

Abstract
When populations of cells are subjected to nonlethal selection, mutations arise in the absence of cell division, a phenomenon that has been called “adaptive mutation.” In a strain of Escherichia coli that cannot metabolize lactose (Lac−) but that reverts to lactose utilization (Lac+) when lactose is its sole energy and carbon source, the mutational process consists of two components. (1) A highly efficient, recombination-dependent mechanism giving rise to mutations on the F′ episome that carries the Lac− allele; and (2) a less efficient, unknown mechanism giving rise to mutations elsewhere in the genome. Both selected and nonselected mutations arise in the Lac− population, but nonselected mutations are enriched in Lac+ mutants, suggesting that some Lac+ cells have passed though a transient period of increased mutation. These results have several evolutionary implications. (1) DNA synthesis initiated by recombination could be an important source of spontaneous mutation, particularly in cells that are not undergoing genomic replication. (2) The highly active mutational mechanism on the episome could be important in the horizontal transfer of variant alleles among species that carry and exchange conjugal plasmids. (3) A subpopulation of cells in a state of transient mutation could be a source of multiple variant alleles and could provide a mechanism for rapid adaptive evolution under adverse conditions.
That spontaneous mutations arise at random during the nonselective growth of a population was established by classic papers published in the 1940s and 1950s.1–4 With some dissent,5,6 the reigning dogma for 40 years was that most spontaneous mutations arise as random errors during genomic replication. In 1988 Cairns and his collaborators7 published a paper that changed our thinking about the nature of spontaneous mutation. They confirmed and extended previous results showing that mutations arise in nondividing bacterial cells when they are subjected to nonlethal selective pressure. In addition, Cairns et al. suggested that only selected mutations, not deleterious or neutral mutations, appeared in a population during selection. This phenomenon was named “directed mutation” by the editors of Nature, but the name “adaptive mutation” is most widely used today.
The phenomenon of adaptive mutation has generated a wealth of research in the subsequent years (reviewed in refs. 8–10). The Lamarckian idea of a reverse information flow from the environment back to the genome was eliminated early on.11 The remaining explanations for the phenomenon all incorporate some form of a reversible process of “trial and error.” That is, during selection a random mutational process affecting the whole genome occurs, but the process is adaptive because the variants (or the cells bearing them) are transient until a mutation arises that allows growth.7,12–14 However, not all cases of mutation in nondividing cells are examples of adaptive mutation, as in some cases it was shown that the mutations can arise when the cells are merely starving.
This review focuses on the best studied example of adaptive mutation, reversion to lactose utilization (Lac+) in Escherichia coli strain FC40. We now know that nonselected mutations arise and persist in the population during lactose selection.15 Thus, the mutational process in FC40 does not meet the original definition of adaptive mutation. However, we continue to call the selected mutations “adaptive” to distinguish them from mutations occurring during nonselective growth and from nonselected mutations occurring during selection. This meaning of adaptive mutation is the same as that used by evolutionists to distinguish beneficial from neutral or deleterious mutations. Recently it was found that among Lac+ clones of FC40 the frequency of nonselected mutations appears to be higher than that among the population at large.15–17 These results imply that a subpopulation of stressed cells undergoes some form of transient mutation, as originally suggested by Hall.13
Properties of the Lac− Allele
The Lac− allele in FC40 is derived from fusion of the lacI gene to the lacZ gene that eliminates the coding sequence for the last four residues of lacI, all of lacP and lacO, and the first 23 residues of lacZ. Transcription is initiated from the lacIq promoter, which is 10-fold stronger than the wild-type lacI promoter and results in about 200 Miller units of β-galactosidase, an amount sufficient to make the cells Lac+. The fusion protein also retains LacI repressor activity.18,19 Mutant versions of this fusion constructed by Miller and coworkers have been used for a variety of mutational studies (e.g., see ref. 20).
The Lac− allele carried by FC40, Φ(lacI33-lacZ), has an ICR191-induced +1 frame-shift at the 320th codon of lacI, changing CCC to CCCC.21 The allele is slightly leaky, producing about 2 Miller units of β-galactosidase, which is not sufficient to allow FC40 to grow on lactose.22 The frameshift is polar on lacY; thus, FC40 is also permease-defective.23 However, as stationary-phase cells of FC40 do not revert to Lac+ unless lactose is present,24 the energy provided by the low amount of β-galactosidase may be required for the mutational process. Indeed, Galitski and Roth25 found that adaptive reversion of various Lac− alleles in Salmonella typhimurium was dependent on the allele being leaky, although reversion rates were not well correlated to the degree of leakiness.
DNA Polymerase Errors
Because the lacI coding sequence is not essential, any mutation that restores the reading frame but does not create a nonsense mutation will revert Φ(lacI33-lacZ). The mutational target is 130 base pairs (bp) bounded by stop codons in the −1 and +1 reading frames. Reversions can occur by simple frameshifts or by more complex DNA rearrangements, and about half of the Lac+ mutations that arise in FC40 during nonselective growth are insertions and deletions larger than +2 or −1 bp. By contrast, 75% of the adaptive mutations are −1-bp deletions26–28 (Fig. 1). Thus, complex mutations are frequent in the absence of selection but are rare during selection. Most of the adaptive frameshifts occur in runs of 3 or more bases. This site specificity is typical of −1 frameshift mutations made by DNA polymerases in vitro and can be explained by replication of a misaligned template.29,30 In vivo, −1 frameshifts are enhanced by the absence of the methyl-directed mismatch repair system.31,32 As shown in Figure 2, the adaptive mutational spectrum is dominated by one hotspot at which over half the frameshift mutations occur.28 Why this run of G:C bp is so mutable is not known, but it is the site originally mutated by ICR191 and may have some special structural property.
FIGURE 1.
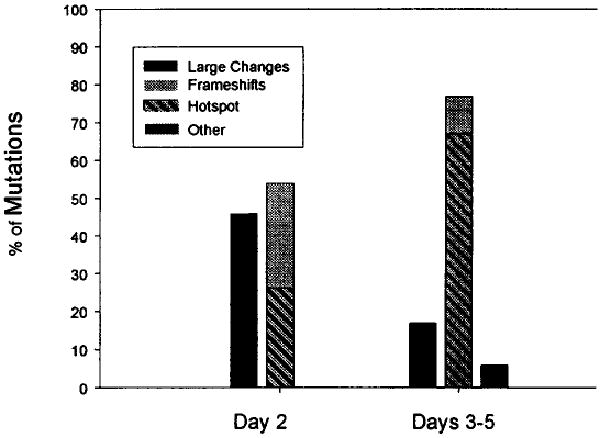
Percent of each class of Lac+ revertants of FC40. Day 2 are mutations that occurred during nonselective growth before plating on lactose medium, whereas days 3–5 are adaptive mutations. Other mutations include unstable revertants and second-site suppressors. (Data are taken from refs. 28 and 46.)
FIGURE 2.
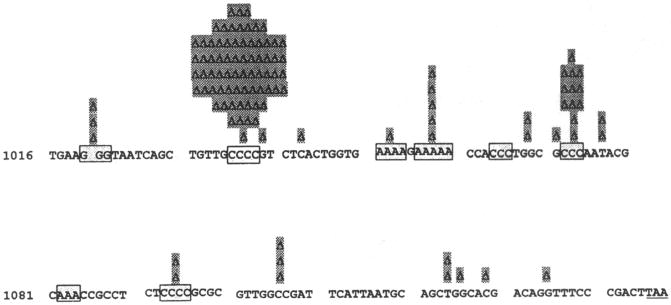
Spectrum of adaptive −1-bp frameshifts in FC40. (Data are taken from refs. 26, 28, and 46.)
E. coli has three DNA polymerases, DNA polymerase I, II, and III, encoded by the genes polA, polB, and dnaE, respectively. Pol I is a repair enzyme, Pol III is the replicative enzyme, and the role of Pol II is unknown. Adaptive mutations in FC40 are greatly decreased by an antimutator allele of dnaE; thus, adaptive mutations are due to errors made by Pol III.28 Surprisingly, if Pol II is inactivated, the rate of adaptive mutation increases about threefold33 but is still decreased by the dnaE915 allele.28 If only the proofreading function of Pol II is inactivated, the rate of adaptive mutation increases sixfold and is unaffected by dnaE915.28 In both cases, the extra mutations are recA-dependent and are dominated by −1 bp-deletions at runs.28 Thus, Pol II synthesizes DNA in nutritionally deprived cells and can make the errors that give rise to adaptive mutations, but normally those errors are corrected by its efficient proofreader. Pol III is also active in nondividing cells and is responsible for the bulk of the mutations that occur.
Recombination and the SOS Response
In our first paper describing FC40, we reported that Lac+ revertants arise by a different mechanism during lactose selection than during nonselective growth.24 Specifically, adaptive but not growth-dependent reversion to Lac+ requires RecA, E. coli's recombinase. In addition to promoting homologous pairing and DNA strand exchange, RecA participates in the cellular response to DNA damage known as the SOS response. The SOS response involves some 20 genes that are repressed by a common repressor, LexA. These genes include umuD and C, which are required for mutagenesis, dinB, a umuC homolog, ruvA and B, which encode branch migration enzymes, polB, which encodes DNA polymerase II, as well as recA and lexA itself. After DNA damage RecA becomes activated to be a “coprotease,” facilitating the cleavage of LexA (inactivating it) and UmuD (activating it).34 We found that adaptive reversion of FC40 was diminished in strains with a defective recA allele or a noncleavable lexA allele, but it was normal in strains defective in umu functions.24 (Recently we found that dinB function also is not required.16) These results established the important point that known SOS mutagenic functions play no role in adaptive reversion of FC40. Because RecA's role in SOS mutagenesis was irrelevant, we concluded that its role as a recombinase was required.8
Adaptive reversion of FC40 is also diminished in recBC− cells.23,36 RecBCD is a multifunctional enzyme with helicase, exonuclease, and endonuclease activities. It participates with RecA in E. coli's recombinational pathway for the repair of double-strand breaks.37 In addition, RecBCD is required for induction of the SOS response when replication is inhibited in the absence of DNA damage.38 But, as our results indicated that SOS mutagenesis is not involved adaptive mutation,24 the requirement for recBC indirectly implicates the double-strand break repair pathway by the process of elimination. Adaptive mutation in FC40 is increased in a recD− mutant,36 but Lac+ mutations occur by a different pathway in recD− cells than in wild-type cells.35
Although the SOS mutagenic functions are not required, they do enhance adaptive reversion to Lac+ if induced.23 This is of concern in certain mutant backgrounds, such as recG−, recD−, and exonuclease defective mutants, in which defects in normal DNA metabolism result in excess single-stranded DNA that can induce the SOS response. Thus, before results obtained in a mutant can be extrapolated to wild-type cells, it has to be demonstrated that the SOS mutagenesis functions are not involved, as we did for recG− cells.39
As just mentioned, LexA represses a variety of genes in addition to recA. To determine if other LexA-repressed genes needed to be derepressed for adaptive reversion of FC40, we tested if a nonrepressable allele of recA would restore the normal level of mutation to a strain with a noncleavable LexA. Although we originally thought that the restoration was complete,24 subsequent experiments showed that only about half the mutation rate was restored.39 This result suggests that at least one other LexA-repressed gene is involved. As RuvAB is uniquely required for adaptive reversion of FC40 (see below), the ruvAB operon is a likely candidate. An interesting alternative hypothesis is that the noncleavable LexA protein itself is inhibitory to adaptive mutation, as in vitro such a LexA mutant protein inhibits RecA-promoted strand exchange.40
Surprisingly, certain recombination functions have different roles in adaptive mutation than they do in normal recombination. E. coli's two enzyme systems for branch migration of recombination intermediates, RuvAB and RecG, are redundant for normal recombination,41 but RuvAB promotes and RecG inhibits adaptive Lac+ mutation.39,42
Conjugal Functions
For ease of genetic manipulation the Lac− allele in FC40 is carried on an F′ episome, and the lac-pro region is deleted from the chromosome. If the Φ(lacI33-lacZ) allele resides on the chromosome instead, the rate of adaptive mutation falls 100-fold43,44 and the remaining mutations do not depend on recombination functions.16,44 Defects in conjugal functions cause a 10-fold reduction in adaptive mutation of the episomal allele,44,45 although in this case the remaining mutations are recA-dependent.46 However, actual episome transfer is not required for adaptive mutation.44,47 These results indicate that recombination-dependent reversion of Φ(lacI33-lacZ) does not require conjugation, but it is enhanced by a process that is closely associated with conjugation.
During conjugation, the episomal DNA is nicked at oriT, the conjugal transfer origin, unwound, and replacement-strand synthesis is initiated. The replaced single-strand is passed though a membrane-spanning structure to the recipient, where it is replicated. All of these events are closely associated with a large membrane-bound complex consisting of both enzymatic and structural proteins.48 Although conjugal replication might give rise to adaptive mutation, it is likely that the only event required is nicking at oriT.39,49 This would explain why adaptive mutations are reduced, but not eliminated, by mutations or treatments that completely inhibit conjugation but may only partially inhibit nicking at oriT.43,44,47
Amplification of the Lac− Allele
Under selection some leaky alleles amplify to give pseudorevertants. These are characteristically unstable because when selection is relieved, the amplified array reverts to a single copy.50,51 We found that 2–3% of the adaptive Lac+ revertants of FC40 are unstable and thus attributable to amplification.22,28 Typically, these revertants appear late in the experiments and are only weakly Lac+, suggesting that they arise in cells that acquired duplications of the lac region during nonselective growth. Because amplification is recombination-dependent, we hypothesized that true revertants might arise during replication of amplified copies of the lac region, which would then be resolved in favor of the Lac+ copy when the cells began to grow.11 This hypothesis has been extended to include a growing subpopulation of cells carrying amplified Lac− alleles.52 However, we found no evidence for a growing subpopulation of FC40 cells during lactose selection,22 nor is amplification of the lac region detectable by Southern hybridization.35 Thus amplification of the Lac− allele appears to play only a minor role in adaptive reversion of FC40.
Mismatch Repair
The mutations that produce adaptive revertants of FC40 are correctable by methyl-directed mismatch repair (MMR). In E. coli, adenines in dGATC sequences are methylated by the dam methylase. Because methylation lags behind replication, newly synthesized DNA is hemimethylated. The MMR enzymes correct mismatches in hemimethylated DNA in favor of the parental methylated strand, achieving a 100- to 1,000-fold increase in the fidelity of DNA replication.53 Defects in MMR result in a 100-fold increase of both the growth-dependent and the adaptive mutation rates of FC4011,39 and of the chromosomal Φ(lacI33-lacZ) allele.16 Thus, MMR is just as efficient in correcting errors during lactose selection as during nonselective growth. Defects in each of the three major MMR proteins, MutS, MutL, and MutH, as well as overproduction of the Dam methylase, have the same phenotype,23,39 indicating that the DNA in nutritionally deprived FC40 cells is methylated as normal.
Similar results have been obtained with other E. coli strains.14,54,55 Thus, MMR activity cannot be globally limiting in nutritionally deprived or stationary-phase cells. Nonetheless, an attractive hypothesis for adaptive mutations is that MMR is depleted in at least a subset of cells during selection,26,27,56 an idea that owes its origin to Stahl.12
There are two experimental results underlying this hypothesis. First, MMR repair capacity is never in great abundance. MMR is rather easily saturated in growing cells,32,57–59 but there is some disagreement about which protein is limiting. MutS is the obvious candidate as it is unstable56 and growth-rate and growth-phase regulated.60,61 Indeed, from theoretical calculations it was postulated that MutS is almost always limiting for MMR.56 However, experiments in which MMR proteins are overproduced from plasmids have separately implicated MutS, MutL, and MutH.59,62,63 As MutS and MutL interact with each other even in the absence of mismatches60 and also interact with other proteins, some of these differences may be attributable to different levels of expression, the different alleles and constructions used, and/or the physiologic state of the cells. We found that overproduction of MutS and MutL together reduced growth-dependent reversion of FC40 about threefold.28
Second, levels of MutS and MutH, but not MutL, appear to decline as cells enter stationary phase or experience nutritional deprivation. The decline in MutH was two to threefold, whereas the decline in MutS varied from 3- to 10-fold among experiments.56,61,64 As just discussed, MMR activity is not globally depleted in nutritionally deprived cells; therefore, despite these declines, the levels of MMR proteins that remain in most cells must be sufficient for the amount of DNA synthesis that occurs. However, we found that overproduction of MutL and MutS together resulted in a three to fivefold reduction in the adaptive reversion rate of FC40,28,39 suggesting that MMR activity can be improved in nutritionally deprived cells (or a subset of them) although not greatly more than in growing cells.
This result was subsequently challenged by Harris et al.64 on two grounds. First, they claimed that excess MutL alone was antimutagenic. In contrast, we found that MutS and MutL each had small effects, but the two together had a larger antimutagenic effect, but only if MutS was in excess.28,46 Harris et al. used different plasmid constructions and their data are highly variable, so they may have missed the contribution of excess MutS. Second, although Harris et al. confirmed our observation that excess MMR proteins reduced the frequency of growth-dependent mutations, they dismissed this result as due to an increase in the time that cells take to form Lac+ colonies when MutS and MutL are overproduced. However, this claim is not supported by their data, as the reported 7 of 58 hours' difference in time to colony formation between experimental and control strains is not significant (p = 0.18).46 Therefore, although the hypothesis that MMR activity is particularly limited in stationary-phase or nutritionally deprived cells is appealing, it has no experimental support.
It should also be noted that overproduction of MMR proteins could reduce mutations by mechanisms unrelated to error correction. For example, MMR proteins bound to mismatched DNA could prevent the recombination required for adaptive mutation. MutS blocks branch migration in vitro, and the blockage is enhanced by the presence of MutL.65,66 That excess MMR proteins might have a similar role in preventing adaptive mutation is suggested by their effects in certain mutant backgrounds. In recC− derivatives of FC40 the adaptive mutation rate is increased 100-fold, yet overproduction of MutS and MutL decreases this rate to below wild-type levels; thus, in recG− cells, MMR proteins in excess prevent or inhibit nearly 100-fold more mutations than in wild-type cells.39 A similar result was obtained with recD− cells.64 Were excess MMR proteins simply correcting mismatches, then Lac+ mutations in wild-type cells should be eliminated entirely when the MMR proteins are overproduced. That recG− and recD− cells are more sensitive to excess MMR proteins than wild-type cells suggests that the MMR proteins can directly inhibit the process that produces the Lac+ mutations. Whether this inhibition occurs when normal levels of MMR proteins are present remains to be seen.
VSR Repair
The five position of the internal Cs of dCC(A/T)GG sites in E. coli are methylated by the Dcm methylase. Deamination of 5-methylcytosine produces thymine, making Dcm recognition sites hotspots for mutation. The frequency of these mutations is reduced by very short-patch repair (VSR) that corrects T:G mismatches to C:G at dCC(A/T)GG and similar sequences. VSR is initiated by the Vsr endonuclease, followed by degradation and resynthesis of the T-containing DNA strand.67
VSR is active in stationary-phase cells,68 and two different hypotheses have been published involving it in adaptive mutation in FC40. First, the repair synthesis could be the source of the polymerase errors that produce the Lac+ mutations.69 Second, efficient VSR requires MutS and MutL,70 and excess Vsr appears to deplete MMR proteins, particularly MutL, resulting in a general mutational state that could produce adaptive mutations.63 Neither of these hypotheses is true because FC40 has a normal level of adaptive mutation in the complete absence of Vsr.71
A Model for the Molecular Mechanism of Adaptive Lac+ Mutation in FC40
The results just summarized suggest that the initiating event for adaptive mutation in FC40 is nicking at the conjugal origin, oriT. It is possible that conjugal replication produces the mutations, but involvement of RecBCD implicates a duplex end, the loading point for this enzyme, and it is not obvious how a duplex end would be created during conjugal replication. In addition, the unusual effects of the branch migration enzymes suggest that the recombination required is special. Kuzminov49 proposed that the duplex end is created when a replication fork initiated at one of the episome's vegetative origins collapses at the nick at oriT.49 The exonuclease and helicase activities of RecBCD then create an invasive single-stranded 3′ end that initiates recombination. After both strands have invaded duplex DNA (of the same or of a different episome), the replication fork is restored and replication resumes. Replication errors produced at this point are in hemimethylated DNA and are corrected by MMR. But the new fork differs from a normal fork in that it is accompanied by a four-stranded recombination intermediate (a Holliday junction). Migration of the Holliday junction towards the fork creates a tract of doubly unmethylated DNA in which polymerase errors will be randomly repaired by MMR. Thus, this tract will contain mutations. Migration of the junction away from the fork, or resolution of the recombination intermediate before DNA synthesis begins, preserves the hemimethylated state of the DNA, allowing polymerase errors to be correctly repaired.39
The opposite effects of the branch migration enzymes in adaptive mutation to Lac+ are accommodated by assuming that RuvAB and RecG promote migration of the Holliday junction away from and towards the replication fork, respectively. Alternatively, RecG may resolve the Holliday junction before replication resumes.39 Both possibilities are consistent with biochemical evidence showing that RuvAB and RecG have different interactions with recombination intermediates.72,73
Several other models are possible,39,42 but this one is the most parsimonious. For example, a model proposed by Harris et al.42 requires the initiation of recombination by single-stranded 5′ ends, for which there is little experimental evidence.74 The Kuzminov model also accounts for the fact that whereas MMR is active in nutritionally deprived cells, the mutational spectrum bears the mark of MMR deficiency.
Nonselected Mutations
Lac+ mutations do not arise when FC40 cells are starved in the absence of lactose, nor do nonselected mutations in the chromosomal rpoB gene giving a rifampicin-resistant (RifR) phenotype appear in the Lac− population during lactose selection.22,24 Thus, mutation to Lac+ in FC40 appeared to be adaptive as originally defined. But because the Lac− allele is on an episome, the mutational process in nondividing cells may affect only the episome, giving the appearance that the mutations were adaptive. To rigorously test this hypothesis we created tetracycline-sensitive (TetS) Tn10 elements close to the lac operon on the episome. The two mutants studied were very similar to the Lac− allele, carrying +1-bp frameshifts in runs of G:C bp in tetA. Unlike the chromosomal rpoB gene, the mutant tetA alleles reverted to TetR when the cells were under lactose selection. TetR mutations accumulated at nearly the same rate and occurred by the same recombination-dependent mechanism as did the Lac+ mutations.15 Because both Lac+ and TetR mutations required that lactose be present, the role of lactose is apparently to provide enough energy for DNA replication and recombination even though the cells are not actively dividing. That TetR mutations appear and persist in the Lac− population eliminates the hypotheses that nonselected mutations are transitory or that the cells (or episomes) bearing them are eliminated from the population. Our previous failure to observe mutants in rpoB was likely due to the lower mutation rate on the chromosome and the relatively small number of mutational events that give a RifR phenotype.
Transient Mutation
In the experiments just described, Lac+ TetR double mutants arose from the Lac− TetS population at about a 50-fold higher frequency than expected.16 Subsequently, Torkelson et al.17 found that Lac+ revertants of FC40 carried additional mutations at other loci on the episome, a plasmid, and the chromosome. However, unlike the case of the episomal TetR mutations, there is no published evidence that these other nonselected mutations are produced by the same mechanism as the Lac+ mutations.
In two previous studies a higher than expected frequency of nonselected mutations was found among selected clones.13,14 This result is a specific prediction of the “hypermutable state model,”13 although it is also implicitly predicted by all the “trial and error” models for adaptive mutation.75 But the frequency of double mutants is most readily accounted for by assuming that a minority of the population has a high mutation rate. Because these mutators produce the selected-for mutants at a higher rate than the rest of the cells, the selected population will be enriched for the mutators. Thus, cells carrying the selected mutation will be far more likely to carry additional mutations than will cells without the selected mutation. Although selection enriches for heritable mutators,76 most of the double mutants that appear in FC40 during lactose selection have normal mutation rates after isolation.16,17 Thus, in the nutritionally deprived population the mutator state must usually be transient.
The contribution of transient mutators to a population's mutation rate has been modeled by Ninio77 and Cairns.9 From the experiments discussed above, R, the ratio of the frequency of a nonselected mutation in the selected population to that in the nonselected population, is estimated to be about 50; then M, the increase in mutation rate of the mutators, must be about 200, and p, the proportion of mutators in the population, must be less than 2%.9 Ninio estimated M to be 300 and p to be 0.05%.77 What is surprising is that when the mutators are less than 1% of the population, even with a high mutation rate, they contribute little to the selected population. Indeed, the mutators have to be more than 0.03% of the population (or have an unrealistically high mutation rate) before they account for more than 10% of the population that carries any single mutation.16 These modeling exercises show that adaptive mutation in FC40 is not explained by the transient mutation hypothesis, because most of the Lac+ mutations do not arise in the mutator population. As pointed out by Ninio,77 in any population, transient mutators would make only a minor contribution to the frequency of single mutations, but they could be responsible for nearly all simultaneous double or greater mutations.
Evolutionary Significance
The research on adaptive mutation in FC40 has revealed mutagenic mechanisms that may play significant roles in evolution. First, recombination is often, perhaps always, associated with DNA synthesis.78 The tracts of DNA that result appear to have a high error rate.79,80 Thus, recombination could be an important source of spontaneous mutation, particularly in cells that are not undergoing genomic replication. Recombination can thus be considered a force increasing variation not only by recombining existing alleles but also by creating new ones. Second, the recombination-dependent mutagenic mechanism is highly active on the F episome. Recently, conjugal plasmids were detected in 15% of natural isolates of E. coli, indicating that such plasmids are more common than previously thought.81 On an evolutionary time scale, F and related plasmids frequently recombine and are passed among the major groups of E. coli and Salmonella enterica.81,82 Because F can recombine with the bacterial chromosome, it can acquire chromosomal genes,48 which would then be exposed to a high mutation rate and be free to diverge from the chromosomal copies. These diverged alleles could spread through the population and even to other species. Thus, the mutational mechanism on the episome may be important in the evolution of species that carry and exchange conjugal plasmids. Third, if a subpopulation of nutritionally deprived cells enters into a state of transient mutation, it could be the source of multiple variant alleles and thus could provide a mechanism for rapid adaptive evolution under adverse conditions.
Acknowledgments
We thank John Cairns for continuing support.
Footnotes
This work was supported by Grant MCB 97838315 from the US National Science Foundation.
References
- 1.Luria SE, Delbrück M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newcombe HB. Origin of bacterial variants. Nature (Lond) 1949;164:150–151. doi: 10.1038/164150a0. [DOI] [PubMed] [Google Scholar]
- 3.Lederberg J, Lederberg E. Replica plating and indirect selection of bacterial mutants. J Bacteriol. 1952;63:399–406. doi: 10.1128/jb.63.3.399-406.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavalli-Sforza LL, Lederberg J. Isolation of preadaptive mutants by sib selection. Genetics. 1956;41:367–381. doi: 10.1093/genetics/41.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryan FJ. Spontaneous mutation in non-dividing bacteria. Genetics. 1955;40:726–738. doi: 10.1093/genetics/40.5.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shapiro JA. Observations on the formation of clones containing araB-lacZ cistron fusions. Molec Gen Genet. 1984;194:79–90. doi: 10.1007/BF00383501. [DOI] [PubMed] [Google Scholar]
- 7.Cairns J, Overbaugh J, et al. The origin of mutants. Nature (Lond) 1988;335:142–145. doi: 10.1038/335142a0. [DOI] [PubMed] [Google Scholar]
- 8.Foster PL. Adaptive mutation: The uses of adversity. Annu Rev Microbiol. 1993;47:467–504. doi: 10.1146/annurev.mi.47.100193.002343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cairns J. Mutation and cancer: The antecedents to our studies of adaptive mutation. Genetics. 1998;148:1433–1440. doi: 10.1093/genetics/148.4.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foster PL. Adaptive mutation: Has the unicorn landed? Genetics. 1998;148:1453–1459. doi: 10.1093/genetics/148.4.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster PL, Cairns J. Mechanisms of directed mutation. Genetics. 1992;131:783–789. doi: 10.1093/genetics/131.4.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stahl FW. A unicorn in the garden. Nature (Lond) 1988;335:112–113. doi: 10.1038/335112a0. [DOI] [PubMed] [Google Scholar]
- 13.Hall BG. Spontaneous point mutations that occur more often when they are advantageous than when they are neutral. Genetics. 1990;126:5–16. doi: 10.1093/genetics/126.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boe L. Mechanism for induction of adaptive mutations in Escherichia coli. Molec Microbiol. 1990;4:597–601. doi: 10.1111/j.1365-2958.1990.tb00628.x. [DOI] [PubMed] [Google Scholar]
- 15.Foster PL. Nonadaptive mutations occur on the F′ episome during adaptive mutation conditions in Escherichia coli. J Bacteriol. 1997;179:1550–1554. doi: 10.1128/jb.179.5.1550-1554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosche, W.A. & P.L. Foster. Unpublished data.
- 17.Torkelson J, Harris RS, et al. Genome-wide hypermutation in a subpopulation of stationary-phase cells underlies recombination-dependent adaptive mutation. EMBO J. 1997;16:3303–3311. doi: 10.1093/emboj/16.11.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Müller-Hill B, Kania J. Lac repressor can be fused to β-galactosidase. Nature (London) 1974;249:561–562. doi: 10.1038/249561a0. [DOI] [PubMed] [Google Scholar]
- 19.Brake AJ, Fowler AV, et al. β-galactosidase chimeras: primary structure of a lac repressor-β-galactosidase protein. Proc Natl Acad Sci USA. 1978;75:4824–4827. doi: 10.1073/pnas.75.10.4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller JH. Mutagenic specificity of ultraviolet light. J Molec Biol. 1985;182:45–65. doi: 10.1016/0022-2836(85)90026-9. [DOI] [PubMed] [Google Scholar]
- 21.Calos MP, Miller JH. Genetic and sequence analysis of frameshift mutations induced by ICR-191. J Molec Biol. 1981;153:39–66. doi: 10.1016/0022-2836(81)90525-8. [DOI] [PubMed] [Google Scholar]
- 22.Foster PL. Population dynamics of a Lac− strain of Escherichia coli during selection for lactose utilization. Genetics. 1994;138:253–261. doi: 10.1093/genetics/138.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foster, P.L. Unpublished data.
- 24.Cairns J, Foster PL. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics. 1991;128:695–701. doi: 10.1093/genetics/128.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galitski T, Roth JR. A search for a general phenomenon of adaptive mutability. Genetics. 1996;143:645–659. doi: 10.1093/genetics/143.2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foster PL, Trimarchi JM. Adaptive reversion of a frameshift mutation in Escherichia coli by simple base deletions in homopolymeric runs. Science. 1994;265:407–409. doi: 10.1126/science.8023164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenberg SM, Longerich S, et al. Adaptive mutation by deletions in small mononucleotide repeats. Science. 1994;265:405–407. doi: 10.1126/science.8023163. [DOI] [PubMed] [Google Scholar]
- 28.Foster PL, Gudmundsson G, et al. Proofreading-defective DNA polymerase II increases adaptive mutation in Escherichia coli. Proc Natl Acad Sci USA. 1995;92:7951–7955. doi: 10.1073/pnas.92.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Streisinger G, Okada Y, et al. Frameshift mutations and the genetic code. Cold Spring Harbor Symp Quant Biol. 1966;31:77–84. doi: 10.1101/sqb.1966.031.01.014. [DOI] [PubMed] [Google Scholar]
- 30.Kunkel TA, Bebenek K. Recent studies of the fidelity of DNA synthesis. Biochim Biophys Acta. 1988;951:1–15. doi: 10.1016/0167-4781(88)90020-6. [DOI] [PubMed] [Google Scholar]
- 31.Schaaper RM, Dunn RL. Spectra of spontaneous mutations in Escherichia coli strains defective in mismatch correction: The nature of in vivo DNA replication errors. Proc Natl Acad Sci USA. 1987;84:6220–6224. doi: 10.1073/pnas.84.17.6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cupples CG, Cabrera M, et al. A set of lacZ mutations in Escherichia coli that allow rapid detection of specific frameshift mutations. Genetics. 1990;125:275–280. doi: 10.1093/genetics/125.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Escarceller M, Hicks J, et al. Involvement of Escherichia coli DNA polymerase II in response to oxidative damage and adaptive mutation. J Bacteriol. 1994;176:6221–6228. doi: 10.1128/jb.176.20.6221-6228.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedberg EC, Walker GC, et al. DNA Repair and Mutagenesis. American Society for Microbiology; Washington, DC: 1995. [Google Scholar]
- 35.Foster PL, Rosche WA. Increased episomal replication accounts for the high rate of adaptive mutation in recD mutants of Escherichia coli. Genetics. 1999;152 doi: 10.1093/genetics/152.1.15. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harris RS, Longerich S, et al. Recombination in adaptive mutation. Science. 1994;264:258–260. doi: 10.1126/science.8146657. [DOI] [PubMed] [Google Scholar]
- 37.Kowalczykowski SD, Dixon DA, et al. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chaudhury AM, Smith GR. Role of Escherichia coli RecBC enzyme in SOS induction. Molec Gen Genet. 1985;201:525–528. doi: 10.1007/BF00331350. [DOI] [PubMed] [Google Scholar]
- 39.Foster PL, Trimarchi JM, et al. Two enzymes, both of which process recombination intermediates, have opposite effects on adaptive mutation in Escherichia coli. Genetics. 1996;142:25–37. doi: 10.1093/genetics/142.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harmon FG, Rehrauer WM, et al. Interaction of Escherichia coli RecA protein with LexA repressor II. inhibition of DNA strand exchange by the uncleavable lexA S119A repressor argues that recombination and SOS induction are competitive processes. J Biol Chem. 1996;271:23874–23883. [PubMed] [Google Scholar]
- 41.West SC. The RuvABC proteins and Holliday junction processing in Escherichia coli. J Bacteriol. 1996;178:1237–1241. doi: 10.1128/jb.178.5.1237-1241.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harris RS, Ross KJ, et al. Opposing roles of the Holliday junction processing systems of Escherichia coli in recombination-dependent adaptive mutation. Genetics. 1996;142:681–691. doi: 10.1093/genetics/142.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radicella JP, Park PU, et al. Adaptive mutation in Escherichia coli: A role for conjugation. Science. 1995;268:418–420. doi: 10.1126/science.7716545. [DOI] [PubMed] [Google Scholar]
- 44.Foster RL, Trimarchi JM. Adaptive reversion of an episomal frameshift mutation in Escherichia coli requires conjugal functions but not actual conjugation. Proc Natl Acad Sci USA. 1995;92:5487–5490. doi: 10.1073/pnas.92.12.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galitski T, Roth JR. Evidence that F plasmid transfer replication underlies apparent adaptive mutation. Science. 1995;268:421–423. doi: 10.1126/science.7716546. [DOI] [PubMed] [Google Scholar]
- 46.Foster PL. Are adaptive mutations due to a decline in mismatch repair? The evidence is lacking. Rev Mutation Res. 1999;436 doi: 10.1016/s1383-5742(98)00023-4. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foster PL, Trimarchi JM. Conjugation is not required for adaptive reversion of an episomal frameshift mutation in Escherichia coli. J Bacteriol. 1995;177:6670–6671. doi: 10.1128/jb.177.22.6670-6671.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holloway B, Low KB. F-prime and R-prime factors. In: Neidhardt FC, Curtiss R III, et al., editors. Escherichia coli and Salmonella: Cellular and Molecular Biology. American Society of Microbiology; Washington, DC: 1996. pp. 2413–2420. [Google Scholar]
- 49.Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Molec Microbiol. 1995;16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- 50.Tlsty DT, Albertini AM, et al. Gene amplification in the lac region of E. coli. Cell. 1984;37:217–224. doi: 10.1016/0092-8674(84)90317-9. [DOI] [PubMed] [Google Scholar]
- 51.Sonti RV, Roth JR. Role of gene duplications in the adaptation of Salmonella typhimurium to growth on limiting carbon sources. Genetics. 1989;123:19–28. doi: 10.1093/genetics/123.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roth JR, Benson N, et al. Rearrangements of the bacterial chromosome: Formation and applications. In: Niedhardt FC, Curtiss R, et al., editors. Escherichia coli and Salmonella: Cellular and Molecular Biology. American Society of Microbiology; Washington, D.C.: 1996. pp. 2256–2276. [Google Scholar]
- 53.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 54.Jayaraman R. Cairnsian mutagenesis in Escherichia coli: Genetic evidence for two pathways regulated by mutS and mutL genes. J Genet. 1992;71:23–41. [Google Scholar]
- 55.Reddy M, Gowrishankar J. A genetic strategy to demonstrate the occurrence of spontaneous mutations in non-dividing cells within colonies of Escherichia coli. Genetics. 1997;147:991–1001. doi: 10.1093/genetics/147.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feng G, Tsui HCT, et al. Depletion of the cellular amounts of the MutS and MutH methyl-directed mismatch repair proteins in stationary-phase Escherichia coli K-12 cells. J Bacteriol. 1996;178:2388–2396. doi: 10.1128/jb.178.8.2388-2396.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Damaonez v, Doutriaux MP, et al. Saturation of mismatch repair in the mutD5 mutator strain of Escherichia coli. J Bacteriol. 1989;171:4494–4497. doi: 10.1128/jb.171.8.4494-4497.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schaaper RM. Escherichia coli mutator mutD5 is defective in the mutHLS pathway of DNA mismatch repair. Genetics. 1989;121:205–212. doi: 10.1093/genetics/121.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maas WK, Wang C, et al. Multicopy single-stranded DNA of Escherichia coli enhances mutation and recombination frequencies by titrating MutS protein. Molec Microbiol. 1996;19:505–509. doi: 10.1046/j.1365-2958.1996.392921.x. [DOI] [PubMed] [Google Scholar]
- 60.Wu TH, Marinus MG. Personal communication. [Google Scholar]
- 61.Tsui HCT, Feng G, et al. Negative regulation of mutS and mutH repair gene expression by the Hfq and RpoS global regulators of Escherichia coli K-12. J Bacteriol. 1997;179:7476–7487. doi: 10.1128/jb.179.23.7476-7487.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schaaper RM, Radman M. The extreme mutator effect of Escherichia coli mutD5 results from saturation of mismatch repair by excessive DNA replication errors. EMBO J. 1989;8:3511–3516. doi: 10.1002/j.1460-2075.1989.tb08516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Macintyre G, Doiron KM, et al. The Vsr endonuclease of Escherichia coli: An efficient DNA repair enzyme and a potent mutagen. J Bacteriol. 1997;179:6048–6052. doi: 10.1128/jb.179.19.6048-6052.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harris RS, Feng G, et al. Mismatch repair protein MutL becomes limiting during stationary-phase mutation. Genes Dev. 1997;11:2426–2437. doi: 10.1101/gad.11.18.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Worth L, Clark S, et al. Mismatch repair proteins MutS and MutL inhibit RecA-cata-lyzed strand transfer between diverged DNAs. Proc Natl Acad Sci USA. 1994;91:3238–3241. doi: 10.1073/pnas.91.8.3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zahrt TC, Maloy SR. Barriers to recombination between closely related bacteria: MutS and RecBCD inhibit recombination between Salmonella typhimurium and Salmonella typhi. Proc Natl Acad Sci USA. 1997;94:9786–9791. doi: 10.1073/pnas.94.18.9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lieb M, Bhagwat SA. Very short patch repair: Reducing the cost of cytosine methylation. Mol Microbiol. 1996;20:467–473. doi: 10.1046/j.1365-2958.1996.5291066.x. [DOI] [PubMed] [Google Scholar]
- 68.Lieb M, Rehmat S. 5-Methylcytosine is not a mutation hot spot in nondividing Escherichia coli. Proc Natl Acad Sci USA. 1997;94:940–945. doi: 10.1073/pnas.94.3.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bhattacharjee SK, Mahajan SK. The origin of adaptive mutations: Explaining the mutational spectra of Lac+ revertants of the Escherichia coli strain FC40. Curr Sci. 1998;74:583–589. [Google Scholar]
- 70.Lieb M. Bacterial genes mutL, mutS, and dcm participate in repair of mismatches at 5-methylcytosine sites. J Bacteriol. 1987;169:5241–5246. doi: 10.1128/jb.169.11.5241-5246.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Foster PL, Rosche WA. Levels of the Vsr endonuclease do not regulate stationary-phase reversion of a Lac− frameshift allele in Escherichia coli. J Bacteriol. 1998;180:1944–1946. doi: 10.1128/jb.180.7.1944-1946.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Whitby MC, Lloyd RG. Branch migration of three-strand recombination intermediates by RecG, a possible pathway for securing exchanges initiated by 3′-tailed duplex DNA. EMBO J. 1995;14:3302–3310. doi: 10.1002/j.1460-2075.1995.tb07337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Whitby MC, Ryder L, et al. Reverse branch migration of Holliday junctions by RecG protein: A new mechanism for resolution of intermediates in recombination and DNA repair. Cell. 1993;75:341–350. doi: 10.1016/0092-8674(93)80075-p. [DOI] [PubMed] [Google Scholar]
- 74.Friedman-Ohana R, Cohen A. Heteroduplex joint formation in Escherichia coli recombination is initiated by pairing of a 3′-ending strand. Proc Natl Acad Sci USA. 1998;95:6909–6914. doi: 10.1073/pnas.95.12.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Foster PL. Directed mutation: Between unicorns and goats. J Bacteriol. 1992;174:1711–1716. doi: 10.1128/jb.174.6.1711-1716.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mao EF, Lane L, et al. Proliferation of mutators in a cell population. J Bacteriol. 1997;179:417–422. doi: 10.1128/jb.179.2.417-422.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ninio J. Transient mutators: A semiquantitative analysis of the influence of translation and transcription errors on mutation rates. Genetics. 1991;129:957–962. doi: 10.1093/genetics/129.3.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kogoma T. Stable DNA replication: Interplay between DNA replication, homologous recombination, and transcription. Microbiol Molec Biol Rev. 1997;61:212–238. doi: 10.1128/mmbr.61.2.212-238.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Demerec M. Selfer mutants of Salmonella typhimurium. Genetics. 1963;48:1519–1531. doi: 10.1093/genetics/48.11.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Strathern JN, Shafer BK, et al. DNA synthesis errors associated with double-strand-break repair. Genetics. 1995;140:965–972. doi: 10.1093/genetics/140.3.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boyd EF, Hill CS, et al. Mosaic structure of plasmids from natural populations of Escherichia coli. Genetics. 1996;143:1091–1100. doi: 10.1093/genetics/143.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boyd EF, Hartl DL. Recent horizontal transmission of plasmids between natural populations of Escherichia coli and Salmonella enterica. J Bacteriol. 1997;179:1622–1627. doi: 10.1128/jb.179.5.1622-1627.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]