

Abstract
A new method for amplification and labeling of RNA is assessed that permits gene expression microarray analysis of formalin-fixed paraffin-embedded tissue (FFPET) samples. Valid biological data were obtained using gene expression microarrays of diffuse large B-cell lymphoma (DLBCL) FFPET samples. We examined 59 matched DLBCL patient samples, FFPET, and fresh/frozen. The samples contained both prognostic subgroups of DLBCL: germinal center B-cell (GCB) and activated B-cell (ABC). Fresh/frozen (FF) samples were amplified by both the traditional Eberwine oligo-dT method and a new method described herein. The matching FFPET samples were also amplified using the new method. Here we detail the comparison of results from all three datasets of matched samples. An established classification model built from previous data accurately classified these new samples. This new method provides a useful technology advance for microarray analysis of FFPET archival samples.
Gene expression profiling with microarrays has led to many valuable insights into the underlying molecular status of disease tissues.1,2,3,4 Most efforts in microarray gene expression profiling have relied on fresh or fresh/frozen (FF) sources of tissue. The use of FF tissue as the sample type of choice is necessitated because traditional methods of RNA amplification (eg, oligo-dT Eberwine-based methods5) require microgram quantities of good quality (ie, nondegraded) RNA. Formalin-fixed paraffin embedded tissue (FFPET) is the method of choice for virtually all routine diagnostic procedures (eg, H&E staining, immunohistochemistry, fluorescent in situ hybridizations, etc) in pathology. Most clinically relevant samples are routinely archived as FFPET blocks, as this represents a minimum requirement for diagnosis. FFPET samples are often the only source of material available to allow correlation with long-term clinical outcome data. Unfortunately, RNA extracted from FFPET archival samples is of poor quality (eg, degraded and cross-linked) and recovered in very small amounts.6,7 To date, robust methods for routine high-density transcriptome microarray analysis of FFPET samples have not been described.
Diffuse large B-cell lymphoma (ie, DLBCL) comprises approximately 40% of all non-Hodgkin Lymphomas. DLBCL is well documented in multiple studies to contain several distinct molecular subtypes of disease, reflected in gene expression. Two of the subtypes, germinal center B cell (GCB) and activated B-cell (ABC), have been correlated with disease prognosis. GCB overall has a better prognosis than the ABC subclass.8,9,10,11,12,13,14,15,16,17,18 Specifically, GCB has a much higher overall survival than the ABC class when treated with CHOP (60% and 30%, respectively19). Lenz et al20 have confirmed that these molecular subtypes maintain their prognostic significance when Rituximab is introduced in the treatment regimen together with standard multiagent chemotherapy (R-CHOP). In the relapse setting, the proteosome inhibitor Bortezimib, in combination with chemotherapy, is associated with a superior OS in patients with the ABC subtype of DLBCL.21 Thus, DLBCL molecular subtypes may be a useful in the initial risk stratification of patients with DLBCL and play a role in therapy selection in the relapse setting. Moreover, cell-of-origin distinctions (GCB versus ABC) may gain an important role in the choice of initial therapy, particularly in the setting of phase III clinical trials testing new targeted agents.
Here we report the use of a novel method of linear amplification (ie, Ribo-SPIA; Nugen Technology22) to produce biologically relevant data from a collection of 59 matched FF and FFPET DLBCL samples.
Materials and Methods
Lymph Node Processing
Samples were obtained with patient informed consent and with IRB approval. Tumor containing lymph nodes were excised and handled using routine diagnostic protocols used by the British Columbia Cancer Agency (BCCA). After excision at the local hospital, the node was processed in a timely manner and fresh tissue was shipped on saline-soaked gauze overnight to BCCA. Fresh material was immediately frozen on arrival at the BCCA. Formalin-fixed paraffin-embedded material was processed in a timely manner at the local site and subsequently shipped to BCCA. This study was approved by the British Columbia Cancer Agency Ethics board and is in accordance to the declaration of Helsinki.
RNA Extraction
RNA from freshly frozen biopsied samples was prepared using TRIzol, following manufacturer's recommendations. RNA was prepared from the FFPET samples using the RNeasy FFPE kit (Qiagen #74404) following manufacturers recommendations. Supplemental Table 1 (see http://jmd.amjpathol.org) contains the RNA recovery from each sample and sample type (eg, FF or FFPET). The range of recovery was 3.94 up to 771 ng/μl of FFPET RNA and 7.19 up to 86.03 ng/μl from fresh frozen samples. All samples yielded sufficient RNA for input to Ribo-SPIA amplification 10 ng for Fresh RNA and 50 ng for FFPET RNA (see Supplemental Table 1 at http://jmd.amjpathol.org).
RNA Amplification and Labeling
Five micrograms of FF RNA was transcribed into double stranded cDNA (GeneChip One-cycle cDNA Synthesis Kit; Affymetrix) and linearly amplified by the traditional method described by Eberwine5 using the GeneChip IVT Labeling Kit (Affymetrix).
Ten nanograms of FF or 50 ng FFPET RNA was transcribed into double-stranded cDNA and linearly amplified using WT-Ovation FFPE System (Nugen). The cDNA was fragmented and biotin labeled using FL-Ovation cDNA Biotin Module V2 (Nugen).
GeneChip Hybridization
Ten micrograms of Eberwine-labeled cRNA or 5 μg of WT-Ovation amplified cDNA was applied to U133plus 2 GeneChips (Affymetrix) and hybridized overnight per manufacturer's recommendations. GeneChips were washed, stained, and scanned using the Fluidic Station 450 and GeneChip Scanner 3000 (Affymetrix) using manufacturer's recommendations. Affymetrix.cel files have been deposited into GEO (http://www.ncbi.nlm.nih.gov/geo/, accession number GSE19246).
Data Analysis and Classification
Microarray signal was generated using Affymetrix MAS 5. A Bayesian model was used for classification. The classification algorithm was based on previously described methods19 and the normalization dataset updated with the use of gene expression data for DLBCL samples.23 As described previously by Wright, 100 genes were selected and weighed based on the DLBCL samples used in the referenced manuscript.19 This updated model was applied to the samples from each of the three datasets in this study as a blinded test.
Results
A collection of 59 matched samples, where RNA was extracted from the same lymph node biopsy but derived from either fresh frozen tissue or FFPET, were used in this study. The initial experiment was performed using 5 μg of total RNA extracted from the fresh frozen samples. The RNA was prepared for microarray analysis using the standard Eberwine-based amplification methodology.5 Amplified cRNA was hybridized to Affymetrix U133plus2 GeneChips. The resulting microarray data served as a means for classification of ABC or GCB DLBCL subclasses. This process was accomplished by using an updated classifier built on previous Affymetrix GeneChip experimental results.23 Gene expression classification of DLBCL using this Bayesian model can result in some samples not exhibiting the distinct signatures of either GCB or ABC, such that there is less than 90% confidence in assigning to either of the two classes. These samples have been called Unclassified (U) samples.19 Table 119,20,21,22,23 lists the 48 samples and the assignment of each sample to GCB, ABC. Those 11 samples that are designated as U (ie, unclassifiable to either class with 90% or greater confidence) are not included in this table. These assignments are considered the true assignment of each of the 48 samples from which further comparisons are made.
Table 1.
Classification Results
| Sample ID | Class FF (Eberwine) | Confidence FF (Eberwine) | Class FF (Ribo SPIA) | Confidence FF (Ribo SPIA) | Class FPET (Ribo SPIA) | Confidence FPET (Ribo SPIA) |
|---|---|---|---|---|---|---|
| Sample_A1 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_A2 | ABC | 99.9% | ABC | 99.5% | ABC | 99.3% |
| Sample_A3 | ABC | 100% | ABC | 100% | ABC | 100% |
| Sample_A4 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_A5 | ABC | 99.9% | ABC | 99.8% | ABC | 95.5% |
| Sample_A6 | GCB | 7.6% | Unclassified | 19.2% | Unclassified | 17.5% |
| Sample_A8 | ABC | 100% | ABC | 100% | ABC | 98.8% |
| Sample_B1 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_B2 | ABC | 100% | ABC | 100% | ABC | 100% |
| Sample_B3 | GCB | 1.3% | Unclassified | 11.5% | Unclassified | 72.6% |
| Sample_B7 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_B8 | GCB | 0 | GCB | 0 | ABC | 93.6% |
| Sample_B9 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_C1 | GCB | 1.1% | GCB | 1.7% | GCB | 0.4% |
| Sample_C3 | ABC | 100% | ABC | 100% | ABC | 100% |
| Sample_C5 | GCB | 0.8% | GCB | 1.5% | GCB | 1.6% |
| Sample_C6 | ABC | 99.8% | ABC | 91.7% | ABC | 98.1% |
| Sample_C7 | ABC | 90.2% | ABC | 97.1% | ABC | 99.4% |
| Sample_C9 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_D1 | ABC | 99.6% | ABC | 93.6% | ABC | 98.7% |
| Sample_D2 | GCB | 0 | GCB | 0 | GCB | 0.1% |
| Sample_D3 | ABC | 94.8% | Unclassified | 60.5% | ABC | 99.6% |
| Sample_D4 | ABC | 99.9% | ABC | 99.8% | ABC | 96.0% |
| Sample_D5 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_D6 | GCB | 0.7% | GCB | 0 | Unclassified | 19.3% |
| Sample_D7 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_D9 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_E1 | ABC | 99.3% | ABC | 99.8% | ABC | 99.9% |
| Sample_E2 | ABC | 96.4% | ABC | 96.8% | ABC | 91.3% |
| Sample_E3 | ABC | 100% | ABC | 100% | ABC | 100% |
| Sample_E4 | ABC | 100% | ABC | 100% | ABC | 99.9% |
| Sample_E5 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_E6 | ABC | 99.6% | ABC | 99.1% | Unclassified | 78.2% |
| Sample_E7 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_E8 | GCB | 0 | GCB | 0 | GCB | 0.1% |
| Sample_E9 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_F1 | GCB | 0 | GCB | 0.2% | GCB | 9.2% |
| Sample_F2 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_F3 | ABC | 99.6% | ABC | 98.6% | ABC | 98.4% |
| Sample_F4 | ABC | 100% | ABC | 100% | ABC | 100% |
| Sample_F5 | GCB | 0 | GCB | 0 | GCB | 0.3% |
| Sample_F6 | ABC | 100% | ABC | 100% | ABC | 100% |
| Sample_F9 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_G1 | GCB | 0.1% | GCB | 0.2% | GCB | 0 |
| Sample_G2 | GCB | 0 | GCB | 0 | GCB | 0 |
| Sample_G3 | ABC | 99.7% | ABC | 99.6% | ABC | 98.6% |
| Sample_G4 | GCB | 1.5% | GCB | 2.3% | GCB | 3.3% |
| Sample_G5 | ABC | 100% | ABC | 100% | ABC | 100% |
Fifty-nine DLBCL fresh frozen RNA samples analyzed by the Eberwine method were classified using a previously described model.19,23 Eleven samples did not have signatures meeting the confidence needed to call ABC or GCB and were placed into the unclassified category.
The Bayesian classifier was used to classify the FF samples amplified by the Ribo-SPIA method [column: Class FFN; the confidence is reported in Confidence (FFN)].
The Bayesian classifier was used to classify the FFPET samples amplified by the Ribo-SPIA method [column: Class FFPET; the confidence is reported in Confidence (FFPET)].
It was important to demonstrate the original prognostic value of these subclasses such that biologically relevant meaning could be attributed to the results. Kaplan–Meier survival curves for the R-CHOP treated patients show GCB patients had better overall survival than the ABC patients (Figure 1; Supplemental Table 2, available at http://jmd.amjpathol.org), consistent with previous CHOP prognostic data. It is interesting to note that overall survival has increased in both groups (GCB and ABC) with the addition of Rituximab compared with the original CHOP alone data. Both subclasses show improved prognosis of about 10 to 15% with the addition of Rituxamab compared with CHOP alone. These data are in complete agreement and verify a study recently published by Lenz et al20 from the LLMPP (Leukemia/Lymphoma Molecular Profiling Project) consortium.
Figure 1.
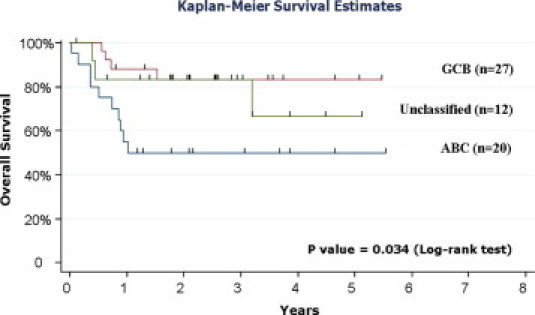
Kaplan-Meier survival plot of DLBCL samples included in this study.
Next, matching samples were amplified by the Ribo-SPIA FFPET method. Ten nanograms of the same fresh-frozen RNA that was used for Eberwine-based amplification (above) was amplified, and 50 ng of FFPET extracted RNA was also amplified. These samples were hybridized to Affymetrix U133plus2 GeneChips. A comparison of gene expression data from all three replicate experiments yielded the following results. A comparison of percent present calls was made for each of the three patient tumor replicates (see Supplemental Figure 1 at http://jmd.amjpathol.org). The Ribo-SPIA amplified fresh-frozen samples consistently had higher present calls (average 55% present calls) when compared with the traditional Eberwine method (average 43% present calls). This is of interest, because the Ribo-SPIA samples used only 10 ng compared with 5 μg of traditional labeled material. The 50 ng RNA extracted from the FFPET matched samples produced lower percent present calls (average 29% present calls), which is expected because the sample RNA is known to be of poorer quality and severely degraded (see Figure 2). Although, the percent present calls from FFPET were lower than those from fresh-frozen, these data still contain an average of 29% present calls out of all of the 54,675 probesets and represents a potential large dataset for analysis. Other metrics commonly used for microarray quality analysis were examined. As expected the scaling factors inversely reflected the percent present calls and no other observations were noteworthy (data not shown). It should be mentioned the Ribo-SPIA FFPET method uses random primers, hence the usual analysis of GAPDH and β-actin 3′/5′ ratios is not as meaningful as with traditional oligo-dT primed Eberwine methods. The 3′/5′ ratios observed from Ribo-SPIA samples were all under 3, even with the highly degraded FFPET RNA.
Figure 2.
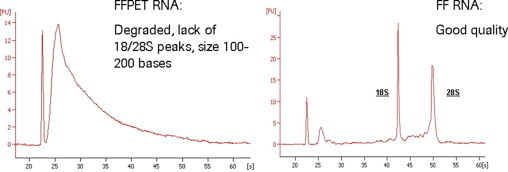
Representative electropherogram of total RNA extracted from FFPET archived lymphoma sample and a freshly frozen lymphoma sample as analyzed on an Agilent BioAnalyzer.
Next, the gene expression signals of the matched samples were compared between methods and sample types (FF Eberwine versus FF Ribo-SPIA, FF Ribo-SPIA versus FFPET Ribo-SPIA, and FF Eberwine versus FFPET Ribo-SPIA) (Figure 3; Supplemental Table 3 available on http://jmd.amjpathol.org). Figure 3 depicts a representative scatter plot, and Supplemental Table 3 (see http://jmd.amjpathol.org) contains the correlation of all 48 samples. R2 signal correlation from scatter plots indicate that FF Ribo-SPIA and FFPET Ribo-SPIA samples demonstrate similar gene expression correlation as between FF Ribo-SPIA and FF Eberwine average of all samples is 0.78 versus 0.77, respectively). The lowest correlation occurs when both the sample type and method (Ribo-SPIA FFPET and Eberwine FF) are compared (∼0.64 average for all samples). Although these R2 correlations are less than what is normally observed when replicates using the same method and same sample technical replicates are compared, these data do indicate that overall gene expression across the matched samples demonstrate reasonable correlation.
Figure 3.
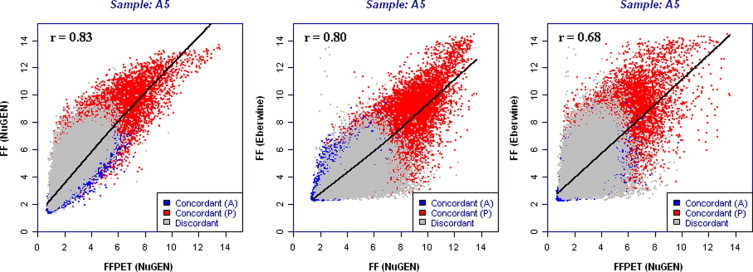
Scatter plots comparing each of the three data sets for a representative sample (FF Nugen versus FFPET Nugen, FF Eberwine versus FF Nugen, and FF Eberwine versus FFPET Nugen) of all probeset signals generated by MAS 5.0 Affymetrix Software.
For subsequent data analysis, only the 48 samples that were previously classified as ABC or GCB were used. The 11 samples called unclassified (U) were removed from further analysis. Genes were selected that statistically (ie, t-test) best separate the 48 samples into their known biological subgroup (ABC or GCB). It was observed that these data from 10 ng of FF Ribo-SPIA samples generated a larger number of genes at each P value level of false-discovery as compared with the 5 μg of FF Eberwine data (Figure 4a). At a FDR of P = 0.05, 1428 genes met this significance for the FF Ribo-SPIA versus 656 genes from FF Eberwine data. The lower quality FFPET RNA resulted in fewer genes detected overall and therefore fewer at this FDR level; 289 genes.
Figure 4.
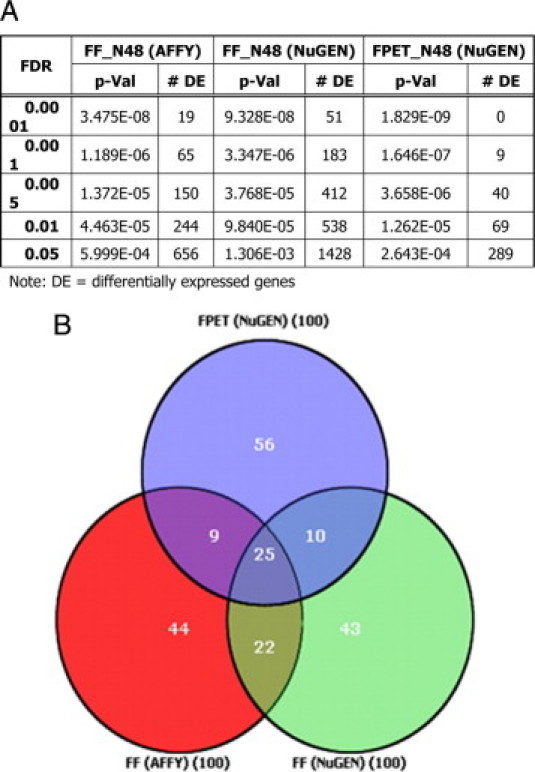
A: Results of t-tests and false discovery rates of the number of genes that statistically separate GCB and ABC sample groups for each dataset: FF Affy (ie, Eberwine), FF Nugen, and FFPET Nugen. B: Venn diagram depicting the top 100 genes with smallest t-test values for each of the three data sets.
When the top 100 significant genes from each of the three datasets were compared for similarity, it was observed that 25% of the genes shared identity between all three datasets (Figure 4b; Supplemental Table 4 available at http://jmd.amjpathol.org).
Next the top 100 selected genes that discriminate ABC from GCB, from each of the three datasets, were examined for biological function. The biological pathways represented by each list of genes were determined. It was observed that 11 of 11 functional pathways identified from the original Eberwine-based classifier were statistically significantly represented in each dataset's gene lists (Figure 5). Several of the pathways center around NF-κB signaling, which is up-regulated in the ABC subclass, consistent with previous observations.24 This result indicates that the underlying biological differences that discriminate ABC from GCB are consistent with these data even though the exact genes selected are not identical.
Figure 5.
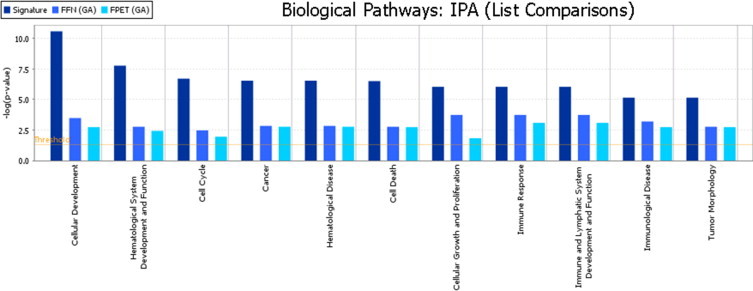
Results of pathway analysis from Ingenuity IPA Software. The horizontal gold bar indicates the threshold of significance. The Signature pathways listed in darkest blue come from the list of genes (published by Wright et al19) that separate ABC from GCB subgroups.
To further assess these data for biological meaning, the FF Ribo-SPIA samples were classified into the subclasses of ABC or GCB based on the updated Bayesian model of classification (updated with data from Dave et al23; see Table 119,20,21,22,23). The majority of the samples were classified as either ABC or GCB (45 of 48). Three samples did not achieve the greater than 90% confidence needed for an ABC/GCB call and were designated U, yielding a call rate of 93.8%. It should be mentioned that the three unclassified samples did trend toward the correct class but failed to meet the required confidence level for classification (Tables 119,20,21,22,23 and 2). All 45 samples classified were correctly classified, resulting in 100% accuracy. These results as well as the pathway results support the assertion that the underlying biology detected in the FF samples is consistent across the two methods, Eberwine and Ribo-SPIA.
Table 2.
Classification Accuracy
| FF (Ribo SPIA) |
|||
|---|---|---|---|
| FF (Eberwine) | ABC | GCB | Unclassified |
| ABC | 20 | 0 | 1 |
| GCB | 0 | 25 | 2 |
| Call rate | 45/48 = 93.8% | ||
| Accuracy | 45/45 = 100% | ||
| Confidence | 91.7%–100% | ||
| FPET (Ribo SPIA) |
|||
|---|---|---|---|
| FF (Eberwine) | ABC | GCB | Unclassified |
| ABC | 20 | 0 | 1 |
| GCB | 1 | 23 | 3 |
| Call rate | 44/48 = 91.7% | ||
| Accuracy | 43/44 = 97.7% | ||
| Confidence | 90.8%–100% | ||
The classification of the FFPET samples is shown in Tables 119,20,21,22,23 and 2. These results show 4 the 48 samples are unclassified yielding a call rate of 91.7% (44 of 48 are classified). It is worth noting that of the 44 samples classified only one sample was miscalled. This results in an overall accuracy rate of 97.7%. This accuracy should be considered in the context of the quality of the starting material, RNA extracted from FFPET samples is known to be highly degraded. It is also known that these FFPET samples produced less robust gene expression data as observed by percent present calls and the number of genes selected with statistical significance for discriminating ABC from GCB. The fact that a classification model based both on a different method (ie, Eberwine) and different sample type (ie, FF sample) can accurately classify the FFPET samples processed with the new Ribo-SPIA method strongly supports the relevance of the underlying biology in FFPET samples.
Discussion
We have described a new method for amplification and labeling of total RNA for microarray analysis. This new method, Ribo-SPIA, provides certain advantages over the traditional Eberwine, oligo-dT based methods. The new method permits very small quantities of FF input RNA (10 ng FF compared with 1–5 μg required for Eberwine). This opens up the potential to use such sample types as fine needle biopsies and purified cell populations (from either flow cytometry or laser capture microdissection). Further, this new method is demonstrated to be amendable to poor quality degraded FFPET RNA. The input requirement for FFPET RNA is 50 ng, which is well within the amount of material obtained from routine 10-micron sections of FFPET blocks.
These data indicate that this new method not only allows for using less input RNA, but also suggests that more data are produced from the microarrays. On average the Ribo-SPIA produced present calls of 55% using 10 ng input RNA. The traditional Eberwine method only produced an average of 43% present calls from 5 μg of the exact same FF samples. The cDNA targets generated by the Ribo-SPIA method have been consistently demonstrated to result in higher percentage calls on Affymetrix GeneChip arrays than cRNA targets. This is true with degraded RNA samples as well as high-quality commercial RNA samples with no evidence of degradation. The reason for this higher present call rate is the improved hybridization specificity of cDNA targets on DNA probe arrays versus cRNA targets. The present calls on GeneChip arrays are influenced by the discrimination of the mismatch probe versus the perfect match probe. cRNA targets have been shown to produce lower perfect match/mismatch discrimination scored than cDNA targets and therefore many elements are not called present with cRNA targets despite having signals significantly above the background. Several publications address this observation including the publications by Barker et al25 and Eklund et al.26 More information is being gained from less input RNA using this new method. This is supported by the higher percent present calls and also the selection of statistically significant genes that discriminate the two biological subtypes of DLBCL (ABC and GCB). The Ribo-SPIA method, when used with fresh-frozen RNA, produced more than twice as many genes meeting statistical significance than did the Eberwine method.
It is important that the genes found using this new method are demonstrated to reflect true biology expressed in the sample. The use of pathway analysis demonstrates that the biological functions separating ABC from GCB subtypes are identical across both methods and sample types (FF or FFPET). Further verification that the new method permits analysis of proper biology is provided by the classification results shown in Tables 119,20,21,22,23 and 2. An established Bayesian classifier built on previous data produced by the Eberwine method accurately classifies the samples processed with the new method regardless if the sample type is FF or FFPET.
These results clearly support the biological validity of data generated from each method (ie, traditional Eberwine or Ribo-SPIA) and sample type (ie, FF or FFPET). The Ribo-SPIA FFPET method can be used to obtain biologically relevant data from valuable archival FFPET samples using microarray gene expression analysis and thus significantly increase available clinical samples for expression profiling studies.
Acknowledgements
We thank Dr. Louis Staudt for his critical review of this manuscript.
Footnotes
Supported by a National Cancer Institute of Canada, Terry Fox Foundation Program Project grant # 19001 (R.G.D.). R.G.D. and G.W. are supported by an NCI Strategic Partnering to Evaluate Cancer Signatures grant (U01-CA-114778).
P.M.W. and R.L. contributed equally to this study.
P.M.W. and R.L. are employed by Roche Molecular Diagnostics. J.-D.H. is employed by Nugen Technologies, the manufacturer of the Ribo-SPIA kit. None of the other authors disclosed any relevant financial relationships.
Supplemental material for this article can be found on http://jmd.amjpathol.org.
Current address for P.M.W.: Patient Characterization Center and Clinical Assay Development Center, SAIC-NCI-Frederick, Frederick, MD.
Web Extra Material
Contains the RNA recovery for each of the 59 samples extracted from FF (freshly frozen lymph nodes) and the matched FFPET (i.e. formalin fixed paraffin embedded lymph nodes).
(additional statistics for Figure 1)
Supporting hazard ratio statistics for Figure 1 Kaplan Meier survival plot.
Correlation of All Samples
Contains the Pearson Correlation
Top 100 t-test genes:
The 48 samples in each method group were classified by the Bayesian classifier as applied to the FF Eberwine samples were assessed by t-tests. This table contains the gene lists for the 100 highest t-test significance of the comparison of ABC and GCB sub-types
{kind=link}
References
- 1.Davies AJ, Rosenwald A, Wright G, Lee A, Last KW, Weisenburger DD, Chan WC, Delabie J, Braziel RM, Campo E, Gascoyne RD, Jaffe ES, Muller-Hermelink K, Ott G, Calaminici M, Norton AJ, Goff LK, Fitzgibbon J, Staudt LM, Andrew Lister T. Transformation of follicular lymphoma to diffuse large B-cell lymphoma proceeds by distinct oncogenic mechanisms. Br J Haematol. 2007;136:286–293. doi: 10.1111/j.1365-2141.2006.06439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 3.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 4.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lonning P, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci USA. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung JY, Braunschweig T, Williams R, Guerrero N, Hoffmann KM, Kwon M, Song YK, Libutti SK, Hewitt SM. Factors in tissue handling and processing that impact rna obtained from formalin-fixed paraffin-embedded tissue. J Histochem Cytochem. 2008;56:1033–1042. doi: 10.1369/jhc.2008.951863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Ahlfen S, Missel A, Bendrat K, Schlumpberger M. Determinants of RNA quality from FFPE samples. PLoS ONE. 2007;2:e1261. doi: 10.1371/journal.pone.0001261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 9.Lawrie CH, Soneji S, Marafioti T, Cooper CD, Palazzo S, Paterson JC, Cattan H, Enver T, Mager R, Boultwood J, Wainscoat JS, Hatton CS. MicroRNA expression distinguishes between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma. Int J Cancer. 2007;121:1156–1161. doi: 10.1002/ijc.22800. [DOI] [PubMed] [Google Scholar]
- 10.Leich E, Hartmann EM, Burek C, Ott G, Rosenwald A. Diagnostic and prognostic significance of gene expression profiling in lymphomas. Apmis. 2007;115:1135–1146. doi: 10.1111/j.1600-0463.2007.apm_867.xml.x. [DOI] [PubMed] [Google Scholar]
- 11.Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, Powell J, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, Campo E, Jaffe ES, Delabie J, Smeland EB, Rimsza LM, Fisher RI, Weisenburger DD, Chan WC, Staudt LM. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA. 2008;105:13520–13525. doi: 10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu X, Nechushtan H, Ding F, Rosado MF, Singal R, Alizadeh AA, Lossos IS. Distinct IL-4-induced gene expression, proliferation, and intracellular signaling in germinal center B-cell-like and activated B-cell-like diffuse large-cell lymphomas. Blood. 2005;105:2924–2932. doi: 10.1182/blood-2004-10-3820. [DOI] [PubMed] [Google Scholar]
- 13.Miyazaki K, Yamaguchi M, Suguro M, Choi W, Ji Y, Xiao L, Zhang W, Ogawa S, Katayama N, Shiku H, Kobayashi T. Gene expression profiling of diffuse large B-cell lymphoma supervised by CD21 expression. Br J Haematol. 2008;142:562–570. doi: 10.1111/j.1365-2141.2008.07218.x. [DOI] [PubMed] [Google Scholar]
- 14.Poulsen CB, Borup R, Nielsen FC, Borregaard N, Hansen M, Gronbaek K, Moller MB, Ralfkiaer E. Microarray-based classification of diffuse large B-cell lymphoma. Eur J Haematol. 2005;74:453–465. doi: 10.1111/j.1600-0609.2005.00429.x. [DOI] [PubMed] [Google Scholar]
- 15.Rosenwald A, Staudt LM. Clinical translation of gene expression profiling in lymphomas and leukemias. Semin Oncol. 2002;29:258–263. doi: 10.1053/sonc.2002.32901. [DOI] [PubMed] [Google Scholar]
- 16.Rosenwald A, Staudt LM. Gene expression profiling of diffuse large B-cell lymphoma. Leuk Lymphoma. 2003;44(Suppl 3):S41–S47. doi: 10.1080/10428190310001623775. [DOI] [PubMed] [Google Scholar]
- 17.Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RC, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, Ray TS, Koval MA, Last KW, Norton A, Lister TA, Mesirov J, Neuberg DS, Lander ES, Aster JC, Golub TR. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. 2002;8:68–74. doi: 10.1038/nm0102-68. [DOI] [PubMed] [Google Scholar]
- 18.Staudt LM, Dave S. The biology of human lymphoid malignancies revealed by gene expression profiling. Adv Immunol. 2005;87:163–208. doi: 10.1016/S0065-2776(05)87005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2003;100:9991–9996. doi: 10.1073/pnas.1732008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, Xu W, Tan B, Goldschmidt N, Iqbal J, Vose J, Bast M, Fu K, Weisenburger DD, Greiner TC, Armitage JO, Kyle A, May L, Gascoyne RD, Connors JM, Troen G, Holte H, Kvaloy S, Dierickx D, Verhoef G, Delabie J, Smeland EB, Jares P, Martinez A, Lopez-Guillermo A, Montserrat E, Campo E, Braziel RM, Miller TP, Rimsza LM, Cook JR, Pohlman B, Sweetenham J, Tubbs RR, Fisher RI, Hartmann E, Rosenwald A, Ott G, Muller-Hermelink HK, Wrench D, Lister TA, Jaffe ES, Wilson WH, Chan WC, Staudt LM. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359:2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunleavy K, Pittaluga S, Czuczman MS, Dave SS, Wright G, Grant N, Shovlin M, Jaffe ES, Janik JE, Staudt LM, Wilson WH. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood. 2009;113:6069–6076. doi: 10.1182/blood-2009-01-199679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurn N, Chen P, Heath JD, Kopf-Sill A, Stephens KM, Wang S. Novel isothermal, linear nucleic acid amplification systems for highly multiplexed applications. Clin Chem. 2005;51:1973–1981. doi: 10.1373/clinchem.2005.053694. [DOI] [PubMed] [Google Scholar]
- 23.Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma EJ, Greiner TC, Weisenburger DD, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Delabie J, Rimsza LM, Braziel RM, Grogan TM, Campo E, Jaffe ES, Dave BJ, Sanger W, Bast M, Vose JM, Armitage JO, Connors JM, Smeland EB, Kvaloy S, Holte H, Fisher RI, Miller TP, Montserrat E, Wilson WH, Bahl M, Zhao H, Yang L, Powell J, Simon R, Chan WC, Staudt LM. Molecular diagnosis of Burkitt's lymphoma. N Engl J Med. 2006;354:2431–2442. doi: 10.1056/NEJMoa055759. [DOI] [PubMed] [Google Scholar]
- 24.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001;194:1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barker CS, Griffin C, Dolganov GM, Hanspers K, Yang JY, Erle DJ. Increased DNA microarray hybridization specificity using sscDNA targets. BMC Genomics. 2005;6:57. doi: 10.1186/1471-2164-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eklund AC, Turner LR, Chen P, Jensen RV, deFeo G, Kopf-Sill AR, Szallasi Z. Replacing cRNA targets with cDNA reduces microarray cross-hybridization. Nature Biotechnol. 2006;24:1071–1073. doi: 10.1038/nbt0906-1071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Contains the RNA recovery for each of the 59 samples extracted from FF (freshly frozen lymph nodes) and the matched FFPET (i.e. formalin fixed paraffin embedded lymph nodes).
(additional statistics for Figure 1)
Supporting hazard ratio statistics for Figure 1 Kaplan Meier survival plot.
Correlation of All Samples
Contains the Pearson Correlation
Top 100 t-test genes:
The 48 samples in each method group were classified by the Bayesian classifier as applied to the FF Eberwine samples were assessed by t-tests. This table contains the gene lists for the 100 highest t-test significance of the comparison of ABC and GCB sub-types