

Abstract
(CGG)n repeat expansion in the FMR1 gene is associated with fragile X syndrome and other disorders. Current methods for FMR1 molecular testing rely on Southern blot analysis to detect expanded alleles too large to be PCR-amplified and to identify female homozygous alleles that often confound interpretations of PCR data. A novel, single-tube CGG repeat primed FMR1 PCR technology was designed with two gene-specific primers that flank the triplet repeat region, as well as a third primer that is complementary to the (CGG)n repeat. This PCR was evaluated with 171 unique DNA samples, including a blinded set of 146 clinical specimens. The method detected all alleles reported by Southern blot analysis, including full mutations in 66 clinical samples and comprised up to 1300 CGG. Furthermore, a blinded cohort of 42 female homozygous and heterozygous specimens, including 21 with full mutation alleles, was resolved with 100% accuracy. Last, AGG interrupter sequences, which may influence the risk of (CGG)n expansion in the children of some carriers, were each correctly identified in 14 male and female clinical samples as referenced to DNA sequencing. As a result, this PCR provides robust detection of expanded alleles and resolves allele zygosity, thus minimizing the number of samples that require Southern blot analysis and producing more comprehensive FMR1 genotyping data than other methods.
Expansion of cytosine-guanine-guanine (CGG) triplet repeats in the 5′-untranslated region of the fragile X mental retardation 1 (FMR1, NM_002024.4) gene is associated with several disorders, including fragile X syndrome, fragile X-associated tremor/ataxia syndrome, and fragile X-associated primary ovarian insufficiency.1,2,3,4 Patients with the FMR1 full mutation (>200 CGG repeats) may be affected by a range of neurological, psychiatric, or emotional challenges, including mental retardation and/or autism.5 Deficits in development and particularly in attention and social communication have also been noted for many children with the FMR1 premutation. Moreover, premutation carriers (55 to 200 CGG repeats) are known to be at risk for fragile X-associated primary ovarian insufficiency and fragile X-associated tremor/ataxia syndrome, and some of these individuals may present additional complications, such as hypothyroidism and fibromyalgia.6 As a result, FMR1 disorders are linked to a range of clinical conditions, necessitating testing patients at different times during their life span.7
Fragile X syndrome molecular diagnosis is usually based on quantification of the (CGG)n repeat elements and the assessment of the methylation state of expanded alleles.5 Although PCR is the preferred approach to determine the (CGG)n repeat length of FMR1 alleles, typically only alleles with less than 100 to 150 CGG have been amenable to PCR amplification. However, a recent publication by our group has demonstrated that full mutations composed of more than 1000 CGG can be reproducibly amplified using a novel PCR technology. In addition, this PCR detected all categories of alleles in concordance with the results of Southern blot analysis from a cohort of nearly 150 blinded clinical samples.8 We also previously described a repeat primed PCR method that can reliably categorize FMR1 alleles throughout the mutation range in both males and females on a number of different DNA templates (including genomic DNA and blood spots).9,10,11 This PCR used a chimeric primer that hybridized internally to the triplet repeat segment and was incorporated into a ladder of products that appeared as a smear on an agarose gel when an expanded allele was present. Here we describe the performance of a three-primer CGG repeat primed (RP) FMR1 PCR method that represents an improved and optimized assay design for use with capillary electrophoresis (CE). This three-primer configuration offers several advantages over the earlier design10 and reveals additional molecular information that can resolve commonly encountered limitations in FMR1 analyses.
Materials and Methods
Clinical and Cell Line DNA Samples
Clinical whole blood specimens were procured from patients evaluated at the M.I.N.D. Institute Clinic, after informed consent was obtained and according to an approved institutional review board protocol. Genomic DNA from clinical samples was isolated from peripheral blood leukocytes using the Gentra Puregene Blood Kit (Qiagen, Germantown, MD). Purified DNA was provided in a coded format before PCR amplification at Asuragen, Inc., and thus the results of any other testing (eg, Southern blot analysis) were unknown to the technician who processed the samples. A total of 146 blinded clinical specimens were sent to Asuragen, Inc. for PCR analysis. All cell line DNA templates were obtained from the Coriell Cell Repositories (Coriell Institute for Medical Research, Camden, NJ). Before PCR, clinical and cell line DNA samples were quantified using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE) and diluted in 10 mmol/L Tris and 0.5 mmol/L EDTA, pH 8.8, to 20 ng/μl before PCR and stored at −15 to −30°C.
CGG RP FMR1 PCR
PCR was performed to interrogate the number of CGG repeats in the 5′-untranslated region of the FMR1 gene, NM_002024.4:c.1–131CGG[1_n], where n represents the variable number of repeats in this region along with interspersed adenine-guanine-guanine (AGG) interruptions. Samples were PCR-amplified by preparing a master mix containing 11.45 μl of GC-rich AMP buffer (no. 49387), 1.5 μl of FAM-labeled FMR1 primers (no. 49386), 0.5 μl of FMR1 CGG primer (no. 49393), 0.5 μl of nuclease-free water, and 0.05 μl of GC-rich polymerase mix (no. 49388) from Asuragen, Inc. (Austin, TX). The gene-specific primer sequences were published previously,8 and the CGG-specific primer followed the design of the sequence provided by Tassone et al10 with a (CGG)5 anchor sequence. A master mix of these reagents was vortexed before dispensing to a microtiter plate (96- or 384-well plates, Phenix Research Products, Chandler, NC). Aliquots of the DNA sample, typically 2 μl at 20 ng/μl, were transferred to the plate. Sealed plates (Aluminum film sheets, Phenix Research Products) were vortexed, centrifuged, and transferred to a thermal cycler (9700, Applied Biosystems, Foster City, CA). Samples were amplified with an initial heat denaturation step of 95°C for 5 minutes, followed by 10 cycles of 97°C for 35 seconds, 62°C for 35 seconds, and 68°C for 4 minutes, and then 20 cycles of 97°C for 35 seconds, 62°C for 35 seconds, and 68°C for 4 minutes with a 20 second autoextension at each cycle. The final extension step was 72°C for 10 minutes. After PCR, samples were stored at −15 to −30°C and either protected from light before analysis or analyzed immediately by CE.
Generation of Model “Unamplifiable” Full-Length CGG Repeat PCR Products
Templates used to model CGG RP PCR amplification in the absence of full-length PCR products were created as follows: i) PCR products from Coriell Cell Repositories genomic DNA templates containing ∼550 CGG (NA07862), ∼645 CGG (NA04025), and ∼940 CGG (NA09237) were prepared by amplification with FMR1 forward primer12 in combination with reverse primer (no. 49386, Asuragen, Inc.) and amplification reagents and conditions described previously.8 ii) The full-length PCR products were then gel-purified and quantified by UV spectrometry and diluted to 6 × 103 copies/μl (ie, a copy number equivalent to 20 ng/μl gDNA). The diluted material, lacking a primer binding site for the forward primer,8 was subsequently used as a template in CGG RP PCR as described above.
Agarose Gel Electrophoresis
A total of 6 μl of the PCR was combined with 3 μl of 3× AGE loading dye (15% glycerol and 0.25% bromphenol blue, Sigma, St. Louis, MO), and all 9 μl was loaded on a 1.75% agarose gel. Gels were stained with SYBR Gold 10000X (Invitrogen, Carlsbad, CA) and imaged with UV light using an Alpha Innotech FluorChem 8800 Imaging Detection System (Alpha Innotech, San Leandro, CA).
Capillary Electrophoresis
All amplicons were evaluated on an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA) as described.8 with the exception that 1 μl of unpurified PCR products was mixed with 12 μl of Hi-Di Formamide (Applied Biosystems) and 2 μl of a ROX 1000 Size Ladder (no. 46083, Asuragen, Inc.). Samples were heat-denatured at 95°C for 2 minutes followed by cooling on ice before transfer to the CE instrument. CE rather than agarose gel electrophoresis is the preferred method for analyzing the FMR1 amplification products of CGG RP PCR.
Data Analysis
PCR products detected by CE were analyzed using GeneMapper 4.0 software (Applied Biosystems) or PeakScanner 1.0 (Applied Biosystems).8 Absolute triplet repeat quantification was accomplished by enumerating CGG-specific peaks beginning with the lowest molecular weight amplicons (corresponding to 5 CGG) and continually counting until the (CGG)n peak morphology was lost. If the full-length amplicon peaks overlapped with the (CGG)n repeat products, the expected spacing of adjacent (CGG)n peaks was used to estimate the corresponding (CGG)n number. Fragment sizing of full-length amplicons in nucleotides was estimated according to 230 + 3 · (CGG)n, where n is the number of CGG repeats. Interspersed AGG sequences were identified in the electropherogram by the relative location of signal “dips” in an otherwise uninterrupted series of (CGG)n repeat product peaks.
DNA Sequencing
Genomic DNA was PCR-amplified with Asuragen GC-Rich AMP buffer (no. 49387) with previously described FMR1 primers.9 The PCR products were purified by a QIAquick PCR Purification Kit (no. 28104, Qiagen), and the DNA concentration was determined with the NanoDrop spectrophotometer. DNA sequencing reactions were completed with 2 to 5 ng of purified PCR products using BigDye Terminator v3.1 Cycle Sequencing Kits (Applied Biosystems).
Southern Blot Analysis
Southern blot analysis was performed as described.10 Results were blinded to Asuragen, Inc. until the files with the PCR results were transferred to the University of California Davis.
Results
Absolute CGG Repeat Quantification in Cell Line Genomic DNA Templates
CGG RP PCR is primarily distinguished from a more conventional two-primer, gene-specific PCR by the addition of a third PCR primer that is complementary to the FMR1 triplet repeat region (Figure 1). Examples of product profiles produced using this approach with cell line genomic DNA (gDNA) are shown in Figure 2. For each template, two distinct populations of amplicons were produced and resolved by CE. The first population represented full-length amplicons generated from the gene-specific forward and reverse primer pair. A second population of amplicons represented a collection of CGG-specific products generated by amplification with the triplet repeat oligodeoxynucleotide primer. For example, the top left electropherogram of Figure 2A profiles both populations of amplicons produced from a normal male hemizygous template with 30 CGG repeats. The first group of peaks on the left corresponds to the shorter PCR products generated by the CGG repeat primer and FAM-labeled reverse primer. In this group, three sets of five or six peaks differing by 3 bp were detected with two interspersed gaps approximately 15 bp wide. In addition, an intense product band corresponding to the full-length amplicon was produced as a result of amplification with the two gene-specific primers. This product encompassed the entire 30 CGG tract, including both 5′ and 3′ flanking regions.
Figure 1.
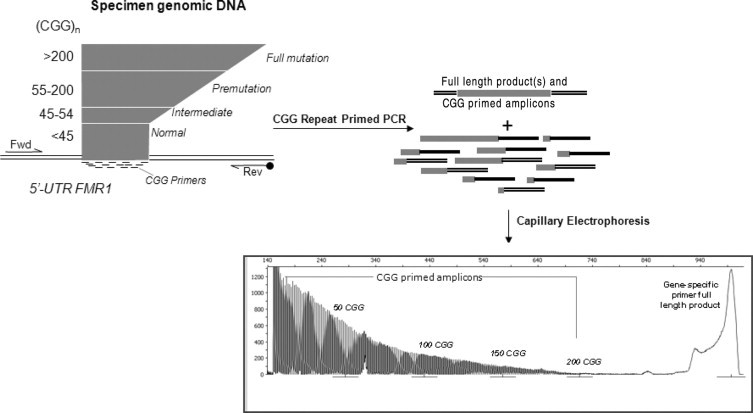
Workflow for amplification and detection of FMR1 amplicons using a three-primer FMR1 PCR. Input gDNA is amplified by two gene-specific primers (forward [Fwd] and reverse [Rev]) and a CGG repeat primer in a single tube. After amplification, the products, which include the full-length amplicon that completely encompasses the triplet repeat region and a multiplicity of CGG repeat primed products, are resolved by CE. The resulting electropherogram supports quantification of the number of CGG repeats, determination of the allele zygosity, and the sequence context of any AGG spacer elements.
Figure 2.
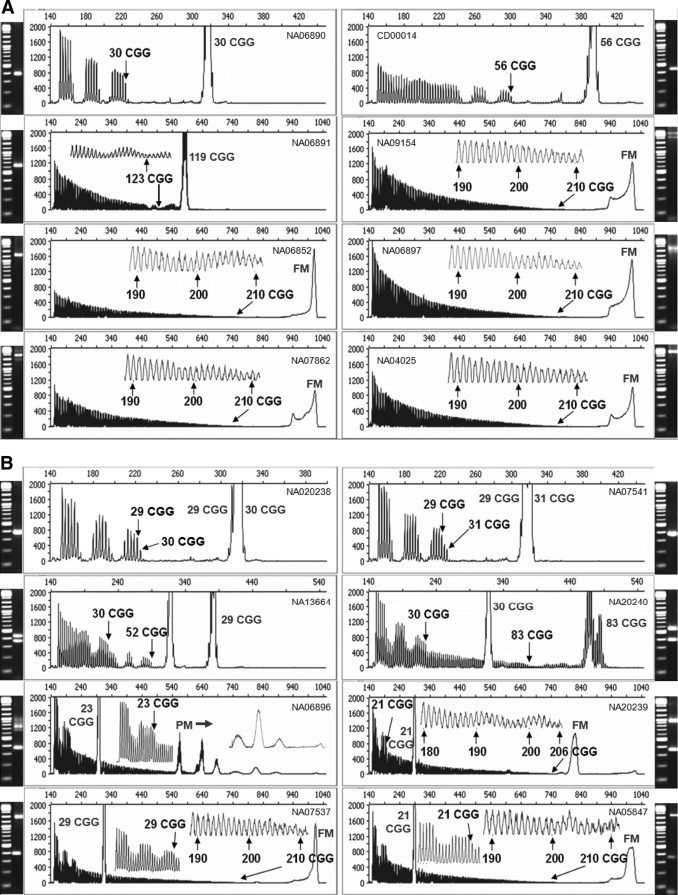
CGG repeat primed PCR produces both full-length gene-specific FMR1 amplicons as well as triplet repeat-specific products that support absolute repeat quantification. A: Comparison of gene-specific PCR (agarose gel image) and repeat primed PCR (CE) of male cell line gDNA templates. Gene-specific PCR was performed as described previously.8B: Comparison of gene-specific PCR (agarose gel image) and repeat primed PCR (CE) of female cell line gDNA templates. Inset: graphs offer a higher resolution view of the underlying data (see arrows). Determination of the number of CGG repeats was provided through absolute quantification, and compared with fragment sizing of the full-length amplicon (Table 1). The Coriell Cell Repositories catalog number for each template is provided on each electropherogram. The CGG number indicated with the corresponding arrow is the repeat number determined by counting the triplet repeat product peaks, whereas the number placed by the prominent gene-specific peak is the repeat number computed from fragment sizing of the full-length amplicon. PM or FM, gene-specific amplicons corresponding to the amplification of premutation or full mutation alleles, respectively.
The PCR products generated with the CGG repeat primer were also used to quantify the number of triplet repeats in each allele. Consider again the example of the 30 CGG male template (Figure 2A). Because the repeat primer comprised 5 complementary CGG repeats, the first product peak corresponded to amplicons with exactly 5 CGG repeats. The gap between the first 5 peaks and the next set of the 5 most intense peaks reflected interference by an intervening AGG sequence (see below). This gap was equivalent to 5 CGG repeats, that is, the span of the repeat primer as it interrogated each possible position for hybridization (which was compromised at each repeat unit in the primer by mismatches of the template AGG with the CGG primer sequence). The next set of 5 peaks revealed a marked increase in signal intensity as the repeat primer bound to sites beyond the “clash” of the AGG interrupter. Another gap in the trace, corresponding to a second AGG, was then observed followed by a third group of amplicons. Counting from left to right and starting with 5 CGG repeats, there were 16 product peaks and two gaps of 5 CGG equivalents corresponding to 30 total triplet repeats. This number of repeat units was identical to the known (CGG)n repeat length for this allele as verified by DNA sequencing and also to the repeat length calculated from the mobility of the full-length product peak after capillary electrophoresis.
A number of other examples of similar CGG RP PCR product profiles, with both male and female samples and including normal, intermediate, and premutation alleles, are shown in Figure 2, A and B. In each case, the gene-specific peak was sized in comparison with a DNA reference (ie, the ROX-labeled standard ladder) and converted to a defined repeat length. Sizing of the full-length peak was in agreement with the (CGG)n product peak counting method that is described above (Table 1). This absolute quantification approach was free from the need for a reference calibration standard, such as the internal ROX standard. The accuracy of triplet repeat quantification using this approach was well correlated to the results of Amos Wilson et al13 using published fragile X consensus materials (Table 1). For example, determination of the (CGG)n repeat length exactly matched 12 of 14 FMR1 alleles whose consensus length were previously determined.13 The remaining two alleles differed by 1 CGG (74 versus 73 CGG) or 3 CGG (83 versus 80 CGG) repeats.
Table 1.
Quantification of CGG Repeats in CCR Lymphoblastoid Cell Lines by CGG Repeat Primed PCR and Comparison with Other Independent Determinations
| CGG repeat primed PCR |
||||||
|---|---|---|---|---|---|---|
| CGG repeats |
||||||
| Cell line | Sex | Submitted CGG repeat number | Fragment sizing* | Absolute quant* | CGG repeats (gene-specific PCR)† | Consensus‡ |
| NA06890 | M | 30 | 30 | 30 | 30 | 30 |
| CD00014 | M | 56 | 56 | 56 | 56 | 56 |
| NA06891 | M | 118 | 119 | 123 | 119 | |
| NA09145 | M | Full | >200 | >200 | >200 | |
| NA06852 | M | >200 | >200 | >200 | >200 | |
| NA06897 | M | 477 | >200 | >200 | >200 | |
| NA07862 | M | 501–550 | >200 | >200 | >200 | |
| NA04025 | M | 645 | >200 | >200 | >200 | |
| NA07538 | F | 29/29 | 29 | 29 | 29 | 29 |
| NA07541 | F | 29/31 | 29 | 29 | ||
| 31 | 31 | 31 | ||||
| NA13664 | F | 28/49 (+/−3) | 30 | 30 | 30 | |
| 51 | 52 | 52 | ||||
| NA20240 | F | 30/80 | 30 | 30 | 30 | 30 |
| 81 | 83 | 83 | 80 | |||
| NA06896 | F | 23/95–140 | 23 | 23 | 23 | |
| 113 | 113 | |||||
| 133–138 | 133–138 | |||||
| 155 | 154 | |||||
| 175 | 176 | |||||
| 198 | 198 | |||||
| >200 | >200 | |||||
| NA20239 | F | 20/183–193 | 20 | 20 | 20 | 20 |
| 198 | 206 | 198 | NC | |||
| >200 | >200 | |||||
| NA07537 | F | 28/336 | 29 | 29 | 29 | |
| >200 | >200 | >200 | ||||
| NAO5847 | F | 21/650 | 20 | 20 | 20 | |
| >200 | >200 | >200 | ||||
| NA20238 | F | 29/30 | 29 | 29 | 29 | 29 |
| 30 | 30 | 30 | 30 | |||
| NA20243 | F | 29/41 | 29 | 29 | 29 | 29 |
| 41 | 41 | 41 | 41 | |||
| NA20235 | F | 29/45 | 29 | 29 | 29 | 29 |
| 45 | 45 | 45 | 45 | |||
| NA20242 | F | 30/73 | 30 | 30 | 30 | 30 |
| 73 | 74 | 74 | 73 | |||
| 105 | 104 | |||||
The “Submitted CGG repeat number” is that provided to and by CCR for the cell line DNA sample indicated. “Fragment sizing” refers to the determination of the CGG repeat number based upon the mobility of the full-length amplicon of the RP PCR reaction. “Absolute quant” indicates the number of CGG repeats based on peak counting alone.
NC, no consensus; M, male; F, female.
This study.
Gene-specific FMR1 PCR (8).
Consensus result from (13).
For longer alleles, individual (CGG)n repeat peaks could be quantified up to at least 200 CGG for each of eight full mutation templates characterized in Figure 2. Absolute quantification was limited to ∼200 to 220 CGG with the CE configuration used, thus permitting identification of expanded repeats into the full mutation range. All full mutation cell line templates were amplified by the flanking gene-specific primers to produce full-length amplicons corresponding to up to at least 940 CGG repeats.
The reproducibility of the CGG RP PCR was assessed using replicate measurements of the same sample with different operators, thermal cyclers, and days of reagent and sample preparation. A DNA template composed of an admixture of 20% 940 CGG gDNA in a background of 80% 23 CGG gDNA8 was evaluated in this repeatability testing. The data were assessed using four criteria: size of the full-length 23 CGG allele, signal of the repeat primed product corresponding to 100 CGG, an assessment of the longest repeat primed amplicons with at least 10 reflectance fluorescence units (rfu) (approximately two to threefold greater than the baseline signal), and the peak height of the full-length 940 CGG allele. Six replicate tests across two operators, two thermal cyclers, and 2 days of testing yielded a 100% pass rate for all data collected, and thus amplicons corresponding to both the 23 and 940 CGG allele were detected in every case. In addition, the following was observed: i) the fragment size of the 23 CGG full-length allele was 298.4 ± 0.2 bp; ii) an average signal of 110 ± 30 rfu was computed for amplicons with 100 CGG repeats; iii) the largest detectable repeat primed products exceeding 10 rfu were 720 ± 30 bp, corresponding to approximately 190 CGG repeats in all replicates; and iv) an average signal of 310 ± 75 rfu was determined for the peak corresponding to the full-length 940 CGG allele. Thus, this method generated highly reproducible and accurate quantification across a range of repeat lengths, with alleles from both male and female samples.
CGG Product Profiles Provide a Unique Signature for Female Heterozygous Alleles That Can Resolve Allele Zygosity
CGG RP PCR generates distinct product profiles for alleles of differing numbers of (CGG)n repeats. As a result, the method can theoretically differentiate heterozygous and homozygous female samples. To demonstrate this capability, 42 blinded female specimens comprising both homozygous and heterozygous samples were assessed by CGG RP PCR. All 42 samples were correctly identified, based on comparisons to Southern blot data (accuracy for detection of homozygous alleles = 100.0% [95% confidence interval 91.6 to 100.0%]). This sample cohort included 21 heterozygous full mutations, 11 heterozygous normal alleles, and 10 homozygous samples with (CGG)n repeat numbers in the normal range. Figure 3 provides results from six representative samples, including four from blinded clinical samples that illustrate the stark differences in the PCR product profiles of alleles of varying zygosity. Homozygous templates presented no CGG-specific product peaks after the longest product corresponding to the two identically sized alleles, whereas a series of distinct CGG-specific peaks that extended beyond the shorter allele were observed whenever heterozygous alleles were present.
Figure 3.
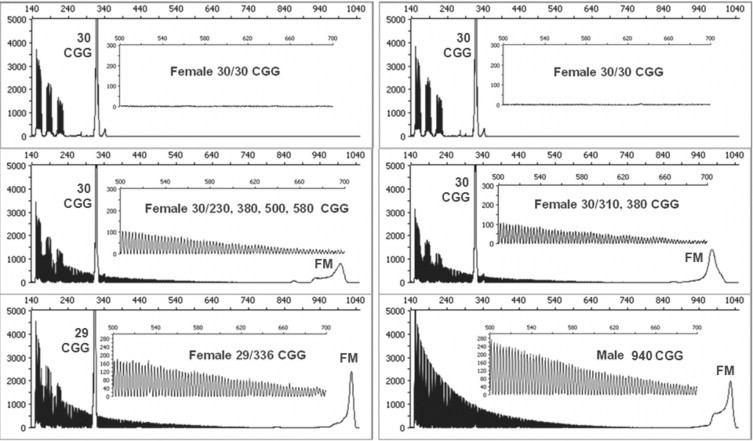
Heterozygous female alleles produce an uninterrupted series of triplet repeat products that indicate the presence of a longer FMR1 allele and thus are readily distinguished from homozygous alleles. Note that PCR/CE of homozygous alleles (top) lack the characteristic and reproducible repeat primed signature (inset) of heterozygous alleles (bottom). FM, gene-specific PCR products that result from amplification of full mutation alleles.
PCR Performance with Clinical Samples: Comparison of CGG RP PCR and Southern Blot Analysis
The CGG RP PCR method was then evaluated with an additional set of 146 blinded clinical specimens that were previously characterized by FMR1 Southern blot at the M.I.N.D. Institute.10 All samples were successfully amplified in the first PCR run, resulting in a 100% pass rate. A total of 42 samples were categorized as normal and 3 as intermediate by both PCR and Southern blot methods. Premutation alleles were also co-identified by CGG RP PCR and Southern blot analysis. In addition, Southern blot analysis revealed 66 full mutation specimens. All 66 of these samples were found to possess full mutation alleles by CGG RP PCR, including alleles with up to 1300 CGG repeats and several others with >1000 CGG. Thus, CGG RP PCR categorized the full range of FMR1 alleles, including full mutations, consistent with the results of Southern blot analysis of the same specimens. In total, 144 of 146 samples were concordant with Southern blot analysis (accuracy for detection of samples with full mutations = 98.6% [95% confidence interval 95.1 to 99.6%]), and 146 of 146 samples were concordant between gene-specific FMR1 PCR8 and CGG RP PCR (accuracy = 100.0% [95% confidence interval 97.4 to 100.0%]). The two discordant samples both presented readily detectable permutation alleles by gene-specific PCR, CGG RP PCR, and Southern blot analysis but also low-intensity full mutations by the two PCR methods only. As discussed previously,8 these results may reflect the increased sensitivity of the PCR reagents for such low-abundance alleles compared with the Southern blot procedure.
Full-Length FMR1 Amplification Is Not Required to Identify Full Mutation Alleles
In theory, the ability of the CGG RP PCR to detect full mutation alleles is not limited by the number of repeats in the DNA template. The combination of the repeat and reverse primer pair interrogate the 3′ end of the repeat region, and as such these primers should produce a series of amplicons with up to hundreds of CGG irrespective of whether the template is 200 CGG or several thousand CGG. To support this idea experimentally, PCR products were prepared from cell line gDNA templates with approximately 550, 645, and 940 CGG using a previously published FMR1 forward primer12 in combination with the gene-specific reverse primer (Figure 4). Purified and diluted full-length PCR products, lacking a primer binding site for the standard forward primer were then input into a CGG RP PCR. As shown in Figure 4, full mutation alleles in all three templates were correctly identified, inasmuch as >220 discrete CGG peaks could be counted in each case even though no full-length amplicon was detected, as expected.
Figure 4.
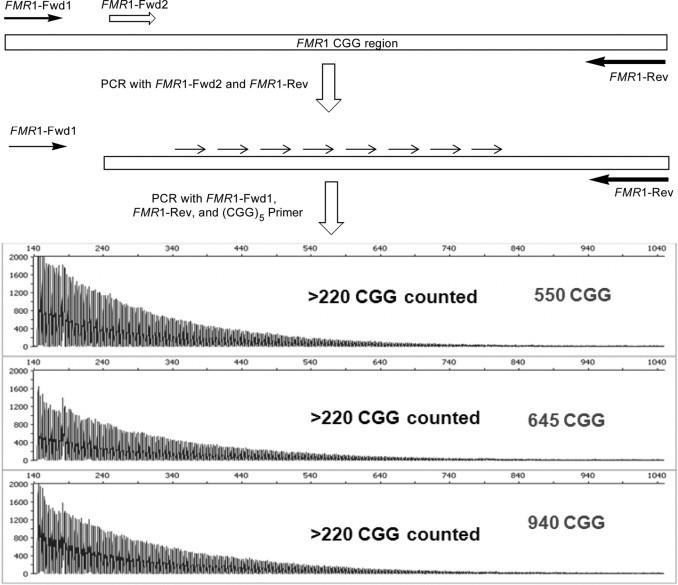
The three-primer repeat primed PCR reports the presence of full mutation alleles even in the absence of a full-length amplicon. Fwd, forward; Rev, reverse.
The Specificity of (CGG)n Primer Binding to FMR1 5′ Untranslated Region Sequences Reveals the Sequence Context of AGG Interruptions
Many FMR1 alleles contain AGG sequences that are interspersed among the (CGG)n repeats, usually in the 5′ region of the repeat segment. Existing methods to map AGG elements include sequencing and restriction mapping.14,15 Both approaches are laborious and are not routinely performed in fragile X diagnostic testing. As described above, the inability of the CGG repeat primer to bind efficiently to AGG sequences in the triplet repeat tract can be exploited to reveal the number and sequence context of such interspersions. An example of the data and subsequent interpretation that yields the AGG interruption sequence is shown in Figure 5. The analysis required to map such AGG sequences in male samples was straightforward, and the results of such AGG assessments were concordant with DNA sequencing in each of 10 representative male clinical samples (Figure 6A, Supplemental Table 1, see http://jmd.amjpathol.org).
Figure 5.
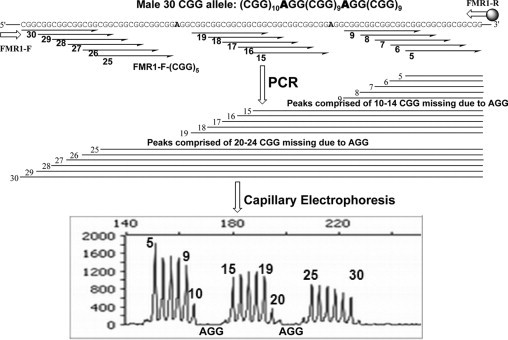
The CGG RP PCR product profile can reveal both the number and sequence context of AGG elements that interrupt the CGG repeat segment. Top: Schematic representation of the impact of defined AGG sequences on the CGG RP PCR product profile for a male 30 CGG allele. Interrupting AGG elements are associated with a loss of product accumulation (and associated signal intensity) for those amplicons whose corresponding CGG primer contacts a templated AGG. Bottom: Annotation of an capillary electropherogram of a male 30 CGG allele with both CGG repeats, and the two interfering AGG sequences.
Figure 6.
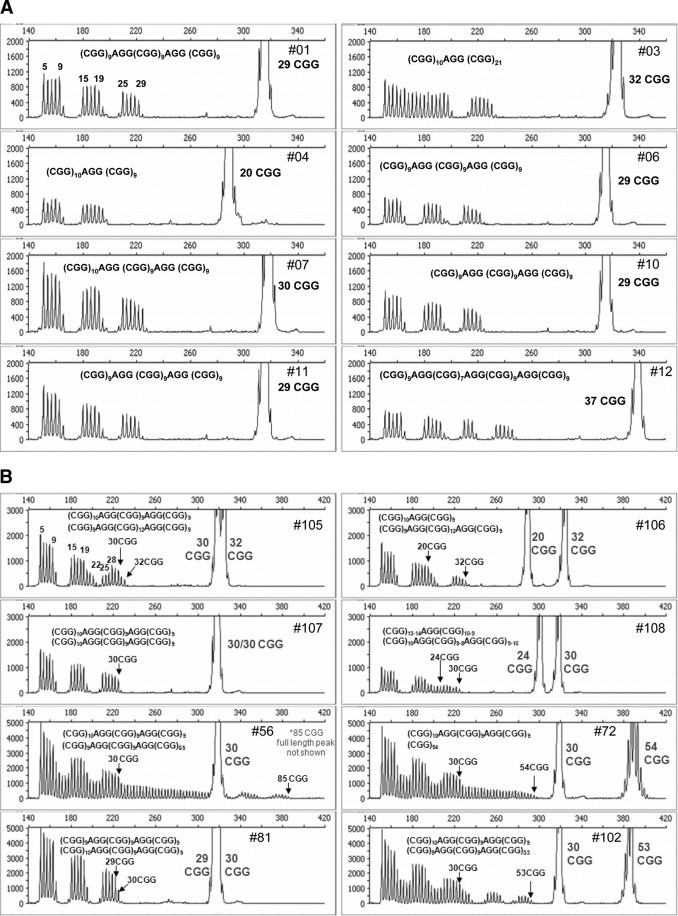
Identification of AGG interspersions in both male and female FMR1 templates. A: Capillary electropherogram and corresponding sequence context of AGG elements in male FMR1 alleles. The CGG repeat number determined from fragment sizing of the full-length, gene-specific peak is indicated for each gDNA template. B: Electropherogram and corresponding sequence context of AGG elements in female FMR1 alleles. The arrows mark the last CGG repeat associated with each of the two FMR1 alleles for each set of female samples. If the interpretation of the AGG sequence is ambiguous, more than one sequence possibility is given.
Figure 6B presents capillary electropherograms of the repeat primed profiles from eight female samples. These results reflected a range of AGG-dependent signal changes, in contrast to the quantitative signal drops observed in AGG elements within male hemizygous templates. The reason for the variability in signals was that female samples are composed of two alleles, and thus the RP PCR profile is a composite of two populations of (CGG)n repeat products. One key difference between the profile of male and female alleles was that this “AGG dip” does not decrease to the level of the baseline noise in female samples unless the AGG positions are exactly overlapping relative to the 3′ end of the amplicon. Nevertheless, the AGG positions could be determined from the relative position and degree of the signal drop in the heterozygous allele RP PCR profile.
As an example, the number and sequence context of AGG interspersions in samples 81, 106, and 107 were unambiguously resolved. Both homozygous alleles in 81 and 107 present overlapping AGG sequence elements and, thus, as observed in male specimens, the signal of the repeat primed product profile was reduced to baseline for each of the two sets of AGG interrupters. However, sample 106, a sample with one allele of 20 repeats and another of 32 repeats, required a more complex analysis. In this case, two AGG dips were observed. The two heterozygous alleles presented one overlapping AGG relative to the 3′ end, as evident by the baseline signal intensity of 5 repeat units. A second dip in signal was also observed, corresponding to another AGG unit. This second dip in signal was beyond the 20 repeats of the first allele; thus, this second AGG could only be contributed by the longer 32 CGG allele. At least for this sample, distinct peak counting allowed unambiguous resolution of not only the presence of AGG elements but also their specific allele haplotype.
The AGG positions for the remaining five samples in Figure 6B could be identified but not definitively sequence-mapped based solely on the empirical CGG repeat primed product profile. In the majority of cases, however, the most likely interpretation was correctly inferred from a knowledge of the most common AGG haplotypes.16,17,18 Because AGG interruptions are clustered near the 5′ end of the repeat segment and are often spaced by 9 or 10 CGG repeats, the AGG interspersions are usually located at either the 10, 20 or 11, 21 positions relative to the starting 5′ CGG repeat. For example, in sample 108, if the observed 4 AGG dips are each assigned to the allele with 85 CGG, then the resulting sequence, (CGG)9AGG(CGG)9AGG(CGG)45AGG(CGG)9 AGG(CGG)9 (ie, a pattern of 10, 20, 66, 76), is inconsistent with known AGG haplotypes. With this logic, the most likely AGG sequence context for each allele of samples 56, 72, 102, and 105 can be deduced. In each case, the most probable genotype was confirmed by other methods, such as DNA sequencing (Supplemental Table 1, see http://jmd.amjpathol.org). Examples of the interpretation of more complex alleles, such as those represented by samples 105 and 108, are provided in Supplemental Figures 1 and 2 (see http://jmd.amjpathol.org), respectively.
Discussion
Quantification of the number of (CGG)n repeats in the 5′ untranslated region of the FMR1 gene is central to fragile X molecular diagnostic testing. Although this information can be extracted by determining the length of the amplified fragment after FMR1 PCR, expanded alleles have been historically refractory to such amplification. As a result, FMR1 analyses also require Southern blot methods to size those alleles too large to support efficient PCR. In this report, we described the performance of a CGG RP FMR1 PCR that reproducibly detected full mutations, enabled absolute quantification of (CGG)n repeats, definitively reconciled allele zygosity, and revealed the sequence context of alleles with AGG interspersions. CGG RP PCR was primarily distinguished from the more conventional two-primer, gene-specific PCR by the addition of a third PCR primer that was complementary to the FMR1 triplet repeat region (Figure 1). This design offered several benefits for FMR1 molecular characterizations compared with current PCR methods.
First, the addition of a repeat primer and analysis using CE permitted absolute quantification of the number of triplet repeats in a given allele. As described for FMR110,19 and other triplet repeat gene targets,20 the repeat primer bound to each corresponding repeat element and produced a discrete PCR product for every such binding event. As a result, a ladder of amplicons was generated that reported both the number and placement of each repeat unit along the gene. When analyzed by agarose gel electrophoresis, these products appeared as a “smear” that extended up to the length of the longest allele present in the reaction.10 When analyzed on high-resolution platforms such as CE, however, these products were visualized as distinct peaks separated by three nucleotides. The number of peaks corresponded to the number of underlying molecular repeat units. Thus, the RP peak profile can theoretically provide very accurate (CGG)n repeat quantification. In our study, (CGG)n repeat lengths for 12 of 14 alleles previously characterized in an interlaboratory study of the Fragile Xperts Working Group13 were identical to those determined using absolute quantification after CGG RP PCR. Discrepancies in the repeat number were 1 or 3 triplet repeats. These differences were observed for both permutation-sized alleles and not for shorter alleles. It is important to note that quantification of the number of (CGG)n repeats by peak counting can provide larger repeat lengths than fragment sizing for alleles greater than about 70 CGG. The reason for this difference is that fragment sizing (like DNA sequencing) reports quantification from the most intense peak in the capillary electropherogram, whereas peak counting reveals the repeat number in the smallest peak that is resolved from the background signal and thus the largest detectable peak present in the data. Because PCR products of templates with (CGG)n repeats manifest prominent n-1, n-2, etc., peaks, we propose that the longest peaks in such RP PCR traces more accurately reflect the underlying repeat size of the FMR1 template. As a result, absolute quantification may prove particularly helpful in characterizing reference materials beyond what DNA sequencing can support and for which consensus testing is the only recourse for the standardization of repeat length.13
Second, the CGG RP PCR repeat profile telegraphed the presence of a longer allele in the amplification, irrespective of whether such an allele could be detected as a full-length product. Again, we note that each of the 66 full mutations assessed in this study was detected as both a full-length amplicon from CGG RP PCR with the two gene-specific primers, as well as through the corresponding pattern of ∼200 discrete CGG RP products. Thus, the RP PCR product profile provided an autonomous data signature for expanded alleles that may not be amplifiable, whether the reason is that the repeat span is too long to efficiently amplify or that the template is partially degraded, or another reason. This point is important because the RP PCR method generates a signature profile for alleles of any CGG repeat length and thus reconciles the interpretation of confounding female homozygous alleles that are often reflexed to Southern blot analysis for resolution.
Third, interspersed AGG elements that are known to be present in many FMR1 alleles were mapped with high precision in many cases (Figures 5 and 6). Because the CGG repeat primer bound poorly to noncomplementary sequences, distinctive allele-specific changes in signal intensity could identify the presence and context of AGG sequence units. Knowledge of the number and spacing of AGG elements has been proposed to be associated with an elevated risk profile in some fragile X carriers, and it has been suggested that the risk profile for mothers with no AGG interspersions and small premutations may be higher than that for mothers with the same number of repeats but with at least one AGG and thus fewer consecutive (CGG)n sequences.14,17,21,22 In fact, Eichler et al14 has previously proposed that an uninterrupted series of 34 to 38 CGG repeats delineates a threshold of allele instability for expansion in future generations. Because the CGG RP PCR provides insights into the AGG interruption pattern, and thus the number of consecutive (CGG)n repeats, on each interrogated sample, the routine use of this assay for FMR1 analysis may support additional clinical research in this area without the need for dedicated DNA sequencing or restriction enzyme mapping assays. Knowledge of the sequence context of AGG elements may also provide insights into the risk prediction or clinical features of fragile X-associated primary ovarian insufficiency and/or fragile X-associated tremor/ataxia syndrome.
It may be instructive to compare the capabilities of the three-primer, CGG RP PCR, with the recently published FMR1 PCR configuration that was evaluated with common reagents, but only two gene-specific primers.8 For example, both technologies generated full-length amplicons that encompass the entire CGG repeat region. Both methods detected all full mutation alleles from blinded clinical specimens, including male and female samples composed of expanded alleles with up to 1300 CGG, and produced concordant results with one another and in excellent agreement with Southern blot analysis. Both methods provided accurate assessments of sample zygosity. However, zygosity was inferred from the two-primer, gene-specific PCR inasmuch as the expanded allele in all female heterozygous samples was detected in every case.8 In contrast, zygosity was independently confirmed for female heterozygous alleles with the RP PCR by virtue of a CGG-specific product profile that was only produced when a longer allele was present, even if that allele could not be amplified as a full-length product (Figure 4). In addition, the RP PCR, but not the gene-specific PCR, enabled absolute triplet repeat quantification up to ∼200 to 220 CGG repeats by producing a series of discrete CGG-specific amplicon peaks. Last, the CGG-specific product profile could be exploited to reveal the number and placement of AGG elements in many samples.
An obvious question is how the novel gene-specific PCR8 and the CGG RP PCR reagents relate to one another to support accurate and efficient FMR1 analysis. One important benefit of the two-primer, gene-specific PCR is that the relatively few products that are generated are amenable to semiautomated allele calls, which can greatly reduce the burden of analysis, particularly for projects associated with large sample volumes. In contrast, RP PCR produces a multiplicity of peaks that require manual, hence more time-consuming, analyses for each sample. RP PCR also provides direct evidence of sample heterozygosity, which must be inferred from the gene-specific PCR. One possibility is to evaluate all incoming samples with the gene-specific PCR and then use CGG RP PCR as a confirmatory method for specific samples, as previously proposed.10 On the other hand, one PCR strategy may be better suited than the other to meet the requirements of different laboratory environments. In either case, the distinctions between the two formats suggest that both PCR technologies can better serve the fragile X community than either one alone.
In summary, the FMR1 CGG RP PCR reagents described here provide detailed genotyping data that may have particular utility for fragile X clinical research. The technology addresses long-standing limitations in FMR1 PCR analysis that have necessitated the use of much lower throughput and laborious Southern blot analysis. As a result, the capabilities of the RP PCR may reduce the burden of Southern blot analysis to only those samples that require methylation information, which are typically ≤2% of the total samples processed by large clinical reference laboratories.23 Moreover, the PCR reagents described both here and previously8 may support PCR-based methylation assessments of FMR1 alleles using methylation-sensitive restriction enzymes. As a result, the RP PCR technology, and the corresponding gene-specific PCR technology,8 represents an important step toward a PCR-only workflow for FMR1 analysis.
Footnotes
Supported in part by awards from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (grants R43-HD060450 to A.H., HD02274 to F.T., and HD040661 to P.J.H.).
L.C., A.H., S.S., S.F.-S., J.K., E.S., T.T.S., and G.J.L. were all employees of Asuragen at the time the work was performed. None of the other authors declare any relevant financial relationships. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health and Human Development or the National Institutes of Health.
This work was presented in part as abstract number G11 at the annual meeting of the Association for Molecular Pathology, Orlando, FL, November 19–22, 2009.
Supplemental material for this article can be found on http://jmd.amjpathol.org.
Current address of E.S.: Luminex Corporation, Austin, TX; of T.T.S.: Quidel Corporation, Sand Diego, CA.
Web Extra Material
AGG Mapping in a Female Heterozygous Sample Comprised of Two Alleles of Similar Size. Three categories of CE peaks indicating four AGG spacers are reflected by the data: 1) High intensity product peaks contributed by both alleles (#5-9,15-19, and 28-30); 2) Low intensity peaks contributed by single allele (20-22, 25-27, and 31-32); and 3) Zero to very low intensity peaks caused by overlapping AGG sequences in both alleles (10-14, and 23-24). Note that the shorter of the two alleles has 30 CGG, thus the 31 and 32 CGG peaks are contributed only by the longer (32 CGG) allele. Since the 10-14 CGG peaks are completely absent across the equivalent of a 5 CGG stretch, both alleles must present an AGG at the same position (e.g., AGG(CGG)9) relative to the 3′ end. Thus, the only remaining issue is how to assign the third and fourth AGG units within the context of peaks 20-24 and 23-27. Since each AGG will cause a reduction in peak intensity across the equivalent of 5 repeats, one possibility is to assign the second AGG to #20-24 in the 30 CGG allele, and #23-27 in the 32 CGG allele (Case 1 scenario at top left). A second possibility is the opposite assignment (Case 2 scenario at top right). Both scenarios will reproduce the observed empirical CE profile. However, Case 1 (with AGG sequences located at positions 11, 21 and 10, 23) is more consistent with known AGG patterns. This result is also in agreement with DNA sequencing (Suppl Table 1).
AGG Mapping in a Complex Female Heterozygous Sample. Three categories of CE peaks indicating three AGG spacers are reflected by the data: 1) High intensity product peaks contributed by both alleles (#5-9,and 16-19); 2) Low intensity peaks contributed by single allele (#10,15, and 20-30); and 3) Zero to very low intensity peaks caused by overlapping AGG sequences in both alleles (#11-14). Note that the shorter of the two alleles has 24 CGG, thus the 25-30 CGG peaks are contributed only by the longer (30 CGG) allele. Since the #20-24 CGG peaks are caused by a single allele, the corresponding AGG must map to the 30 CGG allele. Thus, the question is how to map the reduced peak intensities of #10 and #15. If #10 is assigned to the 24 CGG allele, then #15 must be assigned to the 30 CGG allele since one AGG can only cause a signal loss in 5 consecutive peaks in the CE profile and vice versa. Consequently, there are only two possibilities: 1) #10 assigned to the 24 CGG allele, and #15 assigned to the 30 CGG allele, (CGG)13AGG(CGG)10 /(CGG)10AGG(CGG)9AGG(CGG)9, or 2) #15 assigned to the 24 CGG allele, and #10 assigned to the 30 CGG allele, (CGG)14AGG(CGG)9/ (CGG)10AGG(CGG)8AGG(CGG)10. The first of these two possibilities was consistent with the results of DNA sequencing (Suppl Table 1).
Determination of the AGG Interruption Pattern for a Set of Male and Female Clinical Specimens, and Comparisons with DNA Sequencing. The number of CGG repeats was determined based on fragment sizing using CGG repeat primed PCR. The most likely AGG pattern is indicated by the spacing of AGG interruptors within the context of CGG repeats, i.e., “10, 20” refers to 5′-(CGG)9AGG(CGG)9AGG(CGG)n-3″. ND, Not Determined.
References
- 1.Jacquemont S, Hagerman RJ, Hagerman PJ, Leehey MA. Fragile-X syndrome and fragile X-associated tremor/ataxia syndrome: two faces of FMR1. Lancet Neurol. 2007;6:45–55. doi: 10.1016/S1474-4422(06)70676-7. [DOI] [PubMed] [Google Scholar]
- 2.Hagerman RJ, Hagerman PJ. The fragile X premutation: into the phenotypic fold. Curr Opin Genet Dev. 2002;12:278–283. doi: 10.1016/s0959-437x(02)00299-x. [DOI] [PubMed] [Google Scholar]
- 3.Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, Grigsby J, Gage B, Hagerman PJ. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- 4.Sullivan AK, Marcus M, Epstein MP, Allen EG, Anido AE, Paquin JJ, Yadav-Shah M, Sherman SL. Association of FMR1 repeat size with ovarian dysfunction. Hum Reprod. 2005;20:402–412. doi: 10.1093/humrep/deh635. [DOI] [PubMed] [Google Scholar]
- 5.Hagerman RJ, Hagerman PJ. Fragile X Syndrome: Diagnosis, Treatment, and Research. 3rd ed. The Johns Hopkins University Press; Baltimore: 2002. pp. 3–109. [Google Scholar]
- 6.Coffey SM, Cook K, Tartaglia N, Tassone F, Nguyen DV, Pan R, Bronsky HE, Yuhas J, Borodyanskaya M, Grigsby J, Doerflinger M, Hagerman PJ, Hagerman RJ. Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A. 2008;146A:1009–1016. doi: 10.1002/ajmg.a.32060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hagerman RJ, Hagerman PJ. Testing for fragile X gene mutations throughout the life span. JAMA. 2008;300:2419–2421. doi: 10.1001/jama.2008.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Filipovic-Sadic S, Sah S, Chen L, Krosting J, Sekinger E, Zhang W, Hagerman PJ, Stenzel T, Hadd A, Latham GJ, Tassone F. A novel FMR1 PCR method for the routine detection of low-abundance expanded alleles and full mutations of fragile X syndrome. Clin Chem. 2010;56:399–408. doi: 10.1373/clinchem.2009.136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez-Carvajal I, Walichiewicz P, Xiaosen X, Pan R, Hagerman PJ, Tassone F. Screening for expanded alleles of the FMR1 gene in blood spots from newborn males in a Spanish population. J Mol Diagn. 2009;11:324–329. doi: 10.2353/jmoldx.2009.080173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10:43–49. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cilia R, Kraff J, Canesi M, Pezzoli G, Goldwurm S, Amiri K, Tang HT, Pan R, Hagerman PJ, Tassone F. Screening for the presence of FMR1 premutation alleles in women with parkinsonism. Arch Neurol. 2009;66:244–249. doi: 10.1001/archneurol.2008.548. [DOI] [PubMed] [Google Scholar]
- 12.Saluto A, Brussino A, Tassone F, Arduino C, Cagnoli C, Pappi P, Hagerman P, Migone N, Brusco A. An enhanced polymerase chain reaction assay to detect pre- and full mutation alleles of the fragile X mental retardation 1 gene. J Mol Diagn. 2005;7:605–612. doi: 10.1016/S1525-1578(10)60594-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amos Wilson J, Pratt VM, Phansalkar A, Muralidharan K, Highsmith WE, Jr, Beck JC, Bridgeman S, Courtney EM, Epp L, Ferreira-Gonzalez A, Hjelm NL, Holtegaard LM, Jama MA, Jakupciak JP, Johnson MA, Labrousse P, Lyon E, Prior TW, Richards CS, Richie KL, Roa BB, Rohlfs EM, Sellers T, Sherman SL, Siegrist KA, Silverman LM, Wiszniewska J, Kalman LV. Consensus characterization of 16 FMR1 reference materials: a consortium study. J Mol Diagn. 2008;10:2–12. doi: 10.2353/jmoldx.2008.070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eichler EE, Holden JJ, Popovich BW, Reiss AL, Snow K, Thibodeau SN, Richards CS, Ward PA, Nelson DL. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat Genet. 1994;8:88–94. doi: 10.1038/ng0994-88. [DOI] [PubMed] [Google Scholar]
- 15.Kunst CB, Zerylnick C, Karickhoff L, Eichler E, Bullard J, Chalifoux M, Holden JJ, Torroni A, Nelson DL, Warren ST. FMR1 in global populations. Am J Hum Genet. 1996;58:513–522. [PMC free article] [PubMed] [Google Scholar]
- 16.Eichler EE, Macpherson JN, Murray A, Jacobs PA, Chakravarti A, Nelson DL. Haplotype and interspersion analysis of the FMR1 CGG repeat identifies two different mutational pathways for the origin of the fragile X syndrome. Hum Mol Genet. 1996;5:319–330. doi: 10.1093/hmg/5.3.319. [DOI] [PubMed] [Google Scholar]
- 17.Zhong N, Ju W, Pietrofesa J, Wang D, Dobkin C, Brown WT. Fragile X “gray zone” alleles: AGG patterns, expansion risks, and associated haplotypes. Am J Med Genet. 1996;64:261–265. doi: 10.1002/(SICI)1096-8628(19960809)64:2<261::AID-AJMG5>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 18.Zhong N, Yang W, Dobkin C, Brown WT. Fragile X gene instability: anchoring AGGs and linked microsatellites. Am J Hum Genet. 1995;57:351–361. [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y, Law HY, Boehm CD, Yoon CS, Cutting GR, Ng IS, Chong SS. Robust fragile X (CGG)n genotype classification using a methylation specific triple PCR assay. J Med Genet. 2004;41:e45. doi: 10.1136/jmg.2003.012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warner JP, Barron LH, Goudie D, Kelly K, Dow D, Fitzpatrick DR, Brock DJ. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J Med Genet. 1996;33:1022–1026. doi: 10.1136/jmg.33.12.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fernandez-Carvajal I, Lopez Posadas B, Pan R, Raske C, Hagerman PJ, Tassone F. Expansion of an FMR1 grey-zone allele to a full mutation in two generations. J Mol Diagn. 2009;11:306–310. doi: 10.2353/jmoldx.2009.080174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kunst CB, Warren ST. Cryptic and polar variation of the fragile X repeat could result in predisposing normal alleles. Cell. 1994;77:853–861. doi: 10.1016/0092-8674(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 23.Strom CM, Crossley B, Redman JB, Buller A, Quan F, Peng M, McGinnis M, Fenwick RG, Jr, Sun W. Molecular testing for fragile X syndrome: lessons learned from 119,232 tests performed in a clinical laboratory. Genet Med. 2007;9:46–51. doi: 10.1097/gim.0b013e31802d833c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
AGG Mapping in a Female Heterozygous Sample Comprised of Two Alleles of Similar Size. Three categories of CE peaks indicating four AGG spacers are reflected by the data: 1) High intensity product peaks contributed by both alleles (#5-9,15-19, and 28-30); 2) Low intensity peaks contributed by single allele (20-22, 25-27, and 31-32); and 3) Zero to very low intensity peaks caused by overlapping AGG sequences in both alleles (10-14, and 23-24). Note that the shorter of the two alleles has 30 CGG, thus the 31 and 32 CGG peaks are contributed only by the longer (32 CGG) allele. Since the 10-14 CGG peaks are completely absent across the equivalent of a 5 CGG stretch, both alleles must present an AGG at the same position (e.g., AGG(CGG)9) relative to the 3′ end. Thus, the only remaining issue is how to assign the third and fourth AGG units within the context of peaks 20-24 and 23-27. Since each AGG will cause a reduction in peak intensity across the equivalent of 5 repeats, one possibility is to assign the second AGG to #20-24 in the 30 CGG allele, and #23-27 in the 32 CGG allele (Case 1 scenario at top left). A second possibility is the opposite assignment (Case 2 scenario at top right). Both scenarios will reproduce the observed empirical CE profile. However, Case 1 (with AGG sequences located at positions 11, 21 and 10, 23) is more consistent with known AGG patterns. This result is also in agreement with DNA sequencing (Suppl Table 1).
AGG Mapping in a Complex Female Heterozygous Sample. Three categories of CE peaks indicating three AGG spacers are reflected by the data: 1) High intensity product peaks contributed by both alleles (#5-9,and 16-19); 2) Low intensity peaks contributed by single allele (#10,15, and 20-30); and 3) Zero to very low intensity peaks caused by overlapping AGG sequences in both alleles (#11-14). Note that the shorter of the two alleles has 24 CGG, thus the 25-30 CGG peaks are contributed only by the longer (30 CGG) allele. Since the #20-24 CGG peaks are caused by a single allele, the corresponding AGG must map to the 30 CGG allele. Thus, the question is how to map the reduced peak intensities of #10 and #15. If #10 is assigned to the 24 CGG allele, then #15 must be assigned to the 30 CGG allele since one AGG can only cause a signal loss in 5 consecutive peaks in the CE profile and vice versa. Consequently, there are only two possibilities: 1) #10 assigned to the 24 CGG allele, and #15 assigned to the 30 CGG allele, (CGG)13AGG(CGG)10 /(CGG)10AGG(CGG)9AGG(CGG)9, or 2) #15 assigned to the 24 CGG allele, and #10 assigned to the 30 CGG allele, (CGG)14AGG(CGG)9/ (CGG)10AGG(CGG)8AGG(CGG)10. The first of these two possibilities was consistent with the results of DNA sequencing (Suppl Table 1).
Determination of the AGG Interruption Pattern for a Set of Male and Female Clinical Specimens, and Comparisons with DNA Sequencing. The number of CGG repeats was determined based on fragment sizing using CGG repeat primed PCR. The most likely AGG pattern is indicated by the spacing of AGG interruptors within the context of CGG repeats, i.e., “10, 20” refers to 5′-(CGG)9AGG(CGG)9AGG(CGG)n-3″. ND, Not Determined.