

Abstract
Determination of NPM1 mutation status has become essential for the molecular classification of acute myeloid leukemias (AML). Methods with high clinical sensitivity and specificity adapted to the molecular laboratory workflow are required for the diagnosis, prognosis, and monitoring of AML with normal karyotype. We report here the development and evaluation of a novel, streamlined, RNA-based assay for the rapid multiplex detection of common NPM1 mutations in a 96-well assay format. Using synthetic transcripts and total RNA from leukemic cell lines, we show that the assay can specifically detect NPM1 wild-type and mutants A, B, D, or J transcripts in the same reaction. Dilution experiments indicate an assay dynamic range >4 log units with an analytical sensitivity of approximately 0.01%. Evaluation of 69 clinical specimens at initial diagnosis resulted in 100% agreement with reference methods. Of patients with AML with normal karyotype, 53% carried one of four different mutations. The assay was also combined with other laboratory-developed tests to simultaneously detect NPM1 mutant transcripts and fusion transcripts resulting from t(8;21) or inv(16) in a single reaction well. Overall, these results show that the assay is a versatile and specific tool for the screening of NPM1 mutations in patients with AML. Its high analytical sensitivity further suggests potential utility for the monitoring of residual disease in AML with normal karyotype.
The nucleophosmin gene (NPM1) is thought to be the most frequently mutated gene in de novo acute myeloid leukemias (AMLs), particularly those with normal cytogenetics. In several series, approximately 25 to 35% of adult AMLs demonstrated mutation in one allele of NPM1 (located on chromosome 5q35) at the time of diagnosis.1,2,3 The nucleophosmin protein is a nuclear shuttle protein important in centrosome duplication, ribosome biogenesis, and regulation of transcription factors and also functions as a molecular chaperone.4,5,6,7 Mutations in NPM1 in AML occur characteristically in the last exon of the gene, exon 12, and, although heterogeneous, are most commonly 4-bp insertions that result in frame shifts. These mutations typically do not truncate the protein but create a new nuclear export signal and remove nucleolar-localizing tryptophan residues.8 These changes result in the aberrant cytoplasmic localization of the mutant and remaining wild-type NPM1 protein in a dominant-negative fashion.8
NPM1 mutation has important prognostic implications for patients with AML. Patients with NPM mutations typically do not carry cytogenetic abnormalities and generally respond better to induction chemotherapy than those without mutations.1 Several reports identified a prognostic interrelationship between NPM1 mutation, internal tandem duplication mutation in the FLT3 tyrosine kinase, and, perhaps, mutation in the CCAAT/enhancer binding protein α gene.9,10,11,12 These findings suggest that molecular stratification of AML cases may be important for prognosis and for therapeutic decisions, underlining the need for timely diagnostic testing for these mutations.
A number of methods of identifying NPM1 mutations have been published. Because almost all cases of NPM1 mutation result in length changes to exon 12, PCR amplification of the exon followed by sequencing or sizing by capillary electrophoresis or by high-performance liquid chromatography have all been used.13,14 Both genomic DNA and mRNA have served as substrates for these assays. Locked nucleic acid-clamping of the wild-type allele with chip-based detection of mutants has also been used.3 The mutation spectrum in AML is fairly wide, with almost 40 different variants identified. However, the common mutations are relatively few, with the A mutation (a duplication of a TCTG tetranucleotide at position 956 to 959 of the reference sequence) accounting for 75 to 80% of mutant-bearing AML, the B mutation for approximately 10%, and the D mutation for approximately 5%; other mutations are very rare.15 Because the heterozygous mutations result in 4-nucleotide repeats or insertions in a background of wild-type sequences, sensitive and specific detection of NPM1 mutations using standard PCR methods can be challenging.
We report here the rapid detection of common NPM1 mutations using total RNA purified from cultured cells, bone marrow, or peripheral blood using a laboratory-developed test based on the Signature NPM1 Mutations Research Use Only kit. The assay uses multiplex RT-PCR (RT-PCR) in combination with fluorescent bead-based detection to simultaneously identify transcripts for NPM1 mutations and wild-type targets. The aim of this study was to evaluate the assay workflow and performance in a clinical setting.
Materials and Methods
RNA Specimens
Peripheral blood or bone marrow from patients with AML, acute lymphoid leukemia, chronic myeloid leukemia, acute bilineage leukemia, or myelodysplastic syndrome was collected as part of standard clinical care and following a protocol approved by The Johns Hopkins Medical Institutions Institutional Review Board. Total RNA was isolated from peripheral blood or bone marrow using the RNeasy Mini Kit according to the manufacturer's instructions for blood (Qiagen, Valencia, CA).
OCI-AML3 cells (DSMZ GmbH, Braunschweig, Germany) were cultured according to the supplier's recommendations, and total RNA was isolated with the RNeasy Mini Kit according to the manufacturer's instructions for cultured cells (Qiagen). When indicated, total RNA was diluted in purified HL-60 total RNA (Applied Biosystems, Foster City, CA), keeping the concentration of total RNA constant. Synthetic RNAs corresponding to mutations A, B, D, or J were prepared using standard in vitro transcription methods and diluted in HL-60 total RNA, keeping the concentration of total RNA constant. Purified total RNA was quantified using a NanoDrop ND1000 (NanoDrop Technologies, Wilmington, DE).
Signature Assays
The Signature NPM1 Mutations Research Use Only kit (Asuragen Inc., Austin, TX) was evaluated in combination with Signature General Purpose Reagents (Asuragen Inc.). In brief, total RNA (5 μl) was reverse-transcribed into cDNA in a 20-μl reaction for 45 minutes at 42°C. The resulting cDNA (5 μl) was amplified by multiplex PCR using 1 μl of Signature NPM1 Mutations Primer Set and 2.5 units of AmpliTaq Gold (Applied Biosystems) in a 20-μl reaction (45 cycles consisting of 94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds). The PCR products (5 μl) were then hybridized to 5 μl of Signature NPM1 Mutations Probe Set in a 50-μl reaction for 30 minutes at 52°C. After addition of 25 μl of Reporter Solution, the probe-bound PCR products were detected on a Luminex 200 system (Luminex Corporation, Austin, TX) with subsequent analysis of the median fluorescent intensity (MFI) signal outputs for bead ID #28 (NPM1 wild-type) and #45 (NPM1 mutant) in Excel (Microsoft, Richmond, WA). Positive and negative controls provided with the kit were included in each run.
For co-amplification and codetection of specific AML fusion transcripts, the same protocol was followed except that 1 μl of Signature AML/ETO, inv(16)-ADE Primers (analyte-specific reagent; Asuragen Inc.) was added to the multiplex PCR reaction, and 5 μl of Signature AML/ETO, inv(16)-ADE Probes (analyte-specific reagent) was added to the hybridization reaction. MFI signals corresponding to AML/ETO, inv16-A, inv16-B, and inv16-E fusion transcripts were detected on bead ID #14, #55, #16, and #41, respectively. Positive and negative controls were included in each run; the positive control consisted of pooled, in vitro transcribed RNAs for the pertinent targets in a background of total RNA and thus also controlled for the reverse transcription step. A GeneAmp PCR System 9700 (Applied Biosystems) was used for the RT, PCR, and hybridization steps.
Other Assays
Standard PCR was performed using 5 μl of cDNA generated with the Signature reagents (Asuragen Inc.), pairs of primers flanking the NPM1 exon 12 mutation site (Integrated DNA Technologies, Inc., Coralville, IA), and 35 PCR cycles (94°C for 30 seconds, 55°C for 30 seconds, and 72°C for 60 seconds) followed by a 10-minute elongation step at 72°C. Bidirectional sequencing of the PCR products using the BigDye terminator method was performed by ACGT Inc. (Wheeling, IL). For fragment size analysis, one of the PCR primers was 5′-modified with a FAM dye. The resulting labeled PCR products were diluted 1:50 in water, and 1 μl was mixed with 0.5 μl of GeneScan 500 ROX Size Standard and 13.5 μl of Hi-Di Formamide (Applied Biosystems). After heat denaturation at 95°C for 2 minutes, analysis was performed on a 3130xl Genetic Analyzer (Applied Biosystems) using a 36-cm capillary with POP-7 Polymer and GeneMapper Software v4.0 (Applied Biosystems). Specific run parameters were prerun at 15 kV for 180 seconds, with injection at 1.2 kV for 23 seconds, and run at 15 kV for 30 minutes at a temperature of 60°C.
Results
Assay Design and Workflow
The Signature NPM1 Mutations Research Use Only assay consists of four consecutive steps. Total RNA is first reverse-transcribed (RT step); then cDNA corresponding to NPM1 mutation A, B, D, or J (Figure 1A)15,16 is amplified by PCR using gene-specific primers (PCR step). Labeled amplified products are directly hybridized to target-specific capture probes covalently bound to fluorescent microspheres (hybridization step). After incubation with a reporter molecule, the reaction mixture is directly analyzed by flow cytometry to detect the mutant transcripts on a single bead type (detection step). NPM1 wild-type transcripts are co-amplified in each sample and concurrently detected on a second bead type to serve as endogenous internal controls. The MFI detected on at least 50 beads of each type is reported to determine the presence or absence of the respective NPM1 wild-type or mutant targets in the RNA specimen. For this study, the cutoffs recommended in the package insert were used: a specimen was reported positive if the mutant bead generated a signal ≥400 MFI; a specimen was reported negative if the mutant bead generated a signal <400 MFI with a wild-type bead signal ≥1000 MFI.
Figure 1.
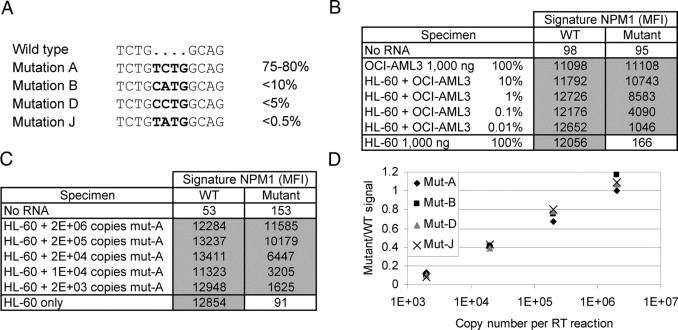
Analytical performance evaluation. A: Wild-type and mutant sequences detected by the assay. Bold indicates insertion mutations. The relative representation of each mutation in the total mutation-positive population is indicated.16,17B: Representative examples of signal (MFI) obtained with a serial dilution of a mutant A-positive cell line RNA (OCI-AML3) in wild-type cell line RNA (HL-60). C: Representative examples of signal (MFI) obtained with a serial dilution of mutant A in vitro transcript (mut-A) in wild-type cell line RNA (HL-60). D: Representative examples of data obtained with a serial dilution of four different in vitro transcripts (mut-A, mut-B, mut-D, or mut-J) in wild-type cell line RNA (HL-60). The graph shows the ratio of mutant to wild-type signal according to the initial copy number input of each in vitro transcript. WT, wild-type.
Positive and negative control samples included in the kit are processed with each batch of specimens to validate the RT, PCR, hybridization, and detection steps. The entire procedure is performed on 96-well plates and requires seven pipetting steps per reaction including two transfer steps, one from the RT plate to the PCR plate and one from the PCR plate to the hybridization plate. With one kit, up to 24 reactions can be performed in less than 6 hours with about 1.5 hours of hands-on time. The assay requires a Luminex 100 or 200 system and RNA isolation kit reagents; both already commonly found and validated in clinical laboratories. Overall, the Signature assay design is compatible with the molecular laboratory workflow, and we undertook evaluation of its analytical and clinical performance for the testing of newly diagnosed leukemia clinical specimens.
Evaluation of Analytical Performance
Assay performance was first evaluated in triplicate using total RNA purified from well characterized cell lines isolated from patients with leukemia. An input mass titration experiment showed that NPM1 mutant A transcripts were reproducibly detected by the assay in 0.1 to 1000 ng of total RNA from the cell line OCI-AML3 heterozygous for the NPM1 mutation A (Supplemental Figure 1A, see http://jmd.amjpathol.org). To assess the analytical sensitivity of the assay, the OCI-AML3 total RNA was serially diluted in a background of total RNA purified from the NPM1 wild-type cell line HL-60 (Figure 1B and Supplemental Figure 1B, see http://jmd.amjpathol.org for all data). NPM1 mutation A transcripts were reproducibly detected at least twofold above the positive cutoff (400 MFI) in as low as 0.1 ng of OCI-AML3 RNA in 1000 ng of HL-60 RNA (0.01% mass equivalent). Above 10% of OCI-AML3 RNA, the mutant NPM1 signal reached a plateau with maximum MFI signals between 10,000 and 12,000 MFI. In contrast, no NPM1 mutant signal ≥400 MFI was observed using up to 1000 ng of HL-60 total RNA. Analytical specificity was also confirmed using synthetic NPM1 mutant transcripts and total RNA samples purified from translocation positive cell lines and whole blood or bone marrow clinical specimens (see below).
To further assess analytical sensitivity, a synthetic NPM1 mutation A transcript (mut-A) was prepared by in vitro transcription, quantified by absorbance, and serially diluted at a specific copy number concentration in a constant background of excess NPM1 wild-type transcripts provided by HL-60 total RNA (10 to 50 million copies/μg). The mut-A transcript was reproducibly detected in as low as 2000 copies per RT reaction (Figure 1C and Supplemental Figure 1C, see http://jmd.amjpathol.org for all data). The MFI response curve was similar to the OCI-AML3 dilution experiment with signal saturation greater than 200,000 mut-A copies per RT reaction. Experiments with mut-B, mut-D, and mut-J synthetic transcripts confirmed that the Signature assay detects all four mutations with similar efficiency and analytical sensitivity (Figure 1D). Together, these data suggest an assay limit of detection at or just below 0.01% dilution or 2000 copies of mutant transcripts.
The assay performance was next compared with that of standard methods based on PCR amplification and size fractionation analysis. The cDNA products from the same RT reactions were either analyzed with the Signature procedure or amplified by PCR using a pair of primers flanking the NPM1 exon 12 mutation site followed by amplicon detection by capillary electrophoresis (Figure 2A). Although NPM1 mutant transcripts were detected in all specimens with the Signature assay, the presence of a robust peak specific for NPM1 mutant PCR products could only be detected at more than 10 ng of OCI-AML3 total RNA in a background of 1000 ng of HL-60 RNA or 1% equivalent (Figure 2B, top right). Likewise, at least 200,000 copies of synthetic mut-A transcript had to be spiked in HL-60 total RNA for positive NPM1 mutant detection using the sizing assay (Figure 2B, bottom right). Similar results were obtained for the other synthetic NPM1 mutant transcripts (Supplemental Figure 2, see http://jmd.amjpathol.org).
Figure 2.
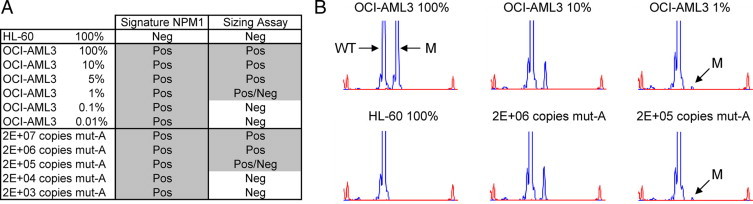
Comparison with a size-based assay. A: Summary of the positive (Pos) and negative (Neg) calls obtained with serial dilutions of a mutant A-positive cell line RNA (OCI-AML3) or in vitro transcript (mut-A) in wild-type cell line RNA (HL-60). A Pos/Neg call indicates that the results were not reproducible (below or at the limit of detection of the method). B: Representative examples of capillary electrophoresis traces. The wild-type (WT)- and mutant (M)-specific peaks are indicated. The small peaks at the beginning and end of each trace correspond to molecular weight markers.
Evaluation of Clinical Specimens
The Signature NPM1 Mutations assay was evaluated using 69 archived total RNA samples isolated from peripheral blood or bone marrow from leukemia patients at initial diagnosis (Supplemental Table 1 at http://jmd.amjpathol.org). The clinical set consisted of 48 AML specimens: 17 with normal karyotype (NK), 2 positive for inv(16), 4 positive for t(8;21), and 25 with various chromosomal abnormalities other than inv(16) or t(8;21), including 2 with acute bilineage leukemia. In addition, 21 non-AML leukemia samples consisting of 9 acute lymphoid leukemia, 9 chronic myeloid leukemia and 3 myelodysplastic syndrome were included. Approximately 53% (9 of 17) of the AML NK specimens were positive for NPM1 mutation with the Signature assay (Table 1). Of the 52 other specimens, only 1 AML specimen with a deletion of chromosome 7q22 was positive for NPM1 mutation. All positive signals were at least 20-fold above the cutoff (400 MFI) for the various RNA inputs tested (Table 1). In contrast, the NPM1 mutant probe signal was well below the cutoff for the 59 negative specimens evaluated, with a mean value of 42 MFI and a SD of 26 MFI. Mixing experiments with representative clinical specimens confirmed that an analytical sensitivity of at least 0.01% could be reached by dilution of NPM1-positive RNA in a background of NPM1-negative RNA (Supplemental Figure 3, see http://jmd.amjpathol.org).
Table 1.
Summary of Clinical Results
| Signature NPM1 result |
||
|---|---|---|
| Positive (Mutant) | Negative (WT) | |
| AML normal karyotype (n = 17) | 9 | 8 |
| AML t(8;21) or inv(16) (n = 6) | 0 | 6 |
| Other AML (n = 25) | 1 | 24 |
| Non-AML (n = 21) | 0 | 21 |
| Total (n = 69) | 10 | 59 |
| Mean NPM1 mutant signal (MFI) | 9849 | 42 |
| Minimum NPM1 mutant signal (MFI) | 8014 | 0 |
| Maximum NPM1 mutant signal (MFI) | 11,416 | 87 |
| Mean RNA concentration (ng/μl) | 239 | 257 |
| Minimum RNA concentration (ng/μl) | 54 | 8 |
| Maximum RNA concentration (ng/μl) | 732 | 825 |
WT, wild-type.
The same specimen set was also independently analyzed with the capillary electrophoresis assay described above (Table 2). There was 100% agreement between the two methods (95% confidence intervals, 94.7 to 100%). The 10 positive specimens and approximately 15% of the negative specimen set were further confirmed by bidirectional sequencing (Table 2). The four NPM1 mutant transcripts A, B, D, and J were detected among the NPM1-positive specimens, with 60% (6 of 10) of the specimens containing mutation A.
Table 2.
Method Comparison
| Signature NPM1 result |
||
|---|---|---|
| Positive (Mutant) | Negative (WT) | |
| Positive by CE | 10 | 0 |
| Negative by CE | 0 | 59 |
| No. cases sequenced | 10 | 9 |
| Sequencing results | 6A, 1B, 1D, 2J | 9 wild type |
WT, wild-type.
A protocol based on the Signature NPM1 Mutations assay and using additional Signature reagents was also developed to combine detection of NPM1 mutations with detection of AML/ETO and inv(16) type A, D, or E fusion transcripts in a single multiplex assay (see Materials and Methods). Serial dilution of total RNA purified from OCI-AML3 or translocation-positive cell lines in a background of HL-60 total RNA showed that the individual NPM1 assay and the combined assay are specific for NPM1 mutations, AML/ETO and inv(16) targets with an analytical sensitivity of at least 0.1% for each target (Supplemental Figure 4, see http://jmd.amjpathol.org). Although the NPM1 mutant signal was reduced by up to 30%, the combined assay still detected 0.01% or 2000 copies of NPM1 mutant transcripts (Table 3). A subset of the clinical set, namely six AML NK, six AML positive for inv(16) or t(8:21), seven other AML, and seven non-AML, was also evaluated with this laboratory-developed test. All of the expected targets were specifically detected in the 26 specimens with 100% agreement between Signature and CE/sequencing for NPM1 status and 100% agreement between Signature and fluorescence in situ hybridization/sequencing for fusion transcript detection (Table 4 and Supplemental Table 1, see http://jmd.amjpathol.org). Mixing experiments with clinical specimens also demonstrated that both NPM1 mutations and AML fusion transcripts could be simultaneously detected in the same RNA sample (data not shown).
Table 3.
Comparison of Analytical Sensitivity for the Signature NPM1 Mutations Assay Alone or Combined with the Signature AML/ETO, inv(16)-ADE Primers and Probes
| Signature NPM1 (MFI)* |
Signature NPM1 + Signature AML/ETO, inv(16) ADE (MFI)* |
|||||||
|---|---|---|---|---|---|---|---|---|
| NPM1-WT | NPM1-Mut | NPM1-WT | NPM1-Mut | Inv16-A | Inv16-D | Inv16-E | AML1/ETO | |
| No RNA | 87 | 87 | 42 | 81 | 86 | 64 | 43 | 57 |
| 100% OCI-AML3 | 13,019 | 12,604 | 9745 | 10,719 | 110 | 67 | 83 | 64 |
| HL-60 + 10% OCI-AML3 | 11,523 | 11,839 | 9745 | 10,263 | 65 | 44 | 77 | 92 |
| HL-60 + 1% OCI-AML3 | 14,023 | 9097 | 10,579 | 7623 | 134 | 78 | 52 | 37 |
| HL-60 + 0.1% OCI-AML3 | 14,025 | 4455 | 9881 | 3408 | 25 | 73 | 30 | 36 |
| HL-60 + 0.01% OCI-AML3 | 13,745 | 1088 | 10,166 | 785 | 159 | 81 | 81 | 74 |
| 100% HL-60 | 12,465 | 159 | 9236 | 77 | 2 | 66 | 24 | 17 |
| HL-60 + 2E+07 copies | 10,980 | 12,416 | 10,388 | 11,638 | 97 | 87 | 102 | 144 |
| HL-60 + 2E+06 copies | 10,245 | 10,718 | 9108 | 10,432 | 115 | 134 | 127 | 97 |
| HL-60 + 2E+05 copies | 11,593 | 8566 | 10,564 | 8151 | 83 | 87 | 100 | 57 |
| HL-60 + 2E+04 copies | 11,511 | 4158 | 10,044 | 3033 | 112 | 79 | 140 | 82 |
| HL-60 + 2E+03 copies | 10,588 | 866 | 10,129 | 664 | 107 | 122 | 77 | 97 |
| HL-60 + 2E+02 copies | 11,287 | 322 | 10,352 | 190 | 88 | 104 | 78 | 91 |
Mut, mutant; WT, wild-type.
Values above the cutoff are in bold.
Table 4.
Codetection of NPM1 Mutations and AML-Specific Fusion Transcripts in Seven Representative Clinical Specimens
| MFI signal* |
||||||
|---|---|---|---|---|---|---|
| NPM1-WT | NPM1-Mut | Inv16-A | Inv16-D | Inv16-E | AML1/ETO | |
| NPM1 positive† | 12,567 | 11,284 | 39 | 53 | 18 | 67 |
| NPM1 negative† | 10,415 | 107 | 35 | 79 | 59 | 33 |
| No RNA† | 36 | 50 | 28 | 29 | 63 | 50 |
| AML-NK, 645 ng‡ | 13,094 | 11,095 | 29 | 48 | 41 | 57 |
| AML-NK, 2000 ng | 9800 | 64 | 48 | 85 | 51 | 16 |
| AML t(8;21), 400 ng‡ | 3269 | 60 | 14 | 99 | 58 | 8210 |
| AML t(7;11), 2330 ng | 8367 | 69 | 89 | 68 | 61 | 36 |
| AML del(7), 268 ng‡ | 11,856 | 11,240 | 50 | 65 | 86 | 29 |
| CML t(9;22), 1815 ng | 8560 | 84 | 30 | 61 | 84 | 60 |
| ALL t(9;22), 1210 ng | 9035 | 59 | 84 | 38 | 73 | 41 |
Mut, mutant; WT, wild-type.
Obtained by combination of the Signature NPM1 Mutations assay with the Signature AML/ETO, inv(16)-ADE Primers and Probes (see Materials and Methods). Values above the cutoff are in bold.
Controls provided with the Signature NPM1 Mutations kit.
These specimens were confirmed to be positive by bidirectional sequencing.
Discussion
AML with mutated NPM1 is a leukemia entity with distinct pathological, molecular and prognostic features that was recently included in the 2008 World Health Organization classification of myeloid neoplasms. With a high frequency in translocation-negative AML (50 to 60%), NPM1 mutations are also useful markers for the monitoring of minimal residual disease. However, because of the broad spectrum and nature of each mutation (small repeated or inserted sequences with almost 40 variants identified to date), sensitive molecular detection of multiple NPM1 mutations in a single assay can be challenging. There is currently a need for improved methods with appropriate mutation coverage but without compromising analytical performance or laboratory efficiency.
The Signature technology is a 96-well assay format that enables rapid analysis of several RNA targets in a single reaction with automatic detection by flow cytometry. This step is performed using the Luminex 100 IS or 200 systems, a platform already validated in more than 4700 laboratories worldwide and compatible with a broad assay menu including 42 US Food and Drug Administration cleared tests (http://www.luminexcorp.com/company/ceo.html last accessed March 8, 2010). The streamlined and multiplexed workflow of the NPM1 assay provides detection of an endogenous control, NPM1 wild-type, and four NPM1 mutations: A, B, D, and J (Figure 1). Depending on the study report, these four mutations represent at least 90 to 95% of all NPM1-positive cases.15,16 Although other synthetic mutant sequences were not tested in our study, detection of additional mutations with the Signature assay had not been reported at the time this manuscript was submitted (F. Ye and E. Labourier, unpublished results). Another advantage of the Signature technology platform is that the multiplexing can be increased using beads with a different identity, up to 100, for detection of the various analytes. For example, co-detection of NPM1 mutations and NPM1 wild-type as an internal control together with common AML-specific fusion transcripts in a single assay (Table 3) may be beneficial for the rapid molecular characterization of patients with newly diagnosed AML.
An input range study showed that the Signature NPM1 assay is compatible with a broad range of total RNA purified from the OCI-AML3 cell line (Supplemental Figure 1A, see http://jmd.amjpathol.org). The saturation of mutant signal and lack of wild-type signal interference observed above a 10-ng input of OCI-AML3 RNA, as well as above 10% of OCI-AML3 RNA dilution or above 200,000 copies of mut-A synthetic transcript (Figure 1 and Supplemental Figure 1, see http://jmd.amjpathol.org), suggest that the assay is extremely robust. Indeed, evaluation of 69 clinical specimens using 5 μl of total RNA corresponded to a broad range of input RNA (40 to 4000 ng per RT reaction) and resulted in 100% agreement with reference methods. Although a single commercially available RNA isolation method was used, independent studies showed that the Signature NPM1 assay is also compatible with other affinity column- or phenol-based extraction methods (F. Ye and E. Labourier, unpublished results). In addition, a recent multisite validation study to determine FLT3 and NPM1 mutation status in total nucleic acid samples (DNA and RNA) extracted with the Qiagen BioRobot EZ1 workstation further confirmed the robustness of the Signature assay and its compatibility with a wide range of clinical specimens (G. J. Tsongalis, Ph.D., personal communication).
In the present study, the specific mutant signals obtained with 10 positive clinical specimens and RNA inputs between 270 and 1200 ng were all robust and on average 25-fold above the cutoff set at 400 MFI (Table 1). Although the number of specimens tested is too low to fully assess clinical sensitivity (95% confidence intervals, 72.2 to 100%), we obtained 100% overall agreement with two independent reference methods (Table 2). The proportion of NPM1 mutant-positive cases within the AML NK population was as expected (9 of 17 or 53%). A recent large series of patients with NPM1 mutations showed that approximately 15% carried a karyotypic abnormality17; we observed a single patient with mutated NPM1 and del(7q22). The overlapping biological, pathological, immunophenotypic, and clinical features of such cases, independent of the presence of additional cytogenetic abnormalities, support the provisional diagnostic entity “AML with NPM1 mutation” in the World Health Organization Classification.18 Importantly, the MFI signals obtained with the 59 negative specimens were all very low with a mean value at 42 MFI, almost 10-fold below the 400 MFI cutoff (Table 1). The tight distribution of these negative signals (SD = 26 MFI) is also indicative of a robust clinical specificity (95% confidence intervals, 93.9 to 100%) at that cutoff. Based on the normal-like distribution of negative signals in this sample set, a recalculated cutoff at approximately 200 MFI (mean of negative signals + 6.11 SD) would still correspond to a probability of approximately 1 in 1 billion of obtaining a false positive.
The assay analytical sensitivity was thoroughly investigated using a mutant A-positive cell line and synthetic mutant transcripts corresponding to each of the four targets detected by the assay. Together, these data show that the Signature NPM1 assay specifically detects NPM1 mutations A, B, D, and J with excellent analytical sensitivity, 2 to 3 orders of magnitude better than a standard capillary electrophoresis (CE)-based PCR method (Figures 1 and 2 and Supplemental Figures 1 and 2, see http://jmd.amjpathol.org). In the presence of excess NPM1 wild-type transcripts the assay could reproducibly detect the equivalent of one positive cell in a background of 10,000 negative cells (0.01%). Recent reports showed that the relative or absolute quantitative monitoring of NPM1 mutation A expression levels in AML NK is an accurate method to measure treatment response and detect potential relapse.19,20 Although the Signature NPM1 Mutations assay is not a truly quantitative assay, analysis of the mutant/wild-type MFI ratio showed a semiquantitative linear response for all four NPM1 mutations as the relative copy number of mutant transcripts was decreased by up to 4 log units (Figure 1D and Supplemental Figure 1, see http://jmd.amjpathol.org). The excellent analytical sensitivity (∼0.01%) and dynamic range (∼4 log units) of this multiplex assay warrant further evaluation to determine its potential clinical utility for monitoring of residual disease during treatment of patients with AML.
Footnotes
Supported in part by the National Institutes of Health/National Cancer Institute (Small Business Innovation Research grant R43CA130501).
M.H. and F.Y. contributed equally to this work.
F.Y., Z.Y., and E.L. were employees of Asuragen, Inc. (the manufacturer of the Signature NPM1 Mutations assay) at the time of this work. None of the other authors declare any relevant financial relationships.
Supplemental material for this article can be found on http://jmd.amjpathol.org.
Web Extra Material
Summary of data for the 69 clinical specimens evaluated in this study. ND = sequence not determined.
References
- 1.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, Liso A, Vignetti M, Fazi P, Meani N, Pettirossi V, Saglio G, Mandelli F, Lo-Coco F, Pelicci PG, Martelli MF, GIMEMA Acute Leukemia Working Party Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 2.Suzuki T, Kiyoi H, Ozeki K, Tomita A, Yamaji S, Suzuki R, Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C, Akiyama H, Nishimura M, Motoji T, Shinagawa K, Takeshita A, Ueda R, Kinoshita T, Emi N, Naoe T. Clinical characteristics and prognostic implications of NPM1 mutations in acute myeloid leukemia. Blood. 2005;106:2854–2861. doi: 10.1182/blood-2005-04-1733. [DOI] [PubMed] [Google Scholar]
- 3.Thiede C, Creutzig E, Illmer T, Schaich M, Heise V, Ehninger G, Landt O. Rapid and sensitive typing of NPM1 mutations using LNA-mediated PCR clamping. Leukemia. 2006;20:1897–1899. doi: 10.1038/sj.leu.2404367. [DOI] [PubMed] [Google Scholar]
- 4.Szebeni A, Olson MO. Nucleolar protein B23 has molecular chaperone activities. Protein Sci. 1999;8:905–912. doi: 10.1110/ps.8.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell. 1989;56:379. doi: 10.1016/0092-8674(89)90241-9. [DOI] [PubMed] [Google Scholar]
- 6.Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Fukasawa K. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell. 2000;103:127–140. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 7.Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat Cell Biol. 2002;4:529–533. doi: 10.1038/ncb814. [DOI] [PubMed] [Google Scholar]
- 8.Falini B, Bolli N, Shan J, Martelli MP, Liso A, Pucciarini A, Bigerna B, Pasqualucci L, Mannucci R, Rosati R, Gorello P, Diverio D, Roti G, Tiacci E, Cazzaniga G, Biondi A, Schnittger S, Haferlach T, Hiddemann W, Martelli MF, Gu W, Mecucci C, Nicoletti I. Both the carboxyl terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood. 2006;107:4514–4523. doi: 10.1182/blood-2005-11-4745. [DOI] [PubMed] [Google Scholar]
- 9.Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, Martelli MF, Haferlach T, Hiddemann W, Falini B. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005;106:3733–3739. doi: 10.1182/blood-2005-06-2248. [DOI] [PubMed] [Google Scholar]
- 10.Verhaak RG, Goudswaard CS, van Putten W, Bijl MA, Sanders MA, Hugens W, Uitterlinden AG, Erpelinck CA, Delwel R, Lowenberg B, Valk PJ. Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood. 2005;106:3747–3754. doi: 10.1182/blood-2005-05-2168. [DOI] [PubMed] [Google Scholar]
- 11.Dohner K, Schlenk RF, Habdank M, Scholl C, Rucker FG, Corbacioglu A, Bullinger L, Frohling S, Dohner H. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–3746. doi: 10.1182/blood-2005-05-2164. [DOI] [PubMed] [Google Scholar]
- 12.Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, Habdank M, Spath D, Morgan M, Benner A, Schlegelberger B, Heil G, Ganser A, Dohner H, German-Austrian Acute Myeloid Leukemia Study Group Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 13.Boissel N, Renneville A, Biggio V, Philippe N, Thomas X, Cayuela JM, Terre C, Tigaud I, Castaigne S, Raffoux E, De Botton S, Fenaux P, Dombret H, Preudhomme C. Prevalence, clinical profile, and prognosis of NPM mutations in AML with normal karyotype. Blood. 2005;106:3618–3620. doi: 10.1182/blood-2005-05-2174. [DOI] [PubMed] [Google Scholar]
- 14.Ammatuna E, Noguera NI, Zangrilli D, Curzi P, Panetta P, Bencivenga P, Amadori S, Federici G, Lo-Coco F. Rapid detection of nucleophosmin (NPM1) mutations in acute myeloid leukemia by denaturing HPLC. Clin Chem. 2005;51:2165–2167. doi: 10.1373/clinchem.2005.055707. [DOI] [PubMed] [Google Scholar]
- 15.Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood. 2007;109:874–885. doi: 10.1182/blood-2006-07-012252. [DOI] [PubMed] [Google Scholar]
- 16.Falini B, Sportoletti P, Martelli MP. Acute myeloid leukemia with mutated NPM1: diagnosis, prognosis and therapeutic perspectives. Curr Opin Oncol. 2009;21:573–581. doi: 10.1097/CCO.0b013e3283313dfa. [DOI] [PubMed] [Google Scholar]
- 17.Haferlach C, Mecucci C, Schnittger S, Kohlmann A, Mancini M, Cuneo A, Testoni N, Rege-Cambrin G, Santucci A, Vignetti M, Fazi P, Martelli MP, Haferlach T, Falini B. AML with mutated NPM1 carrying a normal or aberrant karyotype show overlapping biologic, pathologic, immunophenotypic, and prognostic features. Blood. 2009;114:3024–3032. doi: 10.1182/blood-2009-01-197871. [DOI] [PubMed] [Google Scholar]
- 18.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC; Lyon: 2008. pp. 120–121. [Google Scholar]
- 19.Papadaki C, Dufour A, Seibl M, Schneider S, Bohlander SK, Zellmeier E, Mellert G, Hiddemann W, Spiekermann K. Monitoring minimal residual disease in acute myeloid leukaemia with NPM1 mutations by quantitative PCR: clonal evolution is a limiting factor. Br J Haematol. 2009;144:517–523. doi: 10.1111/j.1365-2141.2008.07488.x. [DOI] [PubMed] [Google Scholar]
- 20.Bacher U, Badbaran A, Fehse B, Zabelina T, Zander AR, Kroger N. Quantitative monitoring of NPM1 mutations provides a valid minimal residual disease parameter following allogeneic stem cell transplantation. Exp Hematol. 2009;37:135–142. doi: 10.1016/j.exphem.2008.09.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of data for the 69 clinical specimens evaluated in this study. ND = sequence not determined.