

Abstract
Extraction and purification of nucleic acids from complex biological samples for PCR are critical steps because inhibitors must be removed that can affect reaction efficiency and the accuracy of results. This preanalytical processing generally involves capturing nucleic acids on microparticles that are then washed with a series of buffers to desorb and dilute out interfering substances. We have developed a novel purification method that replaces multiple wash steps with a single pass of paramagnetic particles (PMPs) though an immiscible hydrophobic liquid. Only two aqueous solutions are required: a lysis buffer, in which nucleic acids are captured on PMPs, and an elution buffer, in which they are released for amplification. The PMPs containing the nucleic acids are magnetically transported through a channel containing liquid wax that connects the lysis chamber to the elution chamber in a specially designed cartridge. Transporting PMPs through the immiscible phase yielded DNA and RNA as pure as that obtained after extensive wash steps required by comparable purification methods. Our immiscible-phase process has been applied to targets in whole blood, plasma, and urine and will enable the development of faster and simpler purification systems.
The extraction and purification of nucleic acids (NA) from complex samples (eg, blood, biopsied tissue, cultured cells, food) is an essential prerequisite for many downstream applications including viral/bacterial detection, genotyping, transcriptional analysis, and epigenetic analysis. One of the most prominent advances in NA extraction and purification methodology was Boom's introduction of silica particles1 almost 20 years ago as an alternative to phenol chloroform extraction.2 It was further advanced by the development of paramagnetic particles (PMPs)3,4,5 and improved surface coatings.6,7,8,9,10 These methods have been automated in a number of processing systems, large11,12 and small,13,14,15 which address a broad range of NA testing situations in clinical and research laboratories, and field testing. However, the purification process is still lengthy and consists of multiple steps making automated systems expensive and costly to operate by using large quantities of sterilized consumables such as filter tips. The open-plate format also makes these processes prone to cross-contamination because the samples are exposed to aerosols and environmental contaminants during processing.16
PMPs themselves, however, have several advantages: NAs can be isolated from crude sample materials, wide ranges of sample volumes can be accommodated, and large batches of samples can be processed without centrifugation.17 In PMP-based systems, the clinical sample is subjected to a lysis buffer wherein NAs are released from the cells and bound to PMPs. Multiple wash steps then remove amplification inhibitors, and finally, NAs are eluted from the PMPs yielding a concentrated and purified sample. Most systems process samples in a single well by repeatedly pelleting PMPs, aspirating the liquid, and adding wash solution. The meticulous washing required with conventional PMP-based purification is necessary to remove amplification interferents that adhere to tube surfaces, become entrapped in the magnetically-aggregated PMPs, or remain in the residual volume after the supernatant is removed by aspiration. More washes are required when purifying NA from complex biological matrices such as plasma and blood because the sample viscosity increases on cell lysis making complete removal of fluid more difficult. Alternatively, systems have been developed recently to move PMPs to a new well with magnetic probes.17,18 However, repeated washing is still required because interferents carry over in liquid entrapped in the pellet and in thin films on the probes themselves.
Our strategy to streamline the purification process is to transfer the PMPs between wells in a specially designed cartridge with an externally applied magnetic field eliminating all contact between the processing system and the sample. The wells are connected with a hydrophobic liquid through which PMPs are transported (Figure 1A), and the hydrophobic liquid acts as a barrier between the lysis chamber and the elution chamber, preventing mixing of the two solutions. On application of the magnetic force, the PMPs are moved through the hydrophobic liquid, transporting NAs from the lysis chamber to the elution chamber while the lysis and elution buffers remain stationary. The hydrophobic liquid acts as an immiscible phase filter (IPF), which blocks interferents and reduces processing to only three steps: cell lysis/NA binding, PMP transport, and NA elution. In addition to minimizing the number of steps and time required for NA purification, moving PMPs instead of fluids simplifies the instrumentation and reduces the number of consumables required to process a sample.
Figure 1.
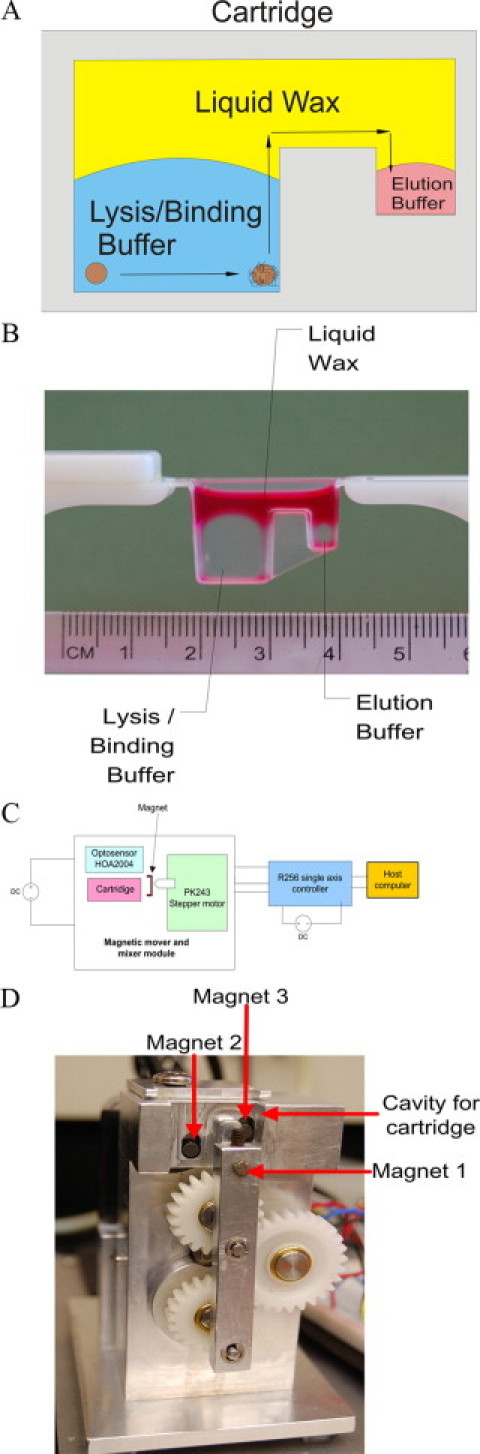
A: Schematic of the IPF process: The PMPs bind to the NA and are moved by an external magnet from the lysis buffer to the elution buffer through liquid wax. B: Molded cartridge containing lysis/binding buffer, elution buffer, and a red colored liquid wax. C: Schematic of the magnetic mixer/mover set-up. D: Photograph of the magnetic mixer/mover.
The efficacy of the IPF NA isolation method was demonstrated in three model systems: quantification of HIV-1 viral RNA from plasma by quantitative reverse transcription–polymerase chain reaction (RT-qPCR), detection of Chlamydia and gonorrhea DNA in urine by quantitative PCR (qPCR), and detection of HIV-1 proviral DNA from lymphocytes in whole blood (WB) by qPCR. An in-depth analysis of variables including the number and size of the PMPs and the composition of the lysis buffer elucidated factors that need to be optimized to adapt this system to different purification processes.
Materials and Methods
Cartridge Manufacture and Sterilization
A cartridge consisting of two chambers was molded out of Pro-Fax PD-702 resin (LyondellBassell, Houston, TX). The fabricated cartridge with a wall thickness of 0.66 mm consists of two chambers with a capacity of 650 μl and 75 μl of fluid, respectively (Figure 1B). To create a sterile RNase free environment for RNA purification, the cartridges were autoclaved and then cleaned with RNase Decontamination Solution (Biosystems, Foster City, CA). They were rinsed twice with water, rinsed once with ethanol, and then dried overnight at 50°C.
Magnetic Mixer/Mover
A magnetic mixer/mover was built to automate the sample purification process establishing uniform performance (Figure, 1C and 1D). The device uses a stepper motor and gears to drive the movement of three axially magnetized Neodymium magnets of diameter 1/8″, thickness 3/16″, and grade N42 (K&J Magnetics, Inc, Jamizon, PA). Magnet 1 rotates on the lever arm at a distance of 0.5 mm from the cartridge wall and is used to aggregate the PMPs within each chamber and then move them from one chamber to the other through the liquid wax. Magnets 2 and 3 can be engaged to bring them in contact with the cartridge or can be moved back, away from the cartridge. The movement of magnet 1, coupled with the engaging and disengaging of magnets 2 and 3, is used to change the direction of the net magnetic field within each chamber. The changing magnetic field causes the particles to agitate and mix the fluid in each chamber, eliminating the need for a vortex mixer. While the current device is controlled by a host computer, the magnetic mixer/mover is capable of stand-alone operation.
RNA Purification from Plasma Using Dextran PMPs
HIV-1 virus, acquired from Rush Virology Quality Assurance Laboratory at 1.5 × 106 copies per milliliter of plasma, was diluted in seronegative plasma to obtain HIV-1 concentrations of 300, 60, and 12 copies per microliter, respectively. HIV-1 viral RNA was extracted and purified from 50 μl of the prepared plasma sample using the Ambion MagMax Viral RNA isolation kit (Applied Biosystem) following the manufacturer's recommended protocol. This protocol utilizes 10 μl of PMPs per reaction and consists of one lysis and binding step, four wash steps, one drying step, and an elution step. In the IPF method, lysis and binding reagents consisting of 200 μl of Ambion Lysis/Binding solution concentrate (Applied Biosystem; Foster City, CA), 200 μl of isopropyl alcohol, 1 μl of carrier RNA (Applied Biosystem), 5 μl of Ambion PMPs, and 5 μl of Binding Enhancer (Applied Biosystem) were mixed and added to the larger chamber of the cartridge. Plasma (50 μl) containing HIV-1 virus was then added to it and mixed for four minutes using the automated system. Elution buffer (50 μl) was aliquoted into the smaller chamber of the IPF cartridge, and the two aqueous fluids were overlaid with Chillout liquid wax (Bio Rad Laboratories; Hercules, CA) as shown in Figure 1B. The automated system aggregated the PMPs for 2 minutes using the external magnet and moved the PMP aggregate from the lysis and binding buffer to the elution buffer. The elution buffer containing the PMPs was heated to 55°C for 10 minutes to elute the RNA. The PMPs were aggregated and removed from the elution buffer. HIV-1 viral load quantification was performed for each sample using the Abbott RealTime HIV-1 Amplification Reagent Kit19 (Abbott Molecular, Des Plaines, IL) in 25-μl reaction volumes with the addition of 0.2 mg/ml bovine serum albumin (B8667, Sigma, St. Louis, MO), 150 mmol/L trehalose (T9531; Sigma) and 0.2% Tween 20 (28320; Pierce Thermo Fisher Scientific, Waltham, MA), and 5 μl template. Amplification reactions were performed in Cepheid SmartCycler II (Sunnyvale, CA).
Purification of Genomic DNA from Whole Blood Using Silica PMPs
Cultured 8E5 cells20 (Rush Virology Quality Assurance Laboratory, Chicago, IL) containing a single copy of the HIV-1 genome per cell added to WB from a seronegative donor was used to simulate infant blood for the proviral DNA assay. The cells were thawed, counted using a hemoctyometer, serially diluted in PBS, and added to WB from a seronegative donor at concentrations of 8000 cells/μl, 1600 cells/μl, 320 cells/μl, and 64 cells/μl. Genomic DNA was extracted and purified from 25 μl of blood sample using the Magnesil Blood Genomic, Max Yield System (Promega Corp., Madison, WI) following the manufacturer's recommended protocol scaled for a 25-μl sample. This protocol was performed manually and consists of seven wash steps, one alcohol drying step, and one elution step. In the IPF method, 25 μl blood was added to 60 μl lysis buffer, agitated for a minute, and incubated for 4 minutes at room temperature. Lysis buffer (44 μl) and 6 μl of PMPs were added, agitated for a minute, and incubated for 4 minutes. Lysis buffer (15 μl) and 200 μl of alcohol wash buffer were added to the solution and the IPF purification was performed as before. The purified DNA from each sample was amplified using the Abbott RealTime HIV-1 Amplification Reagent Kit (Abbott Molecular, Des Plaines, IL)19 in 25-μl reaction volume. Amplification reactions were performed in Cepheid SmartCycler II (Sunnyvale, CA).
Purification of Bacterial DNA from Urine Using Dextran PMPs
The urine samples were prepared by combining Chlamydia: ATCC trachomatis serotype F in McCoy cell culture suspension and lyophilized Neisseria gonorrhoeae resuspended in PBS containing 30% glycerol with control urine (Fisher Scientific, PA). The manual protocol was performed using the Abbott RealTime CT/NG assay as per the manufacturer's protocols. This protocol consists of three wash steps, one drying step, and one elution step. In the IPF method, 200 μl of Ambion Lysis/Binding solution concentrate (Applied Biosystem; Foster City, CA), 200 μl of isopropyl alcohol, 1 μl of carrier RNA (Applied Biosystem; Foster City, CA), 5 μl of Ambion PMPs, and 5 μl of Binding Enhancer (Applied Biosystem; Foster City, CA) were mixed. Urine sample (200 μl) was then added to it. The solution was heated to 55°C for 10 minutes, and the two-step purification was performed as with the plasma samples. The purified DNA was amplified using the Abbott RealTime CT/NG assay in a 50-μl reaction volume.21 Amplification reactions were performed in the Abbott Molecular m2000rt instrument (Abbott Park, IL).
Statistical Analysis
Differences between the IPF and manual purification methods were examined graphically and formally analyzed with paired t-tests according to Bland & Altman.22 Plots were generated with the difference in log10 copy number on the y axis and the mean log10 copy number on the x axis. The log10 copy number of NA was calculated from the quantification cycle (Cq) using the equation of the standard curve. The mean difference and the SD of the differences were calculated, and lines were drawn corresponding to the mean and the mean ± 2SD. Plots were examined for evidence of nonuniform variance and outliers before performing a t-test with the null hypothesis of a mean difference equal to zero. A P value less than 0.05 was considered statistically significant.
Results
Characterization of the IPF Method
Effect of Liquid Wax on PCR
To determine whether passage through the liquid wax affects RT-qPCR performance, PMPs from three different commercial RNA purification kits were transported through liquid wax: Ambion MagMax Total RNA isolation kit (Applied Biosystems), Abbott RealTime RNA assay (Abbott Molecular), and Cortex MagaZorb® RNA isolation kit (Promega Corp.).22,23,24 These kits use, respectively, dextran, iron-oxide and cellulose surface chemistries, and the liquid wax could potentially affect NA binding to or elution from PMPs, or subsequent RT-qPCR amplification. In each case, 50 μl of plasma sample containing 15,000 copies of human immunodeficiency virus type 1 (HIV-1) viral RNA was used. For one set of samples, the RNA was purified using the manufacturer's recommended manual protocol with an elution volume of 50 μl. The second set of samples was treated identically to the first set until the last wash step. The PMPs were then transported from the wash buffer to the elution buffer through liquid wax (Figure 1A). The eluted RNA was quantified by RT-qPCR using the Abbott RealTime HIV-1 Amplification Reagent Kit (Abbott Molecular, Des Plaines, IL).19 No statistically significant differences were observed between the Cq values for the samples purified using the liquid wax and the manual protocol (two-sided P values of 0.76, 0.99, and 0.40, respectively), demonstrating that the movement of PMPs through the wax does not interfere with the binding of the NA to the PMPs or inhibit the subsequent amplification by RT-qPCR (Figure 2A). Of particular interest was that the alcohol evaporation step required by the Ambion protocol could be eliminated. Because alcohol is a potent PCR inhibitor, this study supported our hypothesis that inhibitor carryover with the PMPs can be minimized.
Figure 2.
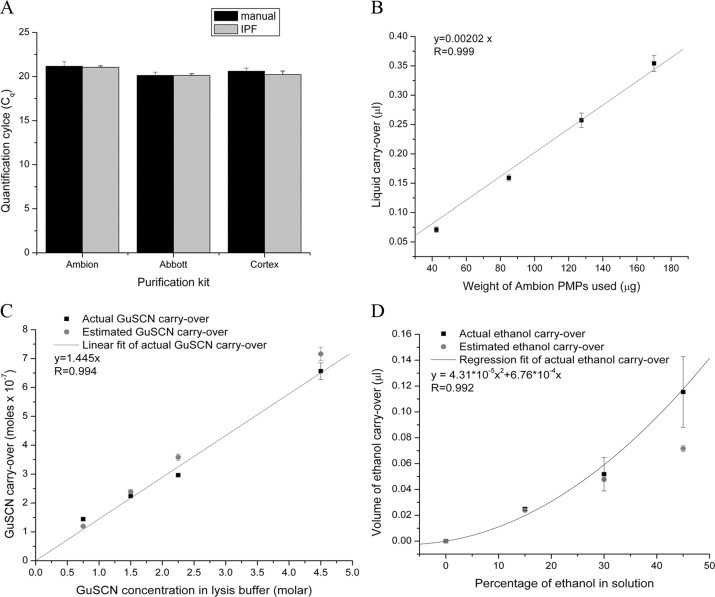
A: Effect of liquid wax on RT-qPCR. The quantification cycle (Cq) values were obtained in triplicate by RT-qPCR of the nucleic acid purified from 15,000 copies of HIV-1 RNA using three different kits. The black bars show the Cq values using the manual method, and the gray bars show the Cq values obtained when the PMPs in each kit were moved from the last wash buffer to the elution buffer through liquid wax. B: Liquid carryover with Ambion PMPs. The volume of liquid carried over measured in triplicate is plotted at four different quantities of Ambion PMPs. The solid black squares show the liquid carryover, and the red line is the linear regression fit (Slope = 0.00202, r = 0.999). C: GuSCN carryover. GuSCN carryover using 85 μg of Ambion PMPs was measured in triplicate at four different concentrations of GuSCN in the lysis chamber of the IPF system. The solid black squares show the liquid carryover. The red line is the regression fit of the carryover. The solid red circles show the expected salt carryover estimated by multiplying the volume of liquid carryover with 85 μg of PMPs with the concentration of GuSCN in the lysis chamber. D: Alcohol carryover. Ethanol carryover was measured in triplicate using 85 μg of Ambion PMPs at four different ethanol concentrations (volume percentage) in the lysis chamber of the IPF system. The solid black squares show the volume of ethanol carryover, and the black line is the regression fit of the carryover. The solid red diamonds show the expected ethanol carryover estimated from the measurement of liquid carryover.
Effect of PMP Pellet Size on Lysis Solution Carryover
Lysis buffers typically contain chaotropic salts such as guanidinium isothiocyanate (GuSCN) and ethanol, which inhibit the PCR. In conventional tube or well-based PMP purification systems, these substances are carried over in the liquid adhering to the PMPs, the walls of the vessel, and the walls of the pipette tips. Since IPF eliminates the walls on which liquid can adhere, the only liquid that is carried across the interface with the PMPs is the liquid entrapped within the PMP aggregate and the fluid film formed around the PMP pellets. We measured the amount of liquid that is carried across the interface by the PMP pellet as a function PMP mass to confirm that the quantity and size of beads affected the amount of carryover. The 450-nm Ambion MagMax PMPs (measured by dynamic light scattering, Zetasizer Nano NS, Malvern, UK) were used for the experiment. The particles were aggregated using a magnetic stand and resuspended in Tris (hydroxymethyl)aminomethane (Tris) buffer containing 10−4 mol/L 5-carboxyfluorescein. The solution was aliquoted into the lysis/binding chamber of the molded cartridge then moved through a layer of liquid wax to 50 μl of Tris buffer in the elution chamber using the automated magnetic mixer/mover. The volume of liquid carried over was estimated by fluorometrically by determining the ratio of 5-carboxyfluorescein concentrations in the elution and lysis chambers. The volume carried over increased linearly with the amount of PMPs (Figure 2B). A carryover of 0.159 μl was observed with 85 μg of PMPs, the quantity used for extraction and purification of viral RNA from plasma and Chlamydia and gonorrhea DNA from urine samples using the IPF method. Using the Abbott RealTime HIV-1 Amplification system as our model system, we found no statistically significant amplification delays when this quantity of Ambion lysis buffer was spiked into 50 μl RT-qPCR reactions. Cq delays >1 or assay failure was observed only when 1 μl of lysis buffer was added to the RT-qPCR reaction. This indicates that carryover of lysis buffer with the PMPs in the IPF method should not be inhibitory to the RT-qPCR assays.
Chaotrope and Alcohol Carryover
Nucleic acid extraction buffers typically contain molar concentrations of known PCR inhibitors such as the chaotrope GuSCN and alcohol, which are carried over to the elution buffer with the PMPs. Assays were developed to measure trace levels of GuSCN and ethanol in the elution buffer. The particles were aggregated using a magnetic stand and resuspended in a solution containing various concentrations of GuSCN or ethanol. The solution was aliquoted into the lysis/binding chamber of the molded cartridge then moved through a layer of liquid wax to 50 μl of water in the elution chamber using the automated magnetic mixer/mover. The quantity of GuSCN carried over to the elution chamber was measured spectrophotometrically at 230 nm and related to a standard curve of absorbance versus GuSCN concentration to quantify the moles of GuSCN transferred with the PMPs (Figure 2C). The amount of GuSCN measured in the elution buffer correlated to liquid carried-over with the PMPs (Figure 2B) and the concentration of GuSCN in the lysis buffer.
Alcohol carryover was determined using an ethanol fluorimetric assay kit according to the manufacturer's instructions (Ethanol Assay Kit, Biovision Inc.; Mountain View, CA) (Figure 2D) and related to a standard curve of relative fluorescence versus ethanol concentration to quantify the volume of ethanol transferred with the PMPs. The ethanol carryover correlated with the liquid carryover (Figure 2B) up to a 30% ethanol solution. The nonlinear increase of alcohol carryover at higher ethanol concentrations may be attributed to the reduction of surface tension between the liquid wax and alcohol-water solution and lower viscosity of the alcohol-water solution leading to increase in liquid carryover. Using the Abbott RealTime HIV-1 Amplification system as our model system, we found that in a 50-μl reaction, no statistically significant difference in Cq was observed at the levels carried over by the IPF method. A Cq delay of ∼1 was observed with as much as 2.5 × 10−6 moles of GuSCN or 4 μl of isoproponal. The quantities at which significant Cq difference is observed are much larger than what is carried over with the IPF method, demonstrating its effectiveness in reducing PCR inhibitors while eliminating multiple wash steps.
Purification of HIV RNA from Plasma Samples
To demonstrate the feasibility of purifying viral RNA with IPF, we extracted HIV-1 RNA from spiked plasma as would be done in measuring viral load. Quantitative measurement of HIV-1 is technically demanding due to the abundance of reverse transcriptase and PCR inhibitors in human blood specimens.25,26 Viral RNA was purified from 50 μl of plasma spiked with HIV-1 virus using 85 μg of PMPs from Ambion. The purified RNA was amplified using the Abbott RealTime HIV-1 Amplification Kit. A PCR efficiency of E = 102% was observed (Figure 3A), suggesting that the inhibitor carryover is minimal even after eliminating the four wash steps and the alcohol evaporation step required for the standard protocol. Differences between the IPF method and a standard protocol for RNA purification using the Ambion MagMax Total RNA isolation kit were compared graphically with a Bland–Altman plot (Figure 3B). A paired t-test indicated that the mean difference, −0.0019 (SD = 0.123), was not significantly different from 0 (two-sided P value = 0.96). A difference of 0.1 - 0.2 Log10 units is usually considered to be due to experimental errors, and only log10 differences greater than 0.5 are judged practically significant.27
Figure 3.
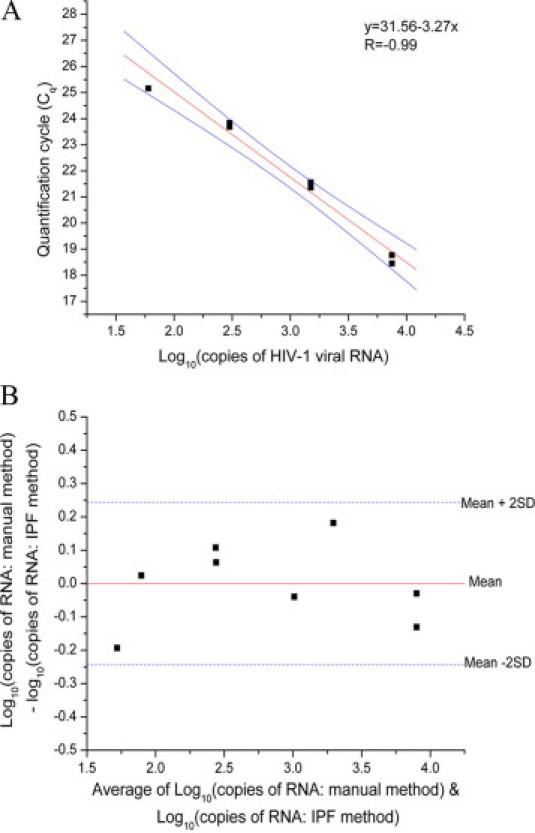
A: IPF RT-qPCR for HIV-1 from plasma. Standard curve of Cq values for four different RNA concentrations run in duplicate plotted versus the log10 of the HIV-1 viral copy number. The solid black squares are the Cq values in duplicates, the red line is the regression fit (R = −0.988) of the Cq values with a slope of −3.27, and the solid blue lines denote the upper and lower 95% confidence limits. The PCR efficiency (E) was 102%, where E = 10(−1/slope) − 1. 60 to 7500 copies were used in a 25-μl RT-qPCR reaction. B: Bland-Altman plot comparing the IPF and manual method of purification. Solid black squares show difference between the two methods, solid red line (y = −0.00772) plots the mean difference between the two methods, and the blue dashes lines show the mean ± 2SD. All of the points lie within the 2-SD intervals.
To determine the limit of detection of viral RNA in 50-μl plasma samples, we purified 10 replicates each with copy numbers ranging from 75 to 25. We detected 100% of samples with 60 copies (1.78 Log10 copies) and above (Table 1). While this limit of detection corresponds to 1200 copies/ml, which is significantly greater than 40 copies/ml reported for the same assay with a commercially available purification system,28 much of the difference can be attributed to a 20-fold difference in sample volumes.
Table 1.
IPF RT-qPCR for HIV-1 from Plasma
| Copies of RNA in PCR tube | Detection (percentage) |
|---|---|
| 75 | 100 |
| 60 | 100 |
| 50 | 90 |
| 40 | 90 |
| 30 | 50 |
| 25 | 30 |
Lower quantification limit of the IPF method was determined by running 10 replicates at levels down to 25 copies. RNA from 50 μl of plasma containing HIV-1 viral RNA was purified with IPF then amplified using the Abbott RealTime HIV-1 Amplification Reagent Kit in a 25-μl reaction with the addition of 0.2 mg/ml bovine serum albumin, 150 mmol/L trehalose, and 0.2% Tween 20 and 5 μl template. Amplification reactions were performed in Cepheid SmartCycler II.
Purification of Chlamydia and Gonorrhea DNA from Urine
To demonstrate that IPF can extract NA from urine, we purified bacterial DNA from Chlamydia trachomatis (CT) and Neisseria gonorrhoeae (NG) using this assay as a model for the diagnosis of sexually transmitted diseases. The purified bacterial DNA samples were quantified using the Abbott RealTime CT/NG assay. The PCR efficiency for the CT and NG assay over seven orders of magnitude was 97.2% and 94.5%, respectively (Figure 4, A–D) suggesting that the inhibitor carryover is minimal. These efficiencies were similar to those obtained from the manual extraction method (87.9% and 87.9%, respectively) using the Abbott Realtime CT/NG kit. The Bland–Altman plots show the differences in the CT and NG assays between DNA purified with Abbott's kit and the IPF method (Figure 4, A–D). Paired t-tests indicated that the mean differences observed for CT, 0.00001 (SD = 0.122), and NG, 0.024 (SD = 0.206) were not significantly different from 0 (two-sided P values ≈ 1 and 0.69, respectively).
Figure 4.
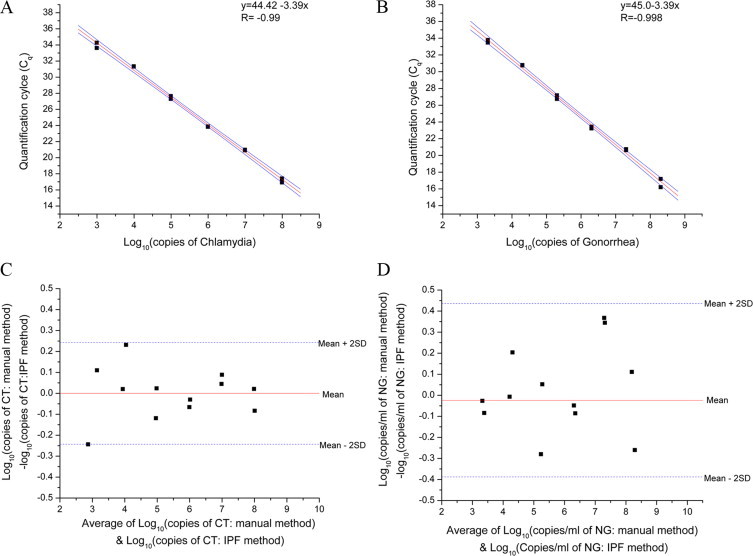
IPF quantification by qPCR of Chlamydia and gonorrhea from urine samples. A: Chlamydia standard curve of Cq values (solid black squares) for seven DNA concentrations run in duplicates plotted versus the log10 of the Chlamydia copy number; red line is the regression fit (R = −0.999) with a slope of −3.39, and solid green lines denote upper and lower 95% confidence limits. PCR efficiency (E) was 97.2%, where E = 10(−1/slope) − 1. B: Gonorrhea standard curve of: Cq values (solid black squares) for seven different DNA concentrations run in duplicate plotted versus the log10 of gonorrhea copy number; red line is the regression fit (R = −0.998) of the Cq values with a slope of −3.46, and solid blue lines denote upper and lower 95% confidence limits. PCR efficiency (E) was 94.5%, where E = 10(−1/slope) − 1. C: Bland-Altman plot comparing the IPF and manual method of purification of Chlamydia. Solid black squares show difference between the two methods, solid red line (y = 0.062) plots the mean difference between the two methods, and the blue dashes lines show the mean ± 2SD. All of the points lie within this interval. D: Bland-Altman plot comparing the IPF and manual method of purification of gonorrhea. Solid black squares show difference between the two methods, solid red line (y = 0.046) plots the mean difference between the two methods, and the blue dashes lines show the mean ± 2SD. All of the points lie within the 2-SD intervals.
Purification of Genomic DNA from Whole Blood and Detection of HIV-1 Provirus
WB from a finger stick can be a ready source of genomic DNA; however, it is an extremely complex medium containing numerous PCR inhibitors in high concentrations. To demonstrate that IPF can process such challenging samples, we developed a qPCR assay to detect proviral HIV-1 DNA integrated into peripheral blood mononuclear cells. Proviral DNA detection is used routinely to diagnose infants with HIV-1.29 We adapted the Promega Magnesil gDNA purification kit, which consists of 10 steps (lysis, seven washes, drying, and elution), for use with the IPF, which consists of three steps (lysis, PMP transport through liquid wax, and elution). HIV-1 proviral DNA was effectively purified from a 25-μl WB sample using the IPF method with 1.11 mg of PMPs from the Promega Magnesil gDNA purification kit. Serial dilutions over four orders of magnitude yielded a standard curve with a slope of −3.15 and PCR efficiency of 108% (Figure 5A). The Bland–Altman plot of the proviral DNA PCR assays shows the differences between the standard method using the Promega purification kit and the IPF method (Figure 5B). A paired t-test indicated that the mean difference, −0.00015 (SD = 0.142), was not significantly different from 0 (two-sided P value ≈ 1).
Figure 5.
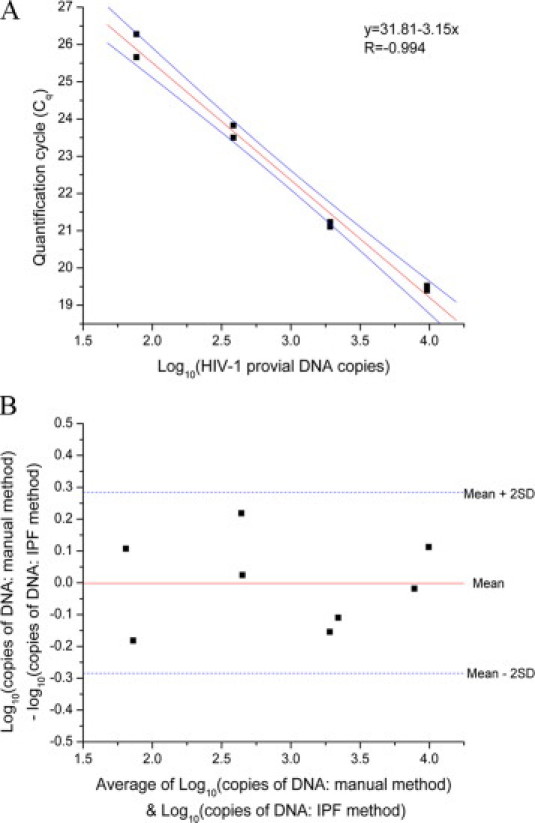
A: IPF PCR for proviral DNA from 25 μl WB. Standard curve of the Cq values for four different DNA concentrations run in duplicates plotted versus the log10 of 8e5 cell copy number. The solid black squares are the Cq values in duplicates, the red line is the regression fit (R = −0.994) of the Cq values with a slope of −3.15, and the solid green lines denote the upper and lower 95% confidence limits. The PCR efficiency (E) was 108%, where E = 10(−1/slope) − 1. B: Bland-Altman plot comparing the IPF and manual method of purification. Solid black squares show difference between the two methods, solid red line (y = 0.002) plots the mean difference between the two methods, and the blue dashed lines show the mean ± 2SD. All of the points lie within the 2-SD intervals.
Discussion
The transfer of PMPs through a hydrophobic liquid using an external magnet provides a simple and reliable way to filter out PCR inhibitors and eliminate extensive wash steps. The IPF also acts as a barrier between the lysis and elution chambers and ambient air preventing environmental or sample cross contamination. Hence, the passage of the PMPs through an IPF has the advantage of providing a closed purification system that requires no fluid pumping and yields highly purified and concentrated NA in only three steps: lysis/binding, PMP transport, and elution.
We have demonstrated that IPF is effective in eliminating wash steps in multiple NA affinity purification schemes and sample types. Conventional methods required between three and seven washes depending on the complexity of the starting matrix (ie, fewer wash steps for urine samples and more for whole blood samples). In addition to reducing the number of steps, and consequently the time, required for NA purification, we have also eliminated the use of multiple pipette tips per sample reducing assay cost and impact on the environment via solid waste disposal.
Conventional NA extraction protocols can be readily converted to IPF by determining the amount of inhibitor carried over with the PMPs that can be tolerated in the amplification assay. Once this limit is known, the optimal lysis buffer composition can be established by determining levels of GuSCN and alcohol, and quantity of PMPs required for the NA extraction that maintain inhibitor carryover below these limits. Protocols that determine the carryover of lysis constituents to the elution buffer with different quantities of PMPs can be used as a guide for the selection of optimal assay conditions. The use of smaller PMPs, which have a greater surface to volume ratio, is highly desirable.
Sample preparation remains a major impediment to nucleic acid testing at point of care because it is time-consuming, complex, and labor-intensive.26 Substituting a single IPF for multiple wash steps would allow for the development of low-cost systems for use in field testing or point-of-care diagnosis, especially in resource limited settings. A major obstacle to testing in these settings is the widespread lack of expertise required to perform venipuncture.30 Blood collection from a finger stick or heel stick provides a lower cost and simpler alternative to venal blood, requires minimal training, and increases success of collection. Because the goal of our project is to develop point-of-care diagnostics, the assays were optimized for small sample volumes that could be readily collected from a finger or heel stick. Using smaller volumes does affect the sensitivity of the test because the number of NA molecules present in the original sample is directly proportional to the sample volume. However, improved outcomes that could result from performing molecular tests, while patients wait, may offset losses in sensitivity. IPF may also be incorporated into other affinity purification protocols potentially reducing or eliminating wash steps required for protein purification, immunoprecipitation of proteins and/or protein complexes, and chromatin immunoprecipitation. It could also greatly enhance the productivity of high-throughput screening systems and at the same time dramatically reduce the amount of consumables used. We envision broad application of this simple method to a wide range of analytical systems.
Footnotes
Supported by the Bill and Melinda Gates Foundation Grand Challenges in Global Health grant number 37774. Abbott Molecular Inc. (Des Plaines, Illinois) provided qPCR and RT-qPCR reagents and Virology Quality Assurance Laboratory (Rush Presbyterian/St. Luke's Medical Center, Chicago, Illinois) provided HIV-1 virus and 8e5 cells.
M.H. and S.S. are employed by Abbott, which has certain rights to commercialize products based on the method described in this article. K.S., S.M., S.J., and D.K. are listed as inventors on a patent application filed by Northwestern University, whose claims include aspects of the method described in this article. None of the other authors declare any relevant financial relationships.
References
- 1.Boom R, Sol CJ, Salimans MM, Jansen CL, Wertheim-van Dillen PM, van der Noordaa J. Rapid and simple method for purification of nucleic acids. J Clin Microbiol. 1990;28:495–503. doi: 10.1128/jcm.28.3.495-503.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 3.Papell SS, inventor; United State of America as Represented by the Administrator of the National Aeronautics and Space Administration, assignee. Low viscosity magnetic fluid obtained by the colloidal suspension of magnetic particles. United States patent US 3,215,572.1965 Nov
- 4.Reimers GW, Khalafalla SE, inventors; United States of America as Represented by the Secretary of the Interior, assignee. Production of magnetic fluids by petization techniques. United States patent US 3,843,540. 1974 Oct
- 5.Landfester K, Ramírez LP. Encapsulated magnetite particles for biomedical application. J Phys Condens Matter. 2003;15:S1345–S1361. [Google Scholar]
- 6.Molday RS, Mackenzie D. Immunospecific ferromagnetic iron-dextran reagents for the labeling and magnetic separation of cells. J Immunol Methods. 1982;52:353. doi: 10.1016/0022-1759(82)90007-2. [DOI] [PubMed] [Google Scholar]
- 7.Sangregorio C, Wiemann JK, O'Connor CJ, Rosenzweig Z. A new method for the synthesis of magnetoliposomes. J Appl Phys. 1999;85:5699. [Google Scholar]
- 8.Pardoe H, Chua-anusorn W, St. Pierre TG, Dobson J. Structural and magnetic properties of nanoscale iron oxide particles synthesized in the presence of dextran or polyvinyl alcohol. J Magn Magn Mater. 2001;225:41–46. [Google Scholar]
- 9.Rembaum A, inventor; California Institute of Technology, assignee. Polyglutaraldehyde synthesis and protein bonding substrates. United States patent US 4,369,226. 1983
- 10.Lee J, Isobe T, Senna M. Magnetic properties of ultrafine magnetite particles and their slurries prepared via in-situ precipitation. Colloids Surf A Physicochem Eng Asp. 1996;109:121–127. [Google Scholar]
- 11.Amellal B, Murphy R, Maiga A, Brucker G, Katlama C, Calvez V, Marcelin AG. Stability of HIV RNA in plasma specimens stored at different temperatures. HIV Med. 2008;9:790–793. doi: 10.1111/j.1468-1293.2008.00632.x. [DOI] [PubMed] [Google Scholar]
- 12.Swanson P, Holzmayer V, Huang S, Hay P, Adebiyi A, Rice P, Abravaya K, Thamm S, Devare SG, Hackett JJ. Performance of the automated Abbott RealTime HIV-1 assay on a genetically diverse panel of specimens from London: comparison to VERSANT HIV-1 RNA 3.0, AMPLICOR HIV-1 MONITOR v1.5, and LCx HIV RNA Quantitative assays. J Virol Methods. 2006;137:184–192. doi: 10.1016/j.jviromet.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 13.Erickson D, Li D. Integrated microfluidic devices. Anal Chim Acta. 2004;507:11–26. [Google Scholar]
- 14.Mitchell P. Microfluidics—downsizing large-scale biology. Nature Biotechnol. 2001;19:717–721. doi: 10.1038/90754. [DOI] [PubMed] [Google Scholar]
- 15.Easley CJ, Karlinsey JM, Bienvenue JM, Legendre LA, Roper MG, Feldman SH, Hughes MA, Hewlett EL, Merkel TJ, Ferrance JP, Landers JP. A fully integrated microfluidic genetic analysis system with sample-in-answer-out capability. Proc Natl Acad Sci USA. 2006;103:19272–19277. doi: 10.1073/pnas.0604663103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Claassen M, van Zyl GU, Preiser W. Extraction buffer contaminated bacterially as a cause of invalid HIV-1 viral load results on the NucliSens EasyQ® system. J Virol Meth. 2008;150:80–81. doi: 10.1016/j.jviromet.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 17.Berensmeier S. Magnetic particles for the separation and purification of nucleic acids. Appl Microbiol Biotechnol. 2006;73:495–504. doi: 10.1007/s00253-006-0675-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang X, Willis RC, Burrell A, Evans K, Hoang Q, Xu W, Bounpheng M. Automation of nucleic acid isolation on kingfisher magnetic particle processors. J Assoc Lab Automat. 2007;12:195–201. [Google Scholar]
- 19.Huang S, Salituro J, Tang N, Luk K-C, Hackett J, Jr, Swanson P, Cloherty G, Mak W-B, Robinson J, Abravaya K. Thermodynamically modulated partially double-stranded linear DNA probe design for homogeneous real-time PCR. Nucl Acids Res. 2007;35:e101. doi: 10.1093/nar/gkm551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Folks TM, Powell D, Lightfoote M, Koenig S, Fauci AS, Benn S, Rabson A, Daugherty D, Gendelman HE, Hoggan MD, Venkatesan S, Martin MA. Biological and biochemical characterization of a cloned Leu-3-cell surviving infection with the acquired immune deficiency syndrome retrovirus. J Exp Med. 1986;164:280–290. doi: 10.1084/jem.164.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marshall R, Chernesky M, Jang D, Hook EW, Cartwright CP, Howell-Adams B, Ho S, Welk J, Lai-Zhang J, Brashear J. Characteristics of the m2000 automated sample preparation and multiplex real-time PCR System for detection of Chlamydia trachomatis and Neisseria gonorrhoeae. J Clin Microbiol. 2007;45:747–751. doi: 10.1128/JCM.01956-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gundling G. Nucleic acid isolation method and kit. Abbott Laboratories; 2005. [Google Scholar]
- 23.Latham G, Fang X, Conrad R, Kemppainen J, Setterquist R, Pasloske B. Modified surfaces as solid supports for nucleic acid purification. USPTO, Ambion Inc.; 2005. [Google Scholar]
- 24.Nargessi, RD, Pourfarzaneh, M, inventors; Cortex Biochem, Inc., assignee. Isolation and purification of nucleic acids. United States patent US 7,264,927. 2007 Sep. 4
- 25.Mylonakis E, Paliou M, Rich J. Plasma viral load testing in the management of HIV infection. Am Fam Physician. 2001;63:483–490. [PubMed] [Google Scholar]
- 26.Dineva MA, Mahilum-Tapay L, Lee H. Sample preparation: a challenge in the development of point-of-care nucleic acid-based assays for resource-limited settings. Analyst. 2007;132:1193–1199. doi: 10.1039/b705672a. [DOI] [PubMed] [Google Scholar]
- 27.Jagodzinski LL, Wiggins DL, McManis JL, Emery S, Overbaugh J, Robb M, Bodrug S, Michael NL. Use of calibrated viral load standards for group M subtypes of human immunodeficiency virus type 1 to assess the performance of viral RNA quantitation tests. J Clin Microbiol. 2000;38:1247–1249. doi: 10.1128/jcm.38.3.1247-1249.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swanson P, Huang S, Holzmayer V, Bodelle P, Yamaguchi J, Brennan C, Badaro R, Brites C, Abravaya K, Devare SG. Performance of the automated Abbott RealTime HIV-1 assay on a genetically diverse panel of specimens from Brazil. J Virol Methods. 2006;134:237–243. doi: 10.1016/j.jviromet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 29.Read JS, Committee on Pediatric AIDS, American Academy of Pediatrics Diagnosis of HIV-1 infection in children younger than 18 months in the United States. Pediatrics. 2007;120:e1547–e1562. doi: 10.1542/peds.2007-2951. [DOI] [PubMed] [Google Scholar]
- 30.Fiscus SA, Cheng B, Crowe SM, Demeter L, Jennings C, Miller V, Respess R, Stevens W. Forum for Collaborative HIV Research Alternative Viral Load Assay Working Group: HIV-1 viral load assays for resource-limited settings. PLoS Med. 2006;3:e47. doi: 10.1371/journal.pmed.0030417. [DOI] [PMC free article] [PubMed] [Google Scholar]