromosomes by Tjio and Levan in 195613 was shortly followed by karyotypic characterizations of various types of cancer. In the 1990s, fluorescence in situ hybridization became a widely used technique for detecting chromosomal aberrations (gains or losses at specific chromosomal regions) in tumor cells examined in either metaphase or interphase state.14 At around the same time, investigators started the routine use of PCR-based microsatellite marker analysis to identify loss of heterozygosity (LOH) in certain chromosomal regions.15 Comparative genomic hybridization (CGH) (wherein tumor and normal genomic DNAs are differentially labeled and co-hybridized onto a metaphase chromosome of normal karyotype) was also introduced in the early 1990s and became a popular technique in genome-wide chromosomal characterization of tumors.16 CGH was then further developed into an array format capable of detecting chromosomal aberrations at a much higher resolution (10-kb range).17 A very significant percentage of publications on cancer cytogenetics from the mid-1990s through the last decade were reports of data generated from CGH and CGH arrays (more than 3000 articles retrieved on entering the key words “cancer” and “comparative genomic hybridization” in a PubMed search).
Another important breakthrough in molecular cancer cytogenetics was the introduction of SNP arrays.18 Like CGH arrays, SNP arrays can be used for high-resolution (albeit more complicated) copy number analysis of cancer genomes. Because of its capability to read SNP copy number and genotype simultaneously, a SNP array can detect a chromosomal region with somatic uniparental disomy (UPD; copy loss LOH followed by the duplication of remaining chromosomal segment), now found to occur frequently in neoplastic transformation.19 In recent years, both CGH array (the oligonucleotide-based platform in particular) and SNP array have become the standard techniques for genome-wide characterization of chromosomal aberrations in various cancer types. Neither of these techniques require metaphase cells, and with their high probe densities (almost two million SNP plus copy number probe sets in the Affymetrix SNP Array 6), have the power to detect very narrow regions of deletions and gains (can be less than 10 kb in size). For the comprehensive comparisons of various cytogenetic techniques used in cancer research, see a recent review by Maciejewski et al.20
Somatic Copy Number Aberrations in CRCs
The genome-wide chromosomal scan of a typical CIN CRC is shown in Figure 1A (generated by SNP array analysis). The copy number changes in this tumor sample (C0114A) include losses in chromosomes 4, 8p, and 18, as well as gains in 7, 8q, 13q, and 20q21. Along with losses in 14q, 17p, and 15q, these are the most commonly occurring chromosomal aberrations in CRCs.22,23,24,25 In contrast, a typical MIN tumor (sample C0323A; Figure 1C is devoid of these chromosomal aberrations and remains diploid. Of all of the common somatic copy number changes in CRCs, it is the loss of 18q24,26,27,28,29,30,31,32,33,34,35,36,37,38,39 that is most clearly associated with poor prognosis (Table 1). This conclusion, which is derived from numerous studies that used varying techniques (microsatellite analysis, CGH, CGH array, SNP array, karyotyping), is actually consistent with the observation that 18q loss is also associated with distant metastasis.40 As indicated in Table 1,24,26 the loss of 8p24,34,35,36,37,38,39,41 or 17p (the location of TP53 tumor suppressor gene)33,34,42 is also linked to lower survival rate.
Figure 1.
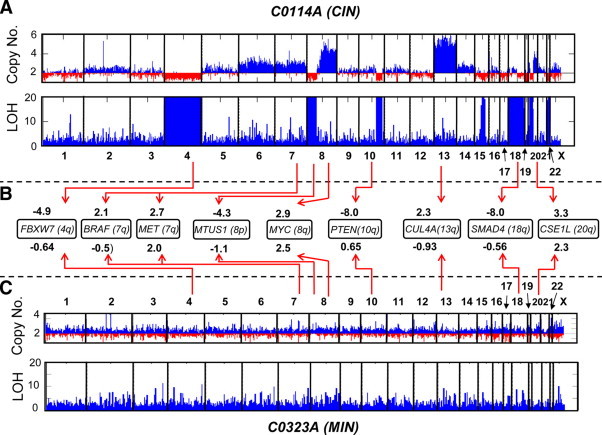
Cytogenetic characterization of CIN (primary tumor sample C0114A; A) and MIN (primary tumor sample C0323A; C) CRC genomes by Affymetrix Xba 240 50K SNP array analysis. As shown here, as well as previously,21 C0114A has acquired copy number losses in chromosomes 4, 22, 8p, and 20p, and gains in chromosomes 7, 8q, 13, and 20q. Also lost were significant portions of 18q and 10q. Unlike C0114A, C0323A did not have noticeable copy number aberrations. B is a comparison of the expression levels of some of the known oncogenes and tumor suppressor genes between the two tumor samples. For example, the expression level (z) of the tumor suppressor gene SMAD4 in C0114A and C0323A (relative to normal colon) are −8.0 and −0.56, respectively. This suggests that the loss of chromosome 18q contributed to C0114A's low mRNA level for SMAD4. Compared with C0323A, C0114A also exhibited lower expression levels for the tumor suppressor genes FBXW7, PTEN, and MTUS1, which are all located in chromosomal regions lost in C0114A. The oncogenes MYC, MET, BRAF, and CUL4A are all located in regions of gain in the C0114A genome. This may explain why C0114A has relatively higher expression levels (at varying degrees) of these genes. ; where It is the normalized, log transformed intensity value (I) of the Affymetrix U133A probe set for the tumor sample, while and σn are the average and SD (respectively) of the I values for 53 normal colon samples. For each gene represented by multiple probe sets (PTEN,CSE1L, MET,MTUS1, CUL4A, SMAD4), the z value indicated in B is actually the average for all probe sets representing the gene. Note: Each genome-wide scan includes a copy number chart (baseline copy number is 2) and LOH chart. High LOH values (for the charts, the LOH value is capped at 20), indicated by tall blue bars represent segments in the chromosome of contiguous homozygous SNPs. Regions of copy loss usually correspond to regions of high LOH.
Table 1.
Studies Which Demonstrated That Certain Chromosomal Aberrations Are Good Prognostic Markers in Colorectal Cancer (Arranged Chronologically)
| Year | Poorer prognosis correlated to | Total number of samples analyzed | Analysis by | Reference no. |
|---|---|---|---|---|
| 1994 | Loss in 18q | 145 (Stages II, III; no HNPCC) | MS markers (18q) | 26 |
| 1997 | Loss in 17p | 141 (had undergone liver resection) | MS markers (5q, 8p, 10q, 15q, 17p, 18p, and 18q) | 42 |
| 1998 | Loss in 18q | 151 (had undergone surgery) | MS markers (18q) | 27 |
| 1998 | Loss in 18q | 125 (no HNPCC) | MS markers (18q) | 28 |
| 1998 | Loss in 18q | 118 (Stages II, III; had undergone surgery) | MS markers (18q) | 29 |
| 1999 | Loss in 18q21 | 195 | MS markers (18q21) | 30 |
| 1999 | Loss in 8p | 508 CRC patients (Stages B2, C) | MS markers (5q, 8p, 15q, 17p, 18q) | 41 |
| 2001 | Losses in 18q, 14q, 8p, 4q, 1p; gain in 20q | 67 (Stages A, B, C, D) | CGH | 31 |
| 2001 | Loss in 18q | 460 (Stage II, III; treated with various combinations of fluorouracil and leucovorin) | MS markers (18q, 17p, and 8p) | 32 |
| 2002 | Losses in 18q, 17p | 228 | MS markers (18q, 17p, and 5q) | 33 |
| 2002 | Losses in 18q, 17p, 8p | 168 (Stage III) | MS markers (to 3p, 4p, 5q, 8p, 9p, 13q, 17p, and 18q) | 34 |
| 2002 | Losses in 18q, 8p | 180 | Digital SNP analysis (8p and 18q) | 35 |
| 2004 | Losses in 18q, 8p | 123 (Stage II, III; had undergone curative resection) | MS markers (18q, 8p, and 4p) | 36 |
| 2004 | Loss in 18q; aberration in 8 | 150 (Stage II, III) | Karyotyping | 37 |
| 2006 | Losses in 18q, 8p, 4p | 70 (had undergone surgery, most without chemotherapy) | Array CGH | 38 |
| 2007 | Losses in 18q12-qter, 8p12- pter; gain in 8q23 and 8q24 | 73 (Stage I, II, III, IV) | CGH | 39 |
| 2009 | Losses in 18q, 15q, 8p, 4p | 182 (Stage I, II, III, IV) | SNP array | 24 |
How Copy Number Aberrations Lead to Dysregulation of Genes Crucial to CRC Progression
Somatic chromosomal copy number changes can confer selective advantages to proliferating cancer cells because these aberrations lead to dysregulation of genes that are important in carcinogenesis (Figure 2, A and B).
Figure 2.
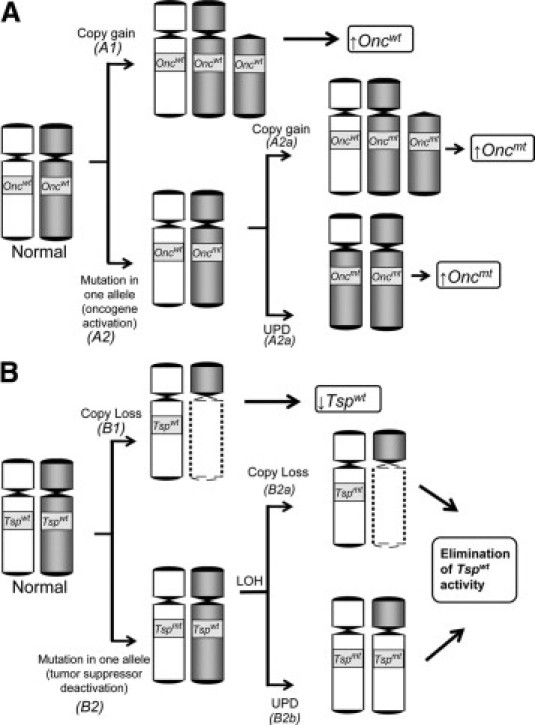
Copy number gain (A) or loss (B) contributes to the dysregulation of an oncogene (Onc) or tumor suppressor gene (Tsp) toward tumor promotion. wt, wild-type; mt, mutant; ↑, higher expression level; ↓, lower expression level.
Dysregulation of Genes in Regions of Chromosomal Gain
In theory, a copy number gain (either interstitial, partial arm, whole arm, or whole chromosome) will result in the elevated expression of its resident genes (Figure 2A). For this reason, we can imagine that within the regions of somatic copy gains are genes whose increased transcriptional level can contribute to the process of cancer progression (see Path A1 in Figure 2A). Examples include the proto-oncogenes MYC (transcription factor)43 and MET (receptor tyrosine kinase),44 located in the often gained 8q and 7q arms, respectively. Both these genes are often up-regulated in CRCs.45,46,47 The 13q and 20q chromosomal arms, both of which harbor a great percentage of genes with up-regulated expression,24,25 are also often gained in CRCs. In our CRC genome-wide expression analysis, one of the 13q genes that turned out to be very highly up-regulated was CUL4A, which has also been found to be copy number-dysregulated (ie, amplified in both copy number and expression) in breast cancer.48, CUL4A is as an important component of an ubiquitin ligase system involved in proteasomal degradation of the tumor suppressor protein p27KIP1.49 Within the often gained 20q arm is CSE1L (coding for a cellular apoptosis susceptibility protein), whose overexpression was recently shown to enhance the invasive potential of cancer cells.50 This is consistent with another recent report of direct correlation between CSE1L expression and lymphatic metastasis among CRC patients.51 In addition, we identified CRC samples with amplifications at narrow regions of chromosomes. One particular sample acquired an amplification (>4 copies) in the region of the 6p arm (which is not commonly gained in CRCs) covering the locus of the oncogene VEGF (unpublished data). Not surprisingly, this sample also registered one of the highest VEGF expression level among our CRC samples. The VEGF overexpression of this tumor sample may have been relevant clinically, since the protein is the direct target of bevacizumab, which in combination with the FOLFOX4 is already an FDA-approved regimen to treat metastatic CRCs.52
Certain CRC-associated proto-oncogenes such as KRAS,53 BRAF,54 and PIK3CA,55 usually need mutational activation (Path A2 in Figure 2A) to be oncogenic. For these aforementioned genes (KRAS is an effector molecule for both BRAF and PIK3CA56), monoallelic mutation without the accompanying copy number increase may suffice due to the dominant nature of the mutation.57 However, a recent report by Soh and colleagues57 demonstrated that a considerable percentage (6/60; 10%) of CRC tumor samples have acquired simultaneous KRAS mutations and copy gains. In our own analysis (unpublished results), we found five of 74 CRC tumor samples (7%) having simultaneous KRAS mutation and copy gain. In theory, the cancer- promoting activity of a mutation-activated oncogene can be further enhanced by gaining an additional copy of the mutated allele (Path A2a in Figure 2A). Alternatively, the oncogenic activity of a mutation-activated oncogene may also be elevated by somatic UPD (Path A2b in Figure 2A). In the same report, Soh and co-workers57 presented evidence that lung adenocarcinoma samples (those with SNP array and sequencing data) can acquire a KRAS mutation and UPD at the same time. It is important to note that the KRAS locus is located on the 12p arm, which rarely gains additional copies in CRCs; thus KRAS copy number gains may just be locus-specific (the authors used quantitative PCR for copy number detection).
Dysregulation of Genes in Regions of Chromosomal Loss (and Somatic UPDs)
By its simplest definition, a tumor suppressor gene codes for a protein that can derail tumor initiation or progression. The deactivation of a tumor suppressor gene is therefore crucial to carcinogenesis. Reduced expression, through copy loss, is one mechanism this can be achieved (Figure 2B, Path B1). For instance, the loss of the 18q arm can result in down-regulation (as exemplified in sample C0114A; see Figure 1B) and subsequent deactivation of the tumor suppressor gene SMAD4 (18q 21.1). An important component of the transforming growth factor-β signaling pathway, SMAD4 forms a complex with phosphorylated SMAD2 and SMAD3. The complex then translocates to the nucleus58 to promote the transcription of genes involved in growth inhibition, such as the cyclin-dependent kinase inhibitors P15INK4b (CDKN2B) and P21CIP1(CDKN1A).59 The SMAD complex may also be involved in the transcriptional regulation of genes that are relevant to cell invasion and metastasis, including MMP9 (matrix metalloproteinase-9),60 which is consistent with SMAD4 down-regulation having been associated with CRC metastasis61 and its poorer clinical outcome.62 Also within the 18q arm is the gene DCC (coding for a netrin-1 receptor), initially identified as a tumor suppressor gene63 and for a long time was considered the primary reason why 1oss of 18q correlates to poor prognosis in CRC.64 Recently, DCC has been shown to induce apoptosis conditionally (ie, when it is not engaged by its ligand Netrin-1) and thus may have some tumor suppressor functionality.65 The analysis of our CRC expression data showed that another frequently down-regulated 18q gene is ATP5A1, which codes for a protein that forms the catalytic subunit of mitochondrial H+-ATP synthase.66 Several groups have described CRC samples exhibiting reduced activity of this enzyme, presumably leading to mitochondrial dysfunction67,68 and thus an elevated rate of glycolysis, which according to Warburg's hypothesis characterizes tumor cells.69 Another study has shown that CRC's resistance to 5-flurouracil (the standard chemotherapeutic drug against CRC) correlated with down-regulation of mitochondrial H+-ATP synthase.70
Chromosome 4, the location of the tumor suppressor gene FBXW7 (or CDC4), which has been shown to be down-regulated in CRCs, is also often lost in CRCs.71 FBXW7 mediates the proteolysis of several oncoproteins and thus may have important role in cancer progression.72 Another recurrent CRC chromosomal aberration is the loss of 17p. This chromosomal arm includes the locus for MAP2K4, a gene with a proven role in metastasis suppression through the p38 and JNK pathways.73 It is not surprising then that our own analysis revealed that MAP2K4 is one of the most highly down-regulated genes within the frequently lost 17p arm. Among the genes we found to be underexpressed in CRCs because of 8p loss is MTUS1 (the down-regulation of this gene in CRCs has also been reported by another group74). MTUS1 codes for a mitotic spindle-associated protein shown to be involved in the eventual reduction of mitotic rate.75
Somatic copy number losses may also occur in narrower regions (may be as narrow as the gene locus) of the chromosomes. We have identified several cases with small deletions in a 10q region, which includes the PTEN locus. The tumor suppressor function of PTEN, a negative regulator of PI3K in the AKT signaling pathway, is well described in the literature.76 Another tumor suppressor gene that is found to exhibit locus-specific LOH is KLF6 (Kruppel-like factor 6) within 10p15 region.77 The transcription factor KLF6 is frequently underexpressed in CRCs.78
In essence, LOH results in the reduced expression of a fully functioning (wild-type) tumor suppressor gene product. For many tumor suppressor genes, however, complete deactivation can be achieved if LOH is preceded by a mutation in one allele (see Path B2 in Figure 2B). This type of gene dysregulation can be observed in the tumor suppressor genes APC and TP53,79 which are among the two most highly mutated genes in CRCs.80 In such cases, LOH is the “second hit” to an earlier somatic mutation (“first hit”) in a manner that is consistent with Knudson's hypothesis.81 In CRCs, the loss of a TP53 allele can result from the loss of the entire 17p arm. Loss of the 5q arm (where APC locus is located) also occurs in CRC samples, but not as common as the losses of 18q, 8p, or 17p.22 As shown in Path B2b (in Figure 2B), LOH may also be acquired through somatic UPD. With the use of SNP arrays, we have identified a case in which the mutated APC gene had acquired a somatic UPD.19 Aside from TP53 and APC, two other tumor suppressor genes located in regions of frequent loss and found to be mutated in CRCs are SMAD4 (18q) and FBXW7 (4q).80,82,83
Integrated Analyses of SNP Array, Expression Array, and Clinical Data Can Identify Prognosis-Associated Genes in Regions of Chromosomal Aberrations
Results from our combined genome-wide expression and molecular cytogenetic analyses point to the up-regulation or down-regulation of a sizeable number of genes25,84 located within the chromosomal regions of gain or loss, respectively (Figure 3). Extensive literature search allows us to make some intelligent suppositions as to which genes within these aberrant arms can possibly contribute to cancer progression and are therefore worthy of further biological studies. If the tumor samples have accompanying clinical records, we can further classify these copy number-dependent genes based on how their expression levels correlate to prognosis. Using this approach, we found that the genes MTUS1, ADAMDEC1 (member of the disintegrin metalloproteinase family of genes), EPHX2 (member of the epoxide hydrolase family), and PPP2CB (catalytic subunit of phosphatase 2A) are among the down-regulated 8p genes whose lower expression level correlated with poorer prognosis (Figure 4, A and C). As stated above, MTUS1 is most likely a tumor suppressor gene.74,75,85 The down-regulation of ADAMDEC1 and EPHX2 were recently associated with colon cancer metastasis,86 and PPP2CB codes for the catalytic component of tumor suppressor protein phosphatase 2A.87 We now hypothesize that the poor CRC prognostication of 8p loss may be explained by the fact that it harbors a number of genes with tumor suppressive properties and which play crucial roles in later stages of carcinogenesis. Likewise, our integrated genome-wide molecular profiling and clinical data analyses indicate that there are 18q genes other than SMAD4 (and possibly DCC) whose lower expression levels are indicative of worse clinical outcome (Figure 4, B and C). One of these is ATP5A1, whose biological function is discussed above. Another possibly relevant gene within the 18q arm is NEDD4L (its down-regulation also correlating to poor prognosis based on our analysis), whose decreasing expression level correlated to increasing Gleason score (a measure of aggressiveness) in prostate cancer.88
Figure 3.
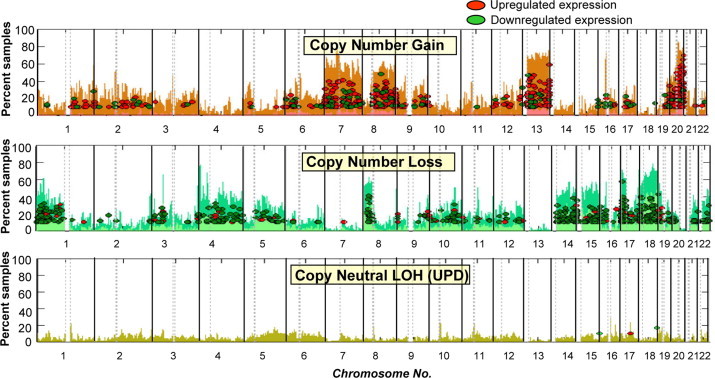
Overall view of gene expression dysregulations within chromosomal aberrations. Shown are the percentages of samples with gains (top chart), losses (middle chart), and copy neutral-LOH or somatic UPDs (bottom chart) in every autosomal chromosome. Each ellipse refers to a U133A probe set representing a gene located in the region of aberration with z ≥ 3 (up-regulated), or z ≤ −3 (down-regulated) in at least 10% of the CRC samples. Note that chromosomal gains and losses are populated by numerous genes that are up-regulated and down-regulated, respectively.
Figure 4.
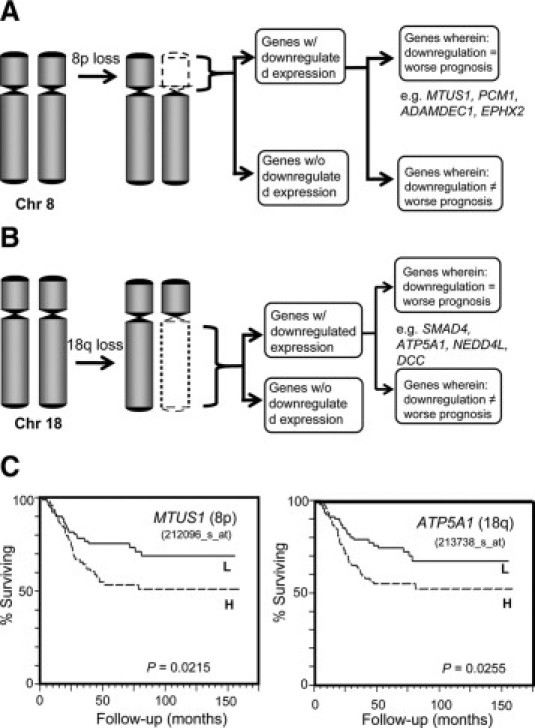
The loss of the 8p (A) or 18q (B) chromosomal arm has been frequently associated with poorer prognosis in CRCs. Our combined SNP and expression array analyses (more than 200 tumor samples) have demonstrated that not all genes within these lost arms have down-regulated expression. A subset of genes with down-regulated expression appear to have prognostic relevance (ie, down-regulation = poor prognosis), including MTUS1, PCM1, ADAMDEC1, and EPHX2 in 8p and ATP5A1, SMAD4, NEDD4L, and DCC in 18q. C: Kaplan-Meier plots based on the expression levels of MTUS1 and ATP5A1 among the primary tumors of 182 CRC patients. In the Kaplan-Meier analysis, the CRC patients were divided into low (L) and high (H) expression groups. Also indicated is the log rank P value for each gene.
Beyond Somatic Copy Number Aberrations: Other Factors That Influence Cancer Gene Dysregulations in Sporadic CRCs
Somatic copy number changes and UPDs are not the only types of chromosomal aberrations a normal cell acquires to dysregulate genes toward tumor promoting activities. Chromosomal translocations (interchange of parts between non-homologous chromosomes) can lead to formation of fusion proteins with oncogenic properties or overexpression of existing proto-oncogenes (eg, when MYC is positioned closer to an enhancer/promoter element of another gene such as IgH).89 Although commonly identified with hematological neoplasms (such as leukemias and lymphomas), a fusion oncogene [EML4-ALK (echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase)] was recently detected in CRC tumors with the use of exon array profiling.90 Promoter hypo/hypermethylation can also dysregulate the expression of cancer-related genes such as MGMT (O-6-methylguanine-DNA methyltransferase), RARB (retinoic acid receptor β) and CDKN2A (cyclin-dependent kinase inhibitor 2A).91,92 In our integrated SNP array/expression array analyses of mostly CIN tumors, we have also identified genes whose up-regulation/down-regulation are not related to copy number changes, thus their changes in expression levels may be epigenetically regulated. One of these genes is CDH3 (P-cadherin), which is located in chromosome 16 and found to be highly expressed in 77% of CRC samples. A recent study has shown that CDH3's overexpression in CRC occurs largely through hypomethylation of CpG sites at its promoter region.93 In theory, a promoter hypermethylation of a tumor suppressor allele can have the same result as a copy loss (ie, lower overall expression) (Figure 5).
Figure 5.
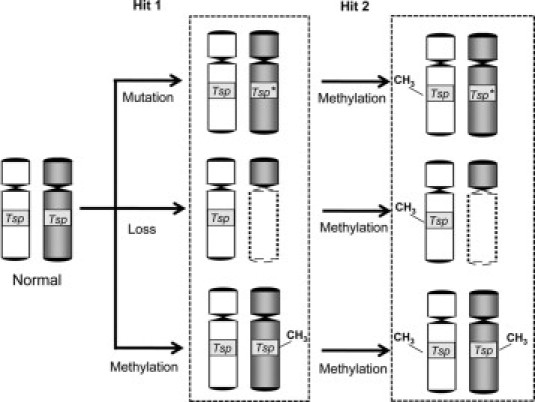
How promoter hypermethylation contributes to dysregulation of a tumor suppressor gene (Tsp). As shown in this diagram, one of the wild-type Tsp alleles of a normal cell gets deactivated (Hit 1) through mutation (Tsp*), copy loss, or promoter hypermethylation. It may then be necessary to deactivate the second allele (Hit 2) for the Tsp to attain its tumor promoting state. As shown in this model, the deactivation of the second allele may also be attained by promoter hypermethylation.
The cancer-promoting activities of tumor suppressor genes and oncogenes may also be regulated posttranscriptionally by non-coding microRNAs, 18- to 25-nucleotide-long RNAs, which can inhibit the translation of their complementary or near-complementary mRNAs.94 Effectively, a microRNA molecule is oncogenic or tumor suppressive if it is inhibitory to the translation of tumor suppressor or oncogene transcript, respectively. In experiments involving CRC cells, the microRNA miR-135a&b was shown to suppress APC translation (thus oncogenic),95 whereas MYC expression can be down-regulated by microRNAs miR-4396 and let-7.97
Future Directions and Concluding Remarks
CRCs (in particular, those classified as CIN) do not just acquire certain chromosomal copy number aberrations randomly. Within regions of gains, copy losses, or UPDs are genes, which on dysregulation, are transformed into cancer-promoting states. These recurrent aberrations, which often involve a whole chromosomal arm (eg, 8p, 8q, 20q), can lead to dysregulation (usually by causing changes in expression level) of numerous genes in either their wild-type or mutated states. Eventually, a single run of next-generation sequencing will be all that is needed to acquire genome-wide mutational, copy number, and LOH profiles of a tumor sample.98 Such a more robust set of information (in addition to genome-wide transcriptional read-out) will certainly strengthen our assessment regarding which genes within the aberrant arms may have some roles in cancer progression. Examples of genes worthy of a closer look would be a mutated, down-regulated gene in a lost region; a mutated (homozygous) gene in a somatic UPD region; and a mutated, up-regulated gene in a region of gain.
The integration of patient survival data to our genome-wide expression analysis led to our observation that among many 18q and 8p genes, lower expression level equates to poor prognosis in CRCs. This may explain why the loss of the 18q or 8p arm has been correlated to worse CRC clinical outcome. We speculate that certain copy number–dependent genes within these arms are essential to the later stages of cancer progression. The next step would be to further evaluate the cancer-related functionalities (such as possible roles in cell cancer proliferation and metastasis) of some of these candidate 8p and 18q genes. In one experimental design, the association of an 8p gene down-regulation to poor CRC prognosis can be tested in vitro by forcibly inhibiting the gene in a cancer cell line, which will then be exposed to a drug specifically used for CRC treatment (such as 5-flurouracil and oxaliplatin). For example, a decrease in the cancer cell-killing effect of 5-flurouracil after PPP2CB (an 8p gene) inhibition will be consistent with PPP2CB down-regulation correlating to poorer clinical outcome, and the potential use of the gene as a prognostic marker may be useful for clinical management of CRC.
Acknowledgements
We thank our colleagues and collaborators who have all made significant contributions in our CRC research over the years: Hanna Pincas, Yu-Wei Cheng, Sarah Giardina, Richard Shattock, Owen Parker, and Jianmin Huang of Weill Cornell Medical College; Dan Notterman and Gunter Schemmann (Princeton and the University of Medicine and Dentistry, New Jersey); Philip Paty, Jinru Shia, and Zhaoshi Zeng from Memorial Sloan-Kettering Cancer Center; Emmanuel Zachariah (UMDNJ); Eytan Domany, Dafna Tsafrir, and Michal Sheffer from Weizmann Institute. We also thank Rodney Wiltshire (Quest Diagnostics) for his very useful suggestions regarding this manuscript.
Footnotes
Supported by National Cancer Institute grants P01-CA65930 and 263MQ610681, the Ludwig Institute for Cancer Research/Conrad N. Hilton Foundation joint Hilton-Ludwig Cancer Metastasis Initiative, and the Gilbert Family Foundation.
Contributor Information
Manny D. Bacolod, Email: mdb2005@med.cornell.edu.
Francis Barany, Email: barany@med.cornell.edu.
References
- 1.Center MM, Jemal A, Ward E. International trends in colorectal cancer incidence rates. Cancer Epidemiol Biomarkers Prev. 2009;18:1688–1694. doi: 10.1158/1055-9965.EPI-09-0090. [DOI] [PubMed] [Google Scholar]
- 2.Boyle P. The globalisation of cancer. Lancet. 2006;368:629–630. doi: 10.1016/S0140-6736(06)69225-8. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 4.Lynch HT, Lynch JF, Lynch PM, Attard T. Hereditary colorectal cancer syndromes: molecular genetics, genetic counseling, diagnosis and management. Fam Cancer. 2008;7:27–39. doi: 10.1007/s10689-007-9165-5. [DOI] [PubMed] [Google Scholar]
- 5.Tenesa A, Dunlop MG. New insights into the aetiology of colorectal cancer from genome-wide association studies. Nat Rev Genet. 2009;10:353–358. doi: 10.1038/nrg2574. [DOI] [PubMed] [Google Scholar]
- 6.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 7.Gervaz P, Bucher P, Morel P. Two colons-two cancers: paradigm shift and clinical implications. J Surg Oncol. 2004;88:261–266. doi: 10.1002/jso.20156. [DOI] [PubMed] [Google Scholar]
- 8.Eshleman JR, Markowitz SD. Microsatellite instability in inherited and sporadic neoplasms. Curr Opin Oncol. 1995;7:83–89. [PubMed] [Google Scholar]
- 9.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- 10.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 11.Bertagnolli MM, Niedzwiecki D, Compton CC, Hahn HP, Hall M, Damas B, Jewell SD, Mayer RJ, Goldberg RM, Saltz LB, Warren RS, Redston M. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: cancer and Leukemia Group B Protocol 89803. J Clin Oncol. 2009;27:1814–1821. doi: 10.1200/JCO.2008.18.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hardy PA, Zacharias H. Reappraisal of the Hansemann-Boveri hypothesis on the origin of tumors. Cell Biol Int. 2005;29:983–992. doi: 10.1016/j.cellbi.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Tjio HJ, Levan A. The chromosome numbers of man. Hereditas. 1956;42:1–6. [Google Scholar]
- 14.Gray JW, Pinkel D. Molecular cytogenetics in human cancer diagnosis. Cancer. 1992;69:1536–1542. doi: 10.1002/1097-0142(19920315)69:6+<1536::aid-cncr2820691306>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 15.Dodson MK, Hartmann LC, Cliby WA, DeLacey KA, Keeney GL, Ritland SR, Su JQ, Podratz KC, Jenkins RB. Comparison of loss of heterozygosity patterns in invasive low-grade and high-grade epithelial ovarian carcinomas. Cancer Res. 1993;53:4456–4460. [PubMed] [Google Scholar]
- 16.Kallioniemi OP, Kallioniemi A, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D. Comparative genomic hybridization: a rapid new method for detecting and mapping DNA amplification in tumors. Semin Cancer Biol. 1993;4:41–46. [PubMed] [Google Scholar]
- 17.Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW, Albertson DG. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 18.Dong S, Wang E, Hsie L, Cao Y, Chen X, Gingeras TR. Flexible use of high-density oligonucleotide arrays for single-nucleotide polymorphism discovery and validation. Genome Res. 2001;11:1418–1424. doi: 10.1101/gr.171101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bacolod MD, Schemmann GS, Giardina SF, Notterman DA, Paty PB, Barany F. Emerging paradigms in cancer genetics: some important findings from high density SNP array studies. Cancer Res. 2009;69:723–727. doi: 10.1158/0008-5472.CAN-08-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maciejewski JP, Tiu RV, O'Keefe C. Application of array-based whole genome scanning technologies as a cytogenetic tool in haematological malignancies. Br J Haematol. 2009;146:479–488. doi: 10.1111/j.1365-2141.2009.07757.x. [DOI] [PubMed] [Google Scholar]
- 21.Bacolod MD, Schemmann GS, Wang S, Shattock R, Giardina SF, Zeng Z, Shia J, Stengel RF, Gerry N, Hoh J, Kirchhoff T, Gold B, Christman MF, Offit K, Gerald WL, Notterman DA, Ott J, Paty PB, Barany F. The signatures of autozygosity among patients with colorectal cancer. Cancer Res. 2008;68:2610–2621. doi: 10.1158/0008-5472.CAN-07-5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diep CB, Kleivi K, Ribeiro FR, Teixeira MR, Lindgjaerde OC, Lothe RA. The order of genetic events associated with colorectal cancer progression inferred from meta-analysis of copy number changes. Genes Chromosomes Cancer. 2006;45:31–41. doi: 10.1002/gcc.20261. [DOI] [PubMed] [Google Scholar]
- 23.Nakao K, Mehta KR, Fridlyand J, Moore DH, Jain AN, Lafuente A, Wiencke JW, Terdiman JP, Waldman FM. High-resolution analysis of DNA copy number alterations in colorectal cancer by array-based comparative genomic hybridization. Carcinogenesis. 2004;25:1345–1357. doi: 10.1093/carcin/bgh134. [DOI] [PubMed] [Google Scholar]
- 24.Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F, Paty PB, Gerald WL, Notterman DA, Domany E. Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proc Natl Acad Sci USA. 2009;106:7131–7136. doi: 10.1073/pnas.0902232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsafrir D, Bacolod M, Selvanayagam Z, Tsafrir I, Shia J, Zeng Z, Liu H, Krier C, Stengel RF, Barany F, Gerald WL, Paty PB, Domany E, Notterman DA. Relationship of gene expression and chromosomal abnormalities in colorectal cancer. Cancer Res. 2006;66:2129–2137. doi: 10.1158/0008-5472.CAN-05-2569. [DOI] [PubMed] [Google Scholar]
- 26.Jen J, Kim H, Piantadosi S, Liu ZF, Levitt RC, Sistonen P, Kinzler KW, Vogelstein B, Hamilton SR. Allelic loss of chromosome 18q and prognosis in colorectal cancer. N Engl J Med. 1994;331:213–221. doi: 10.1056/NEJM199407283310401. [DOI] [PubMed] [Google Scholar]
- 27.Ogunbiyi OA, Goodfellow PJ, Herfarth K, Gagliardi G, Swanson PE, Birnbaum EH, Read TE, Fleshman JW, Kodner IJ, Moley JF. Confirmation that chromosome 18q allelic loss in colon cancer is a prognostic indicator. J Clin Oncol. 1998;16:427–433. doi: 10.1200/JCO.1998.16.2.427. [DOI] [PubMed] [Google Scholar]
- 28.Martinez-Lopez E, Abad A, Font A, Monzo M, Ojanguren I, Pifarre A, Sanchez JJ, Martin C, Rosell R. Allelic loss on chromosome 18q as a prognostic marker in stage II colorectal cancer. Gastroenterology. 1998;114:1180–1187. doi: 10.1016/s0016-5085(98)70423-8. [DOI] [PubMed] [Google Scholar]
- 29.Lanza G, Matteuzzi M, Gafa R, Orvieto E, Maestri I, Santini A, del Senno L. Chromosome 18q allelic loss and prognosis in stage II and III colon cancer. Int J Cancer. 1998;79:390–395. doi: 10.1002/(sici)1097-0215(19980821)79:4<390::aid-ijc14>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 30.Jernvall P, Makinen MJ, Karttunen TJ, Makela J, Vihko P. Loss of heterozygosity at 18q21 is indicative of recurrence and therefore poor prognosis in a subset of colorectal cancers. Br J Cancer. 1999;79:903–908. doi: 10.1038/sj.bjc.6690144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Angelis PM, Stokke T, Beigi M, Mjaland O, Clausen OP. Prognostic significance of recurrent chromosomal aberrations detected by comparative genomic hybridization in sporadic colorectal cancer. Int J Colorectal Dis. 2001;16:38–45. doi: 10.1007/s003840000275. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe T, Wu TT, Catalano PJ, Ueki T, Satriano R, Haller DG, Benson AB, 3rd, Hamilton SR. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001;344:1196–1206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barratt PL, Seymour MT, Stenning SP, Georgiades I, Walker C, Birbeck K, Quirke P. DNA markers predicting benefit from adjuvant fluorouracil in patients with colon cancer: a molecular study. Lancet. 2002;360:1381–1391. doi: 10.1016/s0140-6736(02)11402-4. [DOI] [PubMed] [Google Scholar]
- 34.Choi SW, Lee KJ, Bae YA, Min KO, Kwon MS, Kim KM, Rhyu MG. Genetic classification of colorectal cancer based on chromosomal loss and microsatellite instability predicts survival. Clin Cancer Res. 2002;8:2311–2322. [PubMed] [Google Scholar]
- 35.Zhou W, Goodman SN, Galizia G, Lieto E, Ferraraccio F, Pignatelli C, Purdie CA, Piris J, Morris R, Harrison DJ, Paty PB, Culliford A, Romans KE, Montgomery EA, Choti MA, Kinzler KW, Vogelstein B. Counting alleles to predict recurrence of early-stage colorectal cancers. Lancet. 2002;359:219–225. doi: 10.1016/S0140-6736(02)07448-2. [DOI] [PubMed] [Google Scholar]
- 36.Sarli L, Bottarelli L, Bader G, Iusco D, Pizzi S, Costi R, D'Adda T, Bertolani M, Roncoroni L, Bordi C. Association between recurrence of sporadic colorectal cancer, high level of microsatellite instability, and loss of heterozygosity at chromosome 18q. Dis Colon Rectum. 2004;47:1467–1482. doi: 10.1007/s10350-004-0628-6. [DOI] [PubMed] [Google Scholar]
- 37.Bardi G, Fenger C, Johansson B, Mitelman F, Heim S. Tumor karyotype predicts clinical outcome in colorectal cancer patients. J Clin Oncol. 2004;22:2623–2634. doi: 10.1200/JCO.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 38.Al-Mulla F, Behbehani AI, Bitar MS, Varadharaj G, Going JJ. Genetic profiling of stage I and II colorectal cancer may predict metastatic relapse. Mod Pathol. 2006;19:648–658. doi: 10.1038/modpathol.3800564. [DOI] [PubMed] [Google Scholar]
- 39.Liu XP, Kawauchi S, Oga A, Sato T, Ikemoto K, Ikeda E, Sasaki K. Chromosomal aberrations detected by comparative genomic hybridization predict outcome in patients with colorectal carcinoma. Oncol Rep. 2007;17:261–267. [PubMed] [Google Scholar]
- 40.Kern SE, Fearon ER, Tersmette KW, Enterline JP, Leppert M, Nakamura Y, White R, Vogelstein B, Hamilton SR. Clinical and pathological associations with allelic loss in colorectal carcinoma [corrected] JAMA. 1989;261:3099–3103. doi: 10.1001/jama.261.21.3099. [DOI] [PubMed] [Google Scholar]
- 41.Halling KC, French AJ, McDonnell SK, Burgart LJ, Schaid DJ, Peterson BJ, Moon-Tasson L, Mahoney MR, Sargent DJ, O'Connell MJ, Witzig TE, Farr GH, Jr, Goldberg RM, Thibodeau SN. Microsatellite instability and 8p allelic imbalance in stage B2 and C colorectal cancers. J Natl Cancer Inst. 1999;91:1295–1303. doi: 10.1093/jnci/91.15.1295. [DOI] [PubMed] [Google Scholar]
- 42.Kochhar R, Halling KC, McDonnell S, Schaid DJ, French AJ, O'Connell MJ, Nagorney DM, Thibodeau SN. Allelic imbalance and microsatellite instability in resected Duke's D colorectal cancer. Diagn Mol Pathol. 1997;6:78–84. doi: 10.1097/00019606-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 43.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 44.Mazzone M, Comoglio PM. The Met pathway: master switch and drug target in cancer progression. FASEB J. 2006;20:1611–1621. doi: 10.1096/fj.06-5947rev. [DOI] [PubMed] [Google Scholar]
- 45.Erisman MD, Rothberg PG, Diehl RE, Morse CC, Spandorfer JM, Astrin SM. Deregulation of c-myc gene expression in human colon carcinoma is not accompanied by amplification or rearrangement of the gene. Mol Cell Biol. 1985;5:1969–1976. doi: 10.1128/mcb.5.8.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Di Renzo MF, Olivero M, Giacomini A, Porte H, Chastre E, Mirossay L, Nordlinger B, Bretti S, Bottardi S, Giordano S, Plebani M, Gespach C, Comoglio PM. Overexpression and amplification of the Met/HGF receptor gene during the progression of colorectal cancer. Clin Cancer Res. 1995;1:147–154. [PubMed] [Google Scholar]
- 47.Zeng Z, Weiser MR, D'Alessio M, Grace A, Shia J, Paty PB. Immunoblot analysis of c-Met expression in human colorectal cancer: overexpression is associated with advanced stage cancer. Clin Exp Metastasis. 2004;21:409–417. doi: 10.1007/s10585-005-1617-4. [DOI] [PubMed] [Google Scholar]
- 48.Melchor L, Saucedo-Cuevas LP, Munoz-Repeto I, Rodriguez-Pinilla SM, Honrado E, Campoverde A, Palacios J, Nathanson KL, Garcia MJ, Benitez J. Comprehensive characterization of the DNA amplification at 13q34 in human breast cancer reveals TFDP1 and CUL4A as likely candidate target genes. Breast Cancer Res. 2009;11:R86. doi: 10.1186/bcr2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones KA, Kemp CR. Wnt-induced proteolytic targeting. Genes Dev. 2008;22:3077–3081. doi: 10.1101/gad.1741008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liao CF, Luo SF, Li LT, Lin CY, Chen YC, Jiang MC. CSE1L/CAS, the cellular apoptosis susceptibility protein, enhances invasion and metastasis but not proliferation of cancer cells. J Exp Clin Cancer Res. 2008;27:15. doi: 10.1186/1756-9966-27-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai CS, Chen HC, Tung JN, Tsou SS, Tsao TY, Liao CF, Chen YC, Yeh CY, Yeh KT, Jiang MC. Serum cellular apoptosis susceptibility protein is a potential prognostic marker for metastatic colorectal cancer. Am J Pathol. 2010;176:1619–1628. doi: 10.2353/ajpath.2010.090467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen MH, Gootenberg J, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab plus FOLFOX4 as second-line treatment of colorectal cancer. Oncologist. 2007;12:356–361. doi: 10.1634/theoncologist.12-3-356. [DOI] [PubMed] [Google Scholar]
- 53.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 54.Yuen ST, Davies H, Chan TL, Ho JW, Bignell GR, Cox C, Stephens P, Edkins S, Tsui WW, Chan AS, Futreal PA, Stratton MR, Wooster R, Leung SY. Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res. 2002;62:6451–6455. [PubMed] [Google Scholar]
- 55.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 56.Halilovic E, Solit DB. Therapeutic strategies for inhibiting oncogenic BRAF signaling. Curr Opin Pharmacol. 2008;8:419–426. doi: 10.1016/j.coph.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 57.Soh J, Okumura N, Lockwood WW, Yamamoto H, Shigematsu H, Zhang W, Chari R, Shames DS, Tang X, MacAulay C, Varella-Garcia M, Vooder T, Wistuba II, Lam S, Brekken R, Toyooka S, Minna JD, Lam WL, Gazdar AF. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS One. 2009;4:e7464. doi: 10.1371/journal.pone.0007464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elliott RL, Blobe GC. Role of transforming growth factor Beta in human cancer. J Clin Oncol. 2005;23:2078–2093. doi: 10.1200/JCO.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 59.Peng B, Fleming JB, Breslin T, Grau AM, Fojioka S, Abbruzzese JL, Evans DB, Ayers D, Wathen K, Wu T, Robertson KD, Chiao PJ. Suppression of tumorigenesis and induction of p15(ink4b) by Smad4/DPC4 in human pancreatic cancer cells. Clin Cancer Res. 2002;8:3628–3638. [PubMed] [Google Scholar]
- 60.Xiao DS, Wen JF, Li JH, Wang KS, Hu ZL, Zhou JH, Deng ZH, Liu Y. Effect of DPC4 gene on invasion and metastasis of colorectal carcinoma cells. Acta Biochim Biophys Sin (Shanghai) 2006;38:883–892. doi: 10.1111/j.1745-7270.2006.00233.x. [DOI] [PubMed] [Google Scholar]
- 61.Maitra A, Molberg K, Albores-Saavedra J, Lindberg G. Loss of Dpc4 expression in colonic adenocarcinomas correlates with the presence of metastatic disease. Am J Pathol. 2000;157:1105–1111. doi: 10.1016/S0002-9440(10)64625-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alazzouzi H, Alhopuro P, Salovaara R, Sammalkorpi H, Jarvinen H, Mecklin JP, Hemminki A, Schwartz S, Jr, Aaltonen LA, Arango D. SMAD4 as a prognostic marker in colorectal cancer. Clin Cancer Res. 2005;11:2606–2611. doi: 10.1158/1078-0432.CCR-04-1458. [DOI] [PubMed] [Google Scholar]
- 63.Fearon ER, Cho KR, Nigro JM, Kern SE, Simons JW, Ruppert JM, Hamilton SR, Preisinger AC, Thomas G, Kinzler KW. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science. 1990;247:49–56. doi: 10.1126/science.2294591. [DOI] [PubMed] [Google Scholar]
- 64.Shibata D, Reale MA, Lavin P, Silverman M, Fearon ER, Steele G, Jr, Jessup JM, Loda M, Summerhayes IC. The DCC protein and prognosis in colorectal cancer. N Engl J Med. 1996;335:1727–1732. doi: 10.1056/NEJM199612053352303. [DOI] [PubMed] [Google Scholar]
- 65.Mehlen P, Llambi F. Role of netrin-1 and netrin-1 dependence receptors in colorectal cancers. Br J Cancer. 2005;93:1–6. doi: 10.1038/sj.bjc.6602656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.von Ballmoos C, Wiedenmann A, Dimroth P. Essentials for ATP synthesis by F1F0 ATP synthases. Annu Rev Biochem. 2009;78:649–672. doi: 10.1146/annurev.biochem.78.081307.104803. [DOI] [PubMed] [Google Scholar]
- 67.Cuezva JM, Krajewska M, de Heredia ML, Krajewski S, Santamaria G, Kim H, Zapata JM, Marusawa H, Chamorro M, Reed JC. The bioenergetic signature of cancer: a marker of tumor progression. Cancer Res. 2002;62:6674–6681. [PubMed] [Google Scholar]
- 68.Lopez-Rios F, Sanchez-Arago M, Garcia-Garcia E, Ortega AD, Berrendero JR, Pozo-Rodriguez F, Lopez-Encuentra A, Ballestin C, Cuezva JM. Loss of the mitochondrial bioenergetic capacity underlies the glucose avidity of carcinomas. Cancer Res. 2007;67:9013–9017. doi: 10.1158/0008-5472.CAN-07-1678. [DOI] [PubMed] [Google Scholar]
- 69.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 70.Shin YK, Yoo BC, Chang HJ, Jeon E, Hong SH, Jung MS, Lim SJ, Park JG. Down-regulation of mitochondrial F1F0-ATP synthase in human colon cancer cells with induced 5-fluorouracil resistance. Cancer Res. 2005;65:3162–3170. doi: 10.1158/0008-5472.CAN-04-3300. [DOI] [PubMed] [Google Scholar]
- 71.Iwatsuki M, Mimori K, Ishii H, Yokobori T, Takatsuno Y, Sato T, Toh H, Onoyama I, Nakayama KI, Baba H, Mori M: Loss of FBXW7, a cell cycle regulating gene, in colorectal cancer: clinical significance. Int J Cancer 126:1828–1837 [DOI] [PubMed]
- 72.Tan Y, Sangfelt O, Spruck C. The Fbxw7/hCdc4 tumor suppressor in human cancer. Cancer Lett. 2008;271:1–12. doi: 10.1016/j.canlet.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 73.Robinson VL, Hickson JA, Vander Griend DJ, Dubauskas Z, Rinker-Schaeffer CW. MKK4 and metastasis suppression: a marriage of signal transduction and metastasis research. Clin Exp Metastasis. 2003;20:25–30. doi: 10.1023/a:1022586318678. [DOI] [PubMed] [Google Scholar]
- 74.Zuern C, Heimrich J, Kaufmann R, Richter KK, Settmacher U, Wanner C, Galle J, Seibold S: Down-regulation of MTUS1 in human colon tumors. Oncol Rep 23:183–189 [PubMed]
- 75.Rodrigues-Ferreira S, Di Tommaso A, Dimitrov A, Cazaubon S, Gruel N, Colasson H, Nicolas A, Chaverot N, Molinie V, Reyal F, Sigal-Zafrani B, Terris B, Delattre O, Radvanyi F, Perez F, Vincent-Salomon A, Nahmias C. 8p22 MTUS1 gene product ATIP3 is a novel anti-mitotic protein underexpressed in invasive breast carcinoma of poor prognosis. PLoS One. 2009;4:e7239. doi: 10.1371/journal.pone.0007239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 77.Cho YG, Choi BJ, Kim CJ, Song JW, Kim SY, Nam SW, Lee SH, Yoo NJ, Lee JY, Park WS. Genetic alterations of the KLF6 gene in colorectal cancers. APMIS. 2006;114:458–464. doi: 10.1111/j.1600-0463.2006.apm_431.x. [DOI] [PubMed] [Google Scholar]
- 78.Cho YG, Choi BJ, Song JW, Kim SY, Nam SW, Lee SH, Yoo NJ, Lee JY, Park WS. Aberrant expression of Kruppel-like factor 6 protein in colorectal cancers. World J Gastroenterol. 2006;12:2250–2253. doi: 10.3748/wjg.v12.i14.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249:912–915. doi: 10.1126/science.2144057. [DOI] [PubMed] [Google Scholar]
- 80.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 81.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miyaki M, Iijima T, Konishi M, Sakai K, Ishii A, Yasuno M, Hishima T, Koike M, Shitara N, Iwama T, Utsunomiya J, Kuroki T, Mori T. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene. 1999;18:3098–3103. doi: 10.1038/sj.onc.1202642. [DOI] [PubMed] [Google Scholar]
- 83.Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dafou D, Marth C, Mueller-Holzner E, Corcoran M, Dagnell M, Nejad SZ, Nayer BN, Zali MR, Hansson J, Egyhazi S, Petersson F, Sangfelt P, Nordgren H, Grander D, Reed SI, Widschwendter M, Sangfelt O, Spruck C. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006–9012. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- 84.Platzer P, Upender MB, Wilson K, Willis J, Lutterbaugh J, Nosrati A, Willson JK, Mack D, Ried T, Markowitz S. Silence of chromosomal amplifications in colon cancer. Cancer Res. 2002;62:1134–1138. [PubMed] [Google Scholar]
- 85.Seibold S, Rudroff C, Weber M, Galle J, Wanner C, Marx M. Identification of a new tumor suppressor gene located at chromosome 8p21.3-22. FASEB J. 2003;17:1180–1182. doi: 10.1096/fj.02-0934fje. [DOI] [PubMed] [Google Scholar]
- 86.Macartney-Coxson DP, Hood KA, Shi HJ, Ward T, Wiles A, O'Connor R, Hall DA, Lea RA, Royds JA, Stubbs RS, Rooker S. Metastatic susceptibility locus, an 8p hot-spot for tumour progression disrupted in colorectal liver metastases: 13 candidate genes examined at the DNA, mRNA and protein level. BMC Cancer. 2008;8:187. doi: 10.1186/1471-2407-8-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med. 2008;14:152–160. doi: 10.1016/j.molmed.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 88.Hu XY, Xu YM, Fu Q, Yu JJ, Huang J. Nedd4L expression is downregulated in prostate cancer compared to benign prostatic hyperplasia. Eur J Surg Oncol. 2009;35:527–531. doi: 10.1016/j.ejso.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 89.Nambiar M, Kari V, Raghavan SC. Chromosomal translocations in cancer. Biochim Biophys Acta. 2008;1786:139–152. doi: 10.1016/j.bbcan.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 90.Lin E, Li L, Guan Y, Soriano R, Rivers CS, Mohan S, Pandita A, Tang J, Modrusan Z. Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol Cancer Res. 2009;7:1466–1476. doi: 10.1158/1541-7786.MCR-08-0522. [DOI] [PubMed] [Google Scholar]
- 91.Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004;23:29–39. doi: 10.1023/a:1025806911782. [DOI] [PubMed] [Google Scholar]
- 92.Cheng YW, Pincas H, Bacolod MD, Schemmann G, Giardina SF, Huang J, Barral S, Idrees K, Khan SA, Zeng Z, Rosenberg S, Notterman DA, Ott J, Paty P, Barany F. CpG island methylator phenotype associates with low-degree chromosomal abnormalities in colorectal cancer. Clin Cancer Res. 2008;14:6005–6013. doi: 10.1158/1078-0432.CCR-08-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Milicic A, Harrison LA, Goodlad RA, Hardy RG, Nicholson AM, Presz M, Sieber O, Santander S, Pringle JH, Mandir N, East P, Obszynska J, Sanders S, Piazuelo E, Shaw J, Harrison R, Tomlinson IP, McDonald SA, Wright NA, Jankowski JA. Ectopic expression of P-cadherin correlates with promoter hypomethylation early in colorectal carcinogenesis and enhanced intestinal crypt fission in vivo. Cancer Res. 2008;68:7760–7768. doi: 10.1158/0008-5472.CAN-08-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nagel R, le Sage C, Diosdado B, van der Waal M, Oude Vrielink JA, Bolijn A, Meijer GA, Agami R. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008;68:5795–5802. doi: 10.1158/0008-5472.CAN-08-0951. [DOI] [PubMed] [Google Scholar]
- 96.Chen X, Guo X, Zhang H, Xiang Y, Chen J, Yin Y, Cai X, Wang K, Wang G, Ba Y, Zhu L, Wang J, Yang R, Zhang Y, Ren Z, Zen K, Zhang J, Zhang CY. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene. 2009;28:1385–1392. doi: 10.1038/onc.2008.474. [DOI] [PubMed] [Google Scholar]
- 97.Akao Y, Nakagawa Y, Naoe T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. 2006;29:903–906. doi: 10.1248/bpb.29.903. [DOI] [PubMed] [Google Scholar]
- 98.Schweiger MR, Kerick M, Timmermann B, Albrecht MW, Borodina T, Parkhomchuk D, Zatloukal K, Lehrach H. Genome-wide massively parallel sequencing of formaldehyde fixed-paraffin embedded (FFPE) tumor tissues for copy-number- and mutation-analysis. PLoS One. 2009;4:e5548. doi: 10.1371/journal.pone.0005548. [DOI] [PMC free article] [PubMed] [Google Scholar]