

Abstract
Myotonic dystrophy type 2 (DM2, OMIM #602688) is a multisystemic hereditary degenerative disease caused by a tetranucleotide CCTG expansion in the ZNF9 gene. Routine testing strategies for DM2 require the use of Southern blot or long-range PCR, but the presence of very large expansions and wide somatic mosaicism greatly reduce the sensitivity of these reference techniques. We therefore developed and validated a tetraplet-primed PCR (TP-PCR) method to detect the DM2 mutation by testing 87 DM2-positive and 76 DM2-negative previously characterized patients. The specificity of this technique was evaluated including DNA samples from 39 DM1-positive patients. We then attempted a prospective analysis of 50 patients with unknown genotype who referred to our center for diagnostic or presymptomatic tests. Results show that TP-PCR is a fast, reliable, and flexible technique, whose specificity and sensitivity is almost 100%, with no false positive or negative results either in retrospective and prospective applications. We therefore conclude that using this technique, in combination with the short-range PCR, is sufficient to correctly establish the presence or the absence of ZNF9 expanded alleles in the molecular diagnosis of DM2.
Myotonic dystrophy type 2 (DM2; MIM #602688), or Ricker's disease, is a multisystemic hereditary degenerative pathology, clinically resembling myotonic dystrophy type 1 (DM1; MIM #160900), or Steinert's disease.1,2 The main characteristics in both diseases are clinical or electrical myotonia, dystrophic changes of muscles, bilateral cataract with early onset, together with a variable presence of dismetabolic changes, cardiac rhythm alterations, alopecia, selective IgG and IgM deficits, and decreased fertility.3,4 From a clinical point of view, DM2 affects mainly proximal muscles and hips, a distinctive distribution commonly recognized as PROMM (proximal myotonic myopathy).3,5 Some other features of DM2 patients are the presence of cramps, muscular pain,8 no cognitive involvement, and overall the prognosis is more benign.9 An important element in the differential diagnosis with DM1 is the absence of congenital forms, which are observed only in DM1 families and which are characterized by polyhydramnios, reduced fetal movements, hypotonia, and respiratory distress at birth, with a high rate of perinatal deaths, developmental delay, and mental retardation.10 Histological and immunohistochemical studies of DM2 muscles show the dystrophic phenotype with muscle atrophy, nuclear clumps, and type II fibers atrophy.6,7 Atypical clinical presentations in DM2 have also been described, mimicking spinal muscular atrophy with adult onset,11 fibromyalgia,12 or with hyper-CK-emia as the sole clinical manifestation.13 Because of this wide clinical spectrum DM2 can often be misdiagnosed with nondystrophic myotonias, limb-girdle muscular dystrophies, metabolic disorders, and other myopathies.14
DM1 and DM2 are caused by expansion of (CTG)n and (CCTG)n repeats in the DMPK (MIM #605377) and ZNF9 (MIM #116955) genes, respectively.15,16 The normal number of (CTG) repeats in DMPK gene ranges from 5 to 35,17 while DM1 patients show a number of repeats from 50 to over 4000. In DM1 a correlation between the degree of the expansion and the clinical phenotype has been described, with earlier onset and severity proportional to the (CTG)n repeats number.15,18,19 In DM2, the (CCTG)n tetranucleotide is part of a complex repeated motif [TG]n[TCTG]n[CCTG]n, with (CCTG)n repeats ranging from 11 to 26 in the normal alleles. DM2 patients show a pathological number of repeats starting from 75 to 11,000, with an average of 5000. Despite this large variation, no clear correlation has been demonstrated between the expansion length and the DM2 clinical severity.10 Since the discovery of the DM1/DM2 mutations, molecular analysis of the DMPK/ZNF9 genes confirms the diagnosis in patients and allows presymptomatic genetic tests in their asymptomatic relatives. Molecular protocols to detect DM1 and DM2 mutations are similar and both start with a first round of PCR, called short-range PCR, with primers flanking the repeated regions of DMPK/ZNF9 genes. This step allows the discrimination of different alleles in the normal range after agarose gel or capillary electrophoresis runs. Samples showing only one band or signal can represent true homozygous individuals, or DM-affected patients for which amplification of the expanded allele has failed. For this reason, more extensive tests need to be performed. Southern blot from genomic DNA using labeled DMPK/ZNF9 specific probes, or long-range PCR followed by blotting and hybridization with (CTG)5 or (CCTG)5 probes, are the reference approaches to visualize and measure the DM1/DM2 expansions.10,20 Both techniques are time-consuming and require careful evaluation of results, due to somatic length mosaicism that can give very weak and smeared signals, thus limiting the sensibility of the technique particularly for DM2 tests,10 with the possibility of false negative results. To avoid these difficulties and to make the molecular diagnosis easier and faster, Warner et al21 proposed the use of triplet-primed PCR (TP-PCR) for the detection of repeat expansions in DM1 and in other dynamic mutations diseases. The principle of this technique relies on the use of locus-specific PCR primers in combination with a primer designed across the repeated sequence. After PCR reaction, products of different sizes will be produced, according to the number of repetitions. If an expansion occurs, a continuous ladder of PCR amplification fragments exceeding the normal range will be visualized (Figure 1, A–D). The TP-PCR method allowed a successful discrimination between samples resulted homozygous for a normal allele (normal individuals) and heterozygous sample with an expanded alleles (affected individuals) after short-range PCR.21,22 In this study, we developed a nonradioactive version of the TP-PCR method which specifically detects the (CCTG)n expansion in DM2 patients. Our version of TP-PCR takes advantage of capillary electrophoresis on common automatic sequencers using fluorescent-labeled primers. To establish the specificity and the sensitivity of this technique in the DM2 diagnostic routine tests, we performed TP-PCR on 153 individuals, previously characterized by long-range PCR assay. We also undertook a prospective study on 50 patients with unknown genotype at DM2 locus referred for diagnostic or presymptomatic DM2 molecular test.
Figure 1.
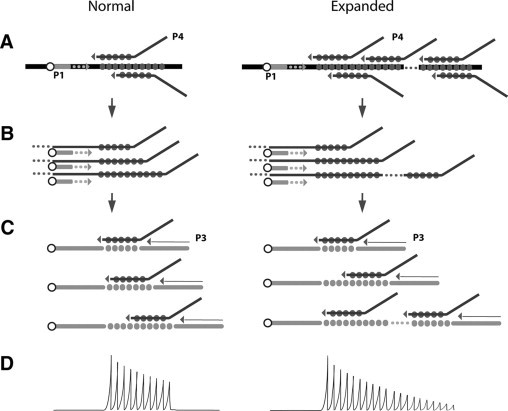
Schematic representation of the TP-PCR principle. Full circles represent tandem repeats. A: Annealing of P4 primer containing complementary repeats. Arrows between panes indicate the passage to subsequent steps. B: P1 FAM-labeled primer (shown as empty circle) permits the formation of fluorescent products of different sizes, according to the number of repeats detected. C: In later cycles, P3 primer enhances selective amplification of repeats-containing products. D: Illustration of electropherogram after capillary electrophoresis on automatic sequencer. Every peak represents a DNA fragment with different number of repeats. Expanded alleles produce a continue ladder of peaks out of the normal range.
Materials and Methods
Patients, Samples, and DNA Extraction
We retrospectively analyzed 153 DNA samples from individuals who underwent molecular diagnosis of DM1 and DM2 in our center from 2001 to 2008. All patients received clinical diagnosis or presented clinical features for which DM was suspected, except for 4 DM1 and 8 DM2, who were referred to us for presymptomatic tests. Each patient gave written informed consent to the use of DNA samples for research purposes. We randomly selected 87 DM2-positive, 39 DM2-negative/DM1-positive, and 37 DM1/DM2-negative DNA samples, previously characterized by short-range PCR and long-range PCR. After selection, all samples were carefully anonymized. To investigate the effects of different DNA extraction procedures on the results, we used DNA, either from fresh (n = 117) and frozen (n = 37) blood samples extracted with EZ1 automatic extraction workstation (Qiagen) and salting-out procedure, respectively.
For prospective analysis, we included 50 patients who referred to our center for DM2 molecular testing between January and June 2009. Except for two patients, who requested presymptomatic test, DM1 genetic analysis was performed before DM2 molecular test. DNA samples were extracted from fresh blood using EZ1 automatic extraction workstation (Qiagen).
Molecular Characterization at DM1 and DM2 Loci
Diagnostic tests for DM1/DM2 were performed following published guidelines.23 Both short-range and long-range PCR across the DM1 and DM2 regions were performed according to reported protocols.20,24 Briefly, PCR fragments obtained after short-range PCR were analyzed on ABI 3130 or ABI 310 automatic sequencers (Applied Biosystems) using ROX500 or LIZ500 (Applied Biosystems) as internal markers. In samples with two alleles in the normal range (corresponding to less than 36 repeats for DM1 and 27 repeats for DM2) the DM1 or DM2 diagnosis was excluded. Samples showing only one signal subsequently underwent long-range PCR and blotting of PCR products for the detection of expanded alleles after hybridization with a radioactive-labeled (CTG)5 or (CCTG)5 probe. DM1 and DM2 expansions were visualized on the Storm 860 Molecular Imager (GMI Inc.) or after exposure on Biomax Light films (Kodak) (Figure 2).
Figure 2.
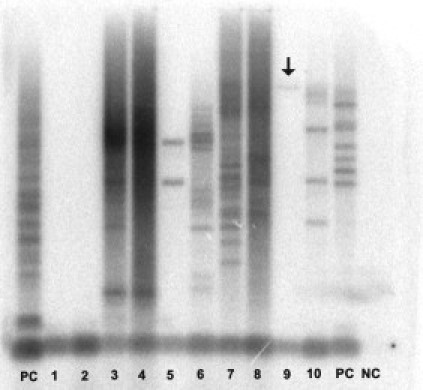
Long-range PCR. CP, Positive control; CN, negative control. Lanes 1–2, Not expanded sample; Lanes 3–10, expanded samples. Arrow in lane 9 indicates a faint band, which could lead to misinterpretation as not expanded samples (false negative).
TP-PCR
Twenty to 200 ng of DNA were amplified in a reaction volume of 30 μl, using 1.5 units of GoTaq Polymerase (Promega), supplied buffer 1×, MgCl2 1.25 mmol/L, DMSO 10%, 7-DeAZA-GTP (Roche) 0.2 mmol/L, primers P1 (Fam-5′-GCCTAGGGGACAAAGTGAGA-3′) 3 pmol, P4 (5′-TACGCATCCGAGTTTGAGACGCCTGCCTGCCTGCCTGCCTG-3′) 0.6 pmol, P3 (5′-TACGCATCCCAGTTTGAGACG-3′) 3 pmol, with the following cycling conditions: initial denaturation 5 minutes/94°C, followed by 35 cycles of (30 seconds/94°C; 30 seconds/56°C; 2 minutes/72°C) and final extension of 10 minutes/72°C, then stored at + 4°C until analyzed. In the prospective study, every analysis included at least one DM2-positive and one DM2-negative sample as controls of PCR reaction.
Ten microliters of each PCR product was run on 3% agarose gel at 100V and checked after 2 hours. For capillary electrophoresis, 2 to 3 μl of the same PCR product were mixed with 16 μl of deionized formamide and loaded on ABI 3130 or ABI 310 automatic sequencers. The analysis of results was performed using GeneMapper 3.7 (Applied Biosystems).
Analysis and Interpretation of TP-PCR Results
Results of capillary electrophoresis for each sample were interpreted by two independent physicians, who were unaware of the patients’ genotypes. When the result was ambiguous or interpretation was not clear for at least one physician, the reason was registered and a new load of the same PCR product was performed on both ABI3130 and ABI310 sequencers. This procedure allows us to establish whether there was a problem in the PCR reaction or in the capillary electrophoresis run. We decided to attempt a new PCR reaction only when no amplification product has been obtained by the first PCR round.
Results
After the first round of TP-PCR on the 153 samples previously characterized, both observers easily identified expanded or normal alleles in 138 samples (90.2%) (Figure, 3A and 3B). Fifteen (9.8%) needed a second capillary run with 3 μl of the same TP-PCR product on automatic sequencer. In seven of these samples (7/153 = 4.6%), the first run failed because too low quantity of the PCR product was loaded on automatic sequencer and, even though peaks over 27 repetitions were visible, it was not possible to establish definitely the presence of expansions in the pathological range. For six samples (6/153 = 3.9%), a high background or very low intensity of fluorescent peaks prevented an unequivocal interpretation of the genotypes. Finally, for two samples (2/153 = 1.3%) the first run failed due to low marker concentration on ABI3130. After the second round of capillary electrophoresis with original PCR products, all remaining 15 genotype statuses were attributed, allowing the definitive classification of all samples as clearly expanded or clearly normal by both physicians. Interpretation of the results was not possible by agarose gel electrophoresis only (Figure 4). At the end of this analysis, we correctly identified all of the 87 DM2-positive and 76 DM2-negative samples. For each DM2-positive sample, we were able to obtain PCR peaks representing tetranucleotide repeats in the pathological range (over 75 CCTG, corresponding to 340 bp), allowing the correct interpretation of all samples with a test sensitivity of 100%. No differences in the electropherograms were noticed between the 39 DM1-positive and the 37 DM1/DM2-negative patients, ruling out the possibility of misinterpretation as false positive due to the presence of CTG repetition in the DMPK gene, resulting in a test specificity of 100%. Use of different DNA quantities, extraction methods, or storage time did not affect the results’ outcome, allowing successful amplification of poor or elder templates conserved up to seven years. Thirty-six of the 50 samples included in the prospective branch of the study resulted heterozygous for normal alleles after short-range PCR, whereas 14 samples showed only one signal. TP-PCR analysis confirmed the absence of expansion for all 36 heterozygous samples. Of the 14 samples with one signal, eight were interpreted as homozygotes for a normal allele and six were interpreted as heterozygotes with one normal and one expanded allele. No sample needed a second run on automatic sequencer, nor TP-PCR repetition. After TP-PCR, all of the 14 samples with one signal underwent long-range PCR that showed the presence of the (CCTG)n expansions in all six samples interpreted as expanded by TP-PCR.
Figure 3.
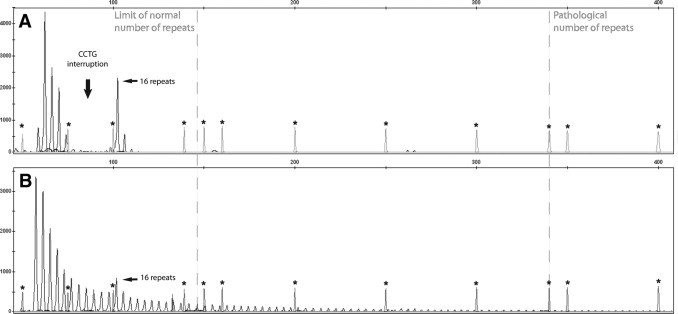
Electropherogram of TP-PCR after capillary electrophoresis. Limits for normal and pathological number of repeats are represented by dashed lines. Asterisks indicate the peaks of ROX500 marker. The continuous ladder of peaks every 4 basepairs represents different fragments generated by random annealing of P4 primer, according to the number of CCTG repeats of the alleles examined. A: Negative heterozygous sample. B: Positive expanded samples, with the ladder of peaks exceeding 340 bp, corresponding to 75 CCTG repeats.
Figure 4.
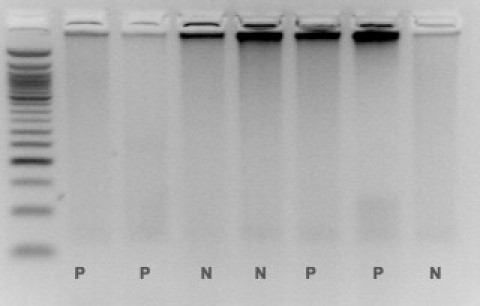
Agarose gel run of TP-PCR products. No clear interpretation is possible. P, Positive (expanded) samples; N, negative (not expanded) samples.
Discussion
Dynamic mutations are a heterogeneous class of pathologies arising from aberrant expansion of normally polymorphic short tandem repeats interspersed in different genes.25,26 Most frequent expansions involve trinucleotides, which can be placed inside exons or in the untranslated portion of the gene, but exceptions occur, as pentanucleotide repeat in SCA10.27 Diagnostic testing for these pathologies has always been challenging, due to extremely wide expansions possible in some of these diseases, as in DM, Friedreich Ataxia, or Fragile-X syndrome.28,29,30 Southern blot is the reference method to detect DNA length variation in dynamic mutations, but it requires a large amount of DNA and is time-consuming with a manual laboratory work-up from four to 13 days to obtain interpretable results. Other conditions make the molecular diagnosis complicated, as in DM2, where the presence of very large expansion and somatic mosaicisms can result in a very smeared and weak signal, limiting the sensibility of this technique to 80% for samples resulted homozygous after short-range PCR.10 To overcome this problem, different strategies have been proposed, as PFGE electrophoresis followed by Southern blot using a combination of ZNF9 and reference probes to compare the intensity of the signal corresponding to the ZNF9 wild-type allele, which should be weaker when an expansion is present.31 Even if somehow useful, all these strategies can still produce ambiguous results and, when adopted, still do not guarantee a correct sizing of (CCTG)n repeated tract. Long-range PCR is an efficient technique to identify expansions up to 2500–3000 repeats but still requires blotting of PCR products and radioactive labeling. Moreover, it has some limitations, because very large expansions could be missed due to failed amplification. After the use of TP-PCR for the detection of (CTG)n expansions in DM1,21 its principle has been successfully adopted to detect a variety of microsatellite expansions, as in SCA2, SCA7, SCA8, SCA10, SCA17, Friedreich's Ataxia, and recently even in Fragile-X Syndrome.32,33,34,35 In 2004, Ranum et al proposed the use of this assay also for DM2, but they consider this procedure only a supportive, and not diagnostic test, due to the presence of 21% false-positive results.10 In their experience, blotting of PCR products and hybridization with radiolabeled-probe is necessary to increase the specificity of this technique, but this process become more time-consuming and expensive.
In this study, we developed a revised version of the TP-PCR assay for the DM2 molecular diagnosis. We validated this technique by evaluating a total of 203 DNA samples. Both retrospective and prospective branches of the study gave no false positives, nor false negatives, without being aware of short-range PCR results. TP-PCR amplification is sufficiently robust to detect all normal and premutated allelic range, and this permits unequivocal interpretation of results. In comparison with the TP-PCR method described by Ranum et al, our protocol used a primer with (CCTG)5 repeats instead of (CCTG)4, together with a higher annealing temperature in the PCR reaction (56°C instead of 50°C). These modifications could be sufficient to explain the higher specificity and sensibility achieved with our method. The compatibility of this technique with common automated DNA extraction methods permits an easy standardization of laboratory work, which reduced to two days the time necessary to complete a molecular diagnosis of DM2, compared with the 6–13 days needed when genomic or PCR blotting and hybridization are adopted. Moreover, no radioactive isotopes are used, leading to an easier management of laboratory work and safety. Our results, moreover, demonstrate that this technique has greater sensitivity and specificity than the combination of conventional short-range and long-range PCR.
However, we have to point out that this method is not quantitative and does not determine the (CCTG)n repeats number. Because no genotype-phenotype correlation has been reported in DM2 patients, this limitation is not relevant for diagnostic purpose. The use of Southern blot and long-range PCR can then be restricted to research applications or as second confirmation techniques in presymptomatic testing. For all these reasons, we propose TP-PCR as a routine diagnostic tool, in combination with short-range PCR, for the molecular diagnosis of DM2.
Footnotes
Supported by Telethon grant No. GPP07250 and AFM grant No. 13360.
References
- 1.Ricker K, Koch MC, Lehmann-Horn F, Pongratz D, Otto M, Heine R, Moxley RT., 3rd Proximal myotonic myopathy: a new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology. 1994;44:1448–1452. doi: 10.1212/wnl.44.8.1448. [DOI] [PubMed] [Google Scholar]
- 2.Thornton CA, Griggs RC, Moxley RT., 3rd Myotonic dystrophy with no trinucleotide repeat expansion. Ann Neurol. 1994;35:269–272. doi: 10.1002/ana.410350305. [DOI] [PubMed] [Google Scholar]
- 3.Ricker K, Koch MC, Lehmann-Horn F, Pongratz D, Speich N, Reiners K, Schneider C, Moxley RT., 3rd Proximal myotonic myopathy: clinical features of a multisystem disorder similar to myotonic dystrophy. Arch Neurol. 1995;52:25–31. doi: 10.1001/archneur.1995.00540250029009. [DOI] [PubMed] [Google Scholar]
- 4.Abbruzzese C, Krahe R, Liguori M, Tessarolo D, Siciliano MJ, Ashizawa T, Giacanelli M. Myotonic dystrophy phenotype without expansion of (CTG)n repeat: an entity distinct from proximal myotonic myopathy (PROMM)? J Neurol. 1996;243:715–721. doi: 10.1007/BF00873977. [DOI] [PubMed] [Google Scholar]
- 5.Meola G, Sansone V. A newly-described myotonic disorder (proximal myotonic myopathy–PROMM): personal experience and review of the literature. Ital J Neurol Sci. 1996;17:347–353. doi: 10.1007/BF01999897. [DOI] [PubMed] [Google Scholar]
- 6.Vihola A, Bassez G, Meola G, Zhang S, Haapasalo H, Paetau A, Mancinelli E, Rouche A, Hogrel JY, Laforet P, Maisonobe T, Pellissier JF, Krahe R, Eymard B, Udd B. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology. 2003;60:1854–1857. doi: 10.1212/01.wnl.0000065898.61358.09. [DOI] [PubMed] [Google Scholar]
- 7.Pisani V, Panico MB, Terracciano C, Bonifazi E, Meola G, Novelli G, Bernardi G, Angelini C, Massa R. Preferential central nucleation of type 2 myofibers is an invariable feature of myotonic dystrophy type 2. Muscle Nerve. 2008;38:1405–1411. doi: 10.1002/mus.21122. [DOI] [PubMed] [Google Scholar]
- 8.George A, Schneider-Gold C, Zier S, Reiners K, Sommer C. Musculoskeletal pain in patients with myotonic dystrophy type 2. Arch Neurol. 2004;61:1938–1942. doi: 10.1001/archneur.61.12.1938. [DOI] [PubMed] [Google Scholar]
- 9.Meola G, Sansone V. Cerebral involvement in myotonic dystrophies. Muscle Nerve. 2007;36:294–306. doi: 10.1002/mus.20800. [DOI] [PubMed] [Google Scholar]
- 10.Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W, Schneider C, Koch MC, Beilman GJ, Harrison AR, Dalton JC, Ranum LP. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology. 2003;60:657–664. doi: 10.1212/01.wnl.0000054481.84978.f9. [DOI] [PubMed] [Google Scholar]
- 11.Rotondo G, Sansone V, Cardani R, Mancinelli E, Krahe R, Stangalini D, Meola G. Proximal myotonic dystrophy mimicking progressive muscular atrophy. Eur J Neurol. 2005;12:160–161. doi: 10.1111/j.1468-1331.2004.01032.x. [DOI] [PubMed] [Google Scholar]
- 12.Auvinen S, Suominen T, Hannonen P, Bachinski LL, Krahe R, Udd B. Myotonic dystrophy type 2 found in two of sixty-three persons diagnosed as having fibromyalgia. Arthritis Rheum. 2008;58:3627–3631. doi: 10.1002/art.24037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merlini L, Sabatelli P, Columbaro M, Bonifazi E, Pisani V, Massa R, Novelli G. Hyper-CK-emia as the sole manifestation of myotonic dystrophy type 2. Muscle Nerve. 2005;31:764–767. doi: 10.1002/mus.20289. [DOI] [PubMed] [Google Scholar]
- 14.Schara U, Schoser BG. Myotonic dystrophies type 1 and 2: a summary on current aspects. Semin Pediatr Neurol. 2006;13:71–79. doi: 10.1016/j.spen.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, Sohn R, Zemelman B, Snell RG, Rundle SA, Crow S, Davies J, Shelbourne P, Buxton J, Jones C, Juvonen V, Johnson K, Harper PS, Shaw DJ, Housman DE. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 16.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 17.Tishkoff SA, Goldman A, Calafell F, Speed WC, Deinard AS, Bonne-Tamir B, Kidd JR, Pakstis AJ, Jenkins T, Kidd KK. A global haplotype analysis of the myotonic dystrophy locus: implications for the evolution of modern humans and for the origin of myotonic dystrophy mutations. Am J Hum Genet. 1998;62:1389–1402. doi: 10.1086/301861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaspert A, Fahsold R, Grehl H, Claus D. Myotonic dystrophy: correlation of clinical symptoms with the size of the CTG trinucleotide repeat. J Neurol. 1995;242:99–104. doi: 10.1007/BF00887824. [DOI] [PubMed] [Google Scholar]
- 19.Hunter A, Tsilfidis C, Mettler G, Jacob P, Mahadevan M, Surh L, Korneluk R. The correlation of age of onset with CTG trinucleotide repeat amplification in myotonic dystrophy. J Med Genet. 1992;29:774–779. doi: 10.1136/jmg.29.11.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonifazi E, Vallo L, Giardina E, Botta A, Novelli G. A long PCR-based molecular protocol for detecting normal and expanded ZNF9 alleles in myotonic dystrophy type 2. Diagn Mol Pathol. 2004;13:164–166. [PubMed] [Google Scholar]
- 21.Warner JP, Barron LH, Goudie D, Kelly K, Dow D, Fitzpatrick DR, Brock DJ. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J Med Genet. 1996;33:1022–1026. doi: 10.1136/jmg.33.12.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falk M, Vojtiskova M, Lukas Z, Kroupova I, Froster U. Simple procedure for automatic detection of unstable alleles in the myotonic dystrophy and Huntington's disease loci. Genet Test. 2006;10:85–97. doi: 10.1089/gte.2006.10.85. [DOI] [PubMed] [Google Scholar]
- 23.Botta A, Bonifazi E, Vallo L, Gennarelli M, Garre C, Salehi L, Iraci R, Sansone V, Meola G, Novelli G. Italian guidelines for molecular analysis in myotonic dystrophies. Acta Myol. 2006;25:23–33. [PubMed] [Google Scholar]
- 24.Gennarelli M, Pavoni M, Amicucci P, Novelli G, Dallapiccola B. A single polymerase chain reaction-based protocol for detecting normal and expanded alleles in myotonic dystrophy. Diagn Mol Pathol. 1998;7:135–137. doi: 10.1097/00019606-199806000-00002. [DOI] [PubMed] [Google Scholar]
- 25.Richards RI, Sutherland GR. Dynamic mutations: a new class of mutations causing human disease. Cell. 1992;70:709–712. doi: 10.1016/0092-8674(92)90302-s. [DOI] [PubMed] [Google Scholar]
- 26.Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 27.Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L, Achari M, Pulst SM, Alonso E, Noebels JL, Nelson DL, Zoghbi HY, Ashizawa T. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26:191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- 28.Harley HG, Rundle SA, MacMillan JC, Myring J, Brook JD, Crow S, Reardon W, Fenton I, Shaw DJ, Harper PS. Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am J Hum Genet. 1993;52:1164–1174. [PMC free article] [PubMed] [Google Scholar]
- 29.Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG, Jr, Warren ST, Oostra BA, Nelson DL, Caskey CT. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 30.Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 31.Jakubiczka S, Vielhaber S, Kress W, Kupferling P, Reuner U, Kunath B, Wieacker P. Improvement of the diagnostic procedure in proximal myotonic myopathy/myotonic dystrophy type 2. Neurogenetics. 2004;5:55–59. doi: 10.1007/s10048-003-0168-6. [DOI] [PubMed] [Google Scholar]
- 32.Cagnoli C, Stevanin G, Michielotto C, Gerbino Promis G, Brussino A, Pappi P, Durr A, Dragone E, Viemont M, Gellera C, Brice A, Migone N, Brusco A. Large pathogenic expansions in the SCA2 and SCA7 genes can be detected by fluorescent repeat-primed polymerase chain reaction assay. J Mol Diagn. 2006;8:128–132. doi: 10.2353/jmoldx.2006.050043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majounie E, Wardle M, Muzaimi M, Cross WC, Robertson NP, Williams NM, Morris HR. Case control analysis of repeat expansion size in ataxia. Neurosci Lett. 2007;429:28–32. doi: 10.1016/j.neulet.2007.09.055. [DOI] [PubMed] [Google Scholar]
- 34.Cagnoli C, Michielotto C, Matsuura T, Ashizawa T, Margolis RL, Holmes SE, Gellera C, Migone N, Brusco A. Detection of large pathogenic expansions in FRDA1. SCA10, and SCA12 genes using a simple fluorescent repeat-primed PCR assay. J Mol Diagn. 2004;6:96–100. doi: 10.1016/S1525-1578(10)60496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10:43–49. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]