

Abstract
Stem cell differentiation and the maintenance of self-renewal are intrinsically complex processes requiring the coordinated dynamic expression of hundreds of genes and proteins in precise response to external signalling cues. Numerous recent reports have used both experimental and computational techniques to dissect this complexity. These reports suggest that the control of cell fate has both deterministic and stochastic elements: complex underlying regulatory networks define stable molecular ‘attractor’ states towards which individual cells are drawn over time, whereas stochastic fluctuations in gene and protein expression levels drive transitions between coexisting attractors, ensuring robustness at the population level.
Stem cells have a crucial role during development, tissue regeneration and healthy homeostatic cell turnover. Collectively, all stem cells share the ability to self-renew and differentiate into various different lineages. Embryonic stem (ES) cells — which are derived from the inner cell mass of the developing blastocyst — are pluripotent, whereas stem cells derived from adult tissues are generally only multipotent, maintaining a limited, tissue- specific, regenerative potential1–3. In culture, ES cells can be propagated indefinitely, whereas adult tissue stem cells are more limited in this capacity.
Owing to their ability to generate tissue de novo following disease or injury there is widespread hope of developing stem cell-based therapies for various degenerative diseases4. Recent reports have indicated that differentiated adult cells can easily be reprogrammed to an embryonic-like pluripotent state5,6 (thus potentially providing a patient-specific source of pluripotent stem cells), as well as be reprogrammed to other adult cell types without intermediate reversion to a pluripotent state7,8. These findings have served to intensify interest in understanding the molecular basis of cell fate regulation and the potential therapeutic uses of stem cells9. However, before such stem cell-based therapies can be routinely and safely developed, numerous crucial issues must be addressed. In particular, although great progress has been made towards understanding the roles of the homeodomain transcription factors OCT4 (also known as POU5F1) and NANOG, as well as SRY box-containing factor 2 (SOX2) in the maintenance of stem cell pluripotency10–16 (BOX 1), the extended molecular mechanisms of ES cell fate control have yet to be fully determined.
Box 1. The core embryonic stem cell transcriptional circuit.
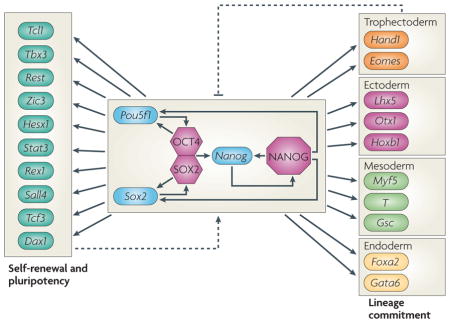
Maintenance of pluripotency and self-renewal in embryonic stem (ES) cells is controlled by a complex interplay between signalling from the extracellular environment and the dynamics of core transcription factors. Although self-renewal signalling pathways differ between mice and humans123, the core transcriptional circuitry seems to be remarkably conserved. In particular, the homeodomain transcription factors OCT4 (also known as POU5F1) and NANOG, as well as SRY box-containing factor 2 (SOX2), form a transcriptional module that has a central role in maintaining ES cell identity both in mice and humans10,11,13–16 (see the figure). This module is rich in positive feedback and feedforward loops (see also BOX 2). In particular, OCT4 and SOX2 form a heterodimer that positively regulates the expression of the Pou5f1 (which encodes OCT4), Sox2 and Nanog10,11,124,125. In addition, NANOG also interacts directly with OCT4 (not shown)17 and positively regulates the expression of all three genes11. Thus, these three transcription factors regulate their own and each other’s expression in a highly coordinated manner, involving positive protein–protein and protein–DNA feedback loop interactions. Furthermore, all three transcription factors co-occupy numerous developmentally important genes and repress the expression of the genes involved in lineage commitment. These include: Hand1 (heart and neural crest derivatives-expressed 1), eomesodermin (Eomes) (both involved in trophectoderm development); Lhx5 (LIM homeobox 5), Otx1 (orthodenticle homologue 1), Hoxb1 (all involved in ectoderm development); Myf5 (myogenic factor 5), T (brachyury protein homologue), Gsc (goosecoid) (all involved in mesoderm development); and Foxa2 (forkhead box A2) and Gata6 (GATA-binding protein 6) (both involved in endoderm development). At the same time, OCT4, NANOG and SOX2 activate genes that are associated with self-renewal and pluripotency, including other ES cell-associated transcription factors such as Tcl (T cell leukaemia/lymphoma), Tbx3, Rest, Zic3, Hesx1 (homeobox expressed in ES cells 1), Stat3 (signal transducer and activator of transcription 3), Rex1 (also known as Zpf42), Sall4, Tcf3 and Dax1 (also known as Nr0b1)11. Thus, OCT4, SOX2 and NANOG are central to the maintenance of ES cell identity; appropriate expression of this protein trio holds the cell in a pluripotent self-renewing state by activating other ES cell-specific genes and repressing genes that are associated with lineage commitment, and loss of expression leads to loss of the self-renewing ES phenotype and commitment to differentiation. Dotted arrows denote potential feedback mechanisms from downstream targets back to the core circuit.
To begin to deconstruct these intrinsically complex regulatory mechanisms it is now common for stem cell studies to combine low-throughput experimental techniques with an ever-increasing range of different high-throughput experimental techniques. Consequently, stem cell studies now often produce large amounts of data, and integrating these data into a coherent quantitative picture of cell fate control at the systems level is an important current research challenge. To address this challenge several groups have begun to apply systems biology approaches to understanding the regulation of stem cell fate decisions11,12,17,18.
Instead of focusing on the role of individual genes, proteins or pathways in biological phenomena, the aim of systems biology is to characterize the ways in which essential molecular parts interact with each other to determine the collective dynamics of the system as a whole19–23. However, it is difficult to understand collective behaviour in complex systems using experimental approaches alone. Therefore systems biology approaches often employ high-throughput experimental techniques alongside theoretical and computational methods, which are specifically designed to dissect collective phenomena in complex systems24–26. Although to date systems biology approaches are mostly successful in lower organisms, such as yeast27,28 and bacteria29,30, the complexity of mammalian stem cell biology as well as the experimental reproducibility of many stem cell systems makes mammalian stem cell biology a good platform for the development of future systems biology techniques. In the context of stem cell biology, the aim of systems biology approaches is to characterize the molecular components involved in stem cell self-renewal and differentiation along specific lineages — from core transcription factors and the genes they regulate to proteins and protein complexes to microRNAs (miRNAs) and other epigenetic marks — and elucidate their functional interactions. The ultimate goal is to understand the dynamic behaviour of the resulting molecular circuits and elucidate how these circuits control cell fate changes. In the context of cellular reprogramming, systems biology approaches aim to use advances in the understanding of the molecular basis of normal cell fate decisions during development to generate strategies for the experimental conversion of adult cells from one type to another.
In this Review we discuss a range of ways in which high-throughput experimental techniques and computational methods are being fruitfully combined towards the development of stem cell systems biology approaches. We begin by outlining how data from high-throughput experiments can be used to reconstruct accurate stem cell regulatory networks. However, as stem cell regulatory circuits are typically intricate and contain highly nested feedback loops and feedforward loops that give rise to complex dynamics, it is difficult to elucidate cell behaviour from this regulatory circuitry. Therefore, we also discuss how computational techniques can be used to relate dynamic cell behaviour to regulatory architecture. In particular, we focus on how cell types can be thought of as balanced states or ‘attractors’ of underlying regulatory networks and the ways in which stochastic and deterministic mechanisms interact to define cell fate. We conclude with some suggestions of directions for future work in this area, including ways in which these notions might be used to better understand cellular reprogramming.
Dissecting stem cell complexity
Molecular biology has entered the high-throughput age. Consequently, it is now typical for stem cell studies to make use of various disparate high-throughput techniques to determine the molecular mechanisms of cell fate specification. These techniques include: micro-arrays to assess genome-wide mRNA expression; high-throughput chromatin immunoprecipitation (ChIP) such as ChIP-on-chip31, ChIP-seq (ChIP-sequencing)32 and ChIP-PET (ChIP-paired-end-ditag)10 to assess protein–DNA interactions; and mass spectrometry proteomics33 and phosphoproteomics34 to assess the protein composition of molecular complexes and global changes in post-translational modifications. Because high-throughput techniques measure system-wide expression patterns, rather than focusing on the behaviour of key molecular elements, their development has driven increasing interest in systems biology approaches to understanding cell behaviour22. An important challenge in this area is how to best integrate the wealth of data that high-throughput studies produce into both a coherent qualitative and quantitative understanding of cell behaviour at the systems level. One approach to dissecting this complexity is to represent the underlying stem cell molecular regulatory mechanisms as ‘networks’.
Building molecular regulatory networks
To make sense of complex biological datasets it is becoming common to represent molecular components and their interactions as networks and apply techniques from the mathematical theory of graphs35 to their analysis (BOX 2). The combination of high-throughput experiments and the representation of high-dimensional data in the form of networks is the basis of much of modern systems biology. This integrated experimental–theoretical approach has greatly enhanced our understanding of a wide range of complex mammalian biochemical systems36, including signalling networks37,38, protein interaction networks39,40 and genetic regulatory networks41. Representing complex biological systems as networks is useful as it provides a formal way to combine different types of biological datasets into a single conceptual framework42.
Box 2. Regulatory networks and graphs.
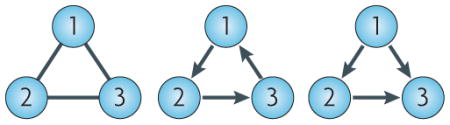
Mathematically, a network is a data structure known as a graph35, consisting of a set of nodes and a set of edges or arcs (which are directed edges) that connect the nodes in the graph. In the context of biological regulatory networks, theNatur nodeReviews set represents the list of molecular components in the network (for example, genes and proteins) and the edge set describes functional relationships between the nodes. So, in a protein–protein interaction network, nodes represent proteins and edges represent physical interactions between proteins, whereas in a transcriptional network nodes represent transcription factors and arcs represent functional regulation of transcription. The figure shows examples of simple directed and undirected graphs. In each case, there are three nodes, labelled 1, 2 and 3, and edges and arcs are coloured black. On the left is an undirected graph on the mutually connected nodes. In this case, the edges have no specified direction and so are drawn without arrowheads. In the middle is a three-node feedback loop. In this case the nodes regulate each other in a directed cyclic manner. On the right is a three-node feedforward loop. In this case, node 1 regulates nodes 2 and 3, and node 2 also regulates node 3. Feedback and feedforward loops such as these are common in transcriptional regulatory networks and can give rise to complex dynamic behaviour. For example, the presence of a positive feedback loop is a necessary condition for the existence of multiple stable stationary states74 (see BOX 3).
Two key elements are required to construct a biological regulatory network: a list of molecular parts (such as sets of genes, proteins or miRNAs) and a set of regulatory interactions between these parts (for example, activation or inhibition of expression). The molecular parts lists that are needed to construct a regulatory network typically come from data derived from high-throughput experiments (for example, sets of genes that are differentially expressed in treated or control conditions). Physical interactions between elements in the molecular parts list can be identified by techniques such as yeast two-hybrid screens or affinity purification followed by mass spectrometry (AP–MS); however, the regulatory nature of such interactions cannot be determined by these methods alone. Extracting putative functional connections between elements in the parts list often requires some computational input, either by reverse engineering regulatory networks from specific experimentally derived datasets using sophisticated computational inference techniques41,43,44 or by comparing experimentally derived expression patterns with databases of interactions collated from the published literature45,46.
Stem cell regulatory networks
Recently, several examples of how high-throughput experimental techniques can be used to infer regulatory networks have been documented in the published literature on stem cells. For example, Wang and co-workers17 derived a high-quality protein–protein interaction network for pluripotency in mouse ES cells that is centred around the core stem cell transcription factor NANOG (FIG. 1a). To construct this network they adopted an iterative proteomics approach in which proteins that physically associate with NANOG and NANOG-associated proteins were identified using AP–MS. By doing so, they identified a complex network that is highly enriched in stem cell-specific transcription factors, many of which transcriptionally regulate the expression of other members of the protein–protein interaction network. This indicates that stem cell fate control is highly combinatorial and involves coordinated interactions between key transcription factors and the genes that encode them.
Figure 1. Stem cell regulatory networks.

a | Schematic showing high-confidence protein–protein interactions between NANOG and NANOG-associated proteins, as derived from rEFS 17,127,128. b | Schematic showing the stem cell transcriptional regulatory circuit. This network was reconstructed from the data presented in various recent high-throughput chromatin immunoprecipitation (ChIP) experiments10–12,31,47,49–51,55–58. Both networks are rich in regulatory loops (see also BOX 2), suggesting a complex system with the ability to exhibit a wide range of context-dependent dynamic behaviours. Factors present in both the NANOG interactome and the core transcriptional network are shown in red. Note that there is great overlap between these two networks (with shared factors being the most central elements of both networks), suggesting that the core transcription factors regulate each other’s expression in a coordinated, combinatorial manner, involving both protein–protein and protein–DNA interactions. See Integrated Stem Cell Molecular Interactions database for an interactive version of this network.
Similarly, numerous groups have used high-throughput ChIP techniques to identify targets of core ES cell transcription factors, including NANOG14,16, OCT4 (REF. 15) and SOX2 (REF. 13), and thereby reconstruct core ES cell-specific transcriptional circuits that are centred around these (and other) factors10–12,47–50 (FIG. 1b). Furthermore, recent reports have also connected miRNAs51 and key stem cell signalling pathways32 to the core ES cell transcriptional circuit. These reports are useful because, by finding functional associates of known core factors, they produce a detailed dissection of the stem cell molecular regulatory core and thus provide the basis of a systematic understanding of the control of stem cell fate. However, by focusing on interactions involving a small number of central transcription factors, these studies are also limited in their ability to elucidate the extended molecular regulatory networks that underpin cell fate decisions. Nevertheless, although techniques such as AP–MS and high-throughput ChIP inevitably identify numerous false positive interactions, as is observed by the often poor overlap of results from comparative studies52,53, the datasets of inferred interactions produced by these techniques can be useful for the generation of hypotheses if used with caution.
In contrast to these focused experimental studies, a recent report by Müller and co-workers18 used a new computational approach to reconstruct an extended stem cell regulatory network. First, they generated a database of global gene expression patterns in approximately 150 samples of pluripotent, multipotent and differentiated human cell types, and named this database the ‘stem cell matrix’. Using a computational clustering technique, they found that undifferentiated pluripotent stem cells samples, including ES cells and induced pluripotent stem (iPS) cells, strongly clustered together on the basis of gene expression. Then, they used a graph theoretic algorithm known as MATISSE (module analysis via topology of interactions and similarity sets)54 and identified a putative pluripotency network, which they named PluriNet. This was achieved by searching for connected sub-networks involving pluripotency-related factors from a previously compiled background network of human protein–protein and protein–DNA interactions, including those in the NANOG interactome described by Wang and co-workers17. PluriNet is an undirected graph (that is, regulatory directions and effects such as activation or inhibition are not specified), and many interactions have yet to be directly experimentally characterized in any specific cell type. Despite this, the approach of Müller and co-workers18 is useful because it provides a formal way to ‘project’ experimentally derived datasets onto previously compiled databases and interpret new findings in the context of known biological processes45,46. Efforts such as PluriNet are inevitably works-in-progress; as our understanding of the molecular mechanisms of cell fate control becomes increasingly detailed, it will become important that prior knowledge is appropriately used, tested for reliability and organized in a coherent, structured and user-friendly manner so that new results can be assessed appropriately in light of previous data.
With this in mind, we have constructed a database of directed transcriptional interactions in ES cells as a supplement to this Review (see Integrated Stem Cell Molecular Interactions database). This repository currently integrates the data presented in 12 recent publications10–12,31,47,49–51,55–58, which collectively report high-throughput ChIP profiling experiments for 20 transcription factors that are known to have a central role in ES cell fate regulation. In total, the repository currently contains 50,250 putative transcription factor–gene interactions that have been identified specifically in ES cells. We connected the 20 core transcription factors to their gene targets and formed a directed background network (BOX 2). This network is highly dense and rich in feedback and feedforward loops; this indicates that many of the 20 core transcription factors share target genes, which suggests combinatorial regulation of gene expression. To generate a more focused network we collated a shortlist of 264 genes that are known to have an important role in the maintenance of ES cell self-renewal, pluripotency, cell cycle progression and differentiation along all three germ layers (mesoderm, endoderm and ectoderm). By searching for shortest paths between nodes in the background network we obtained two subnetworks: a network containing 156 mutual interactions between the 20 core transcription factors (FIG. 1b) and a network containing 1,739 links connecting the 264 shortlisted genes. Although directed, most of the interactions in these subnetworks are not signed (that is, regulatory effects such as activation or inhibition are not provided). However, regulatory effects can be inferred from studies that combine ChIP experiments with mRNA expression profiling that is obtained following loss-of-function experiments.
We have provided this initial database of interactions and the background and focused sub-networks as a web-based resource to accompany this Review (see Integrated Stem Cell Molecular Interactions database). On this site users can navigate from node to node and examine how target genes are co-regulated by core transcription factors. We invite the stem cell community to deposit additional interactions in this repository as they are reported and thereby continually improve this resource.
The work of Wang and co-workers17, our initial transcriptional interaction repository and the work of Müller and co-workers18 illustrate two different (and complementary) approaches to determining stem cell regulatory architecture. In the first approach precise experimentation is used to elucidate high-confidence functional interactions among a limited number of key components. In the second approach extended networks are generated by inferring interactions between numerous components using computational methods without direct experimental validation. It will be important to elucidate the nature of extended regulatory networks while maintaining high confidence in the inferred interactions. Perhaps the most promising way to address this issue is the combination of meticulous experimentation with computational inference, in which computational techniques are used to infer initial interaction networks and experimental techniques are used to validate inferred interactions and refine network structure59. In this regard, integration of large-scale RNA interference screens58,60,61, in which thousands of genes may be systematically and accurately individually silenced in a cell population, with high-throughput ChIP experiments and subsequent genome-wide expression profiling will be particularly useful in validating inferred regulatory interactions and their effects.
In summary, evidence suggests that ES cell fate is controlled by a core transcriptional circuit enriched in feedback and feedforward loops that itself is part of a much more extensive and highly complex dynamic regulatory network involving protein–protein interactions17, additional transcription factors12, signalling pathways32, miRNAs51 and other epigenetic modifiers56. This complexity is central to the cell’s ability to respond in a flexible way to disparate exogenous stimuli; however, it also makes it extremely difficult to determine cell behaviour from the regulatory network structure. Therefore, in the following section we discuss ways in which mathematical models can be used to make sense of this complexity and link molecular regulatory architecture to cell behaviour. In particular, we focus on the ways in which notions from dynamical systems theory can be used to interpret cell types as balanced states or attractors of underlying regulatory networks, and how molecular noise has a role in defining cell fate by triggering stochastic transitions between coexisting attracting states.
The stem cell landscape
Consider the core stem cell transcriptional network given in FIG. 1b. Although this network is small, it is highly complex, containing many feedback and feedforward loops, and this complexity makes it extremely difficult to determine how this network behaves (that is, how it controls stem cell fate). To begin to elucidate how the architecture of this network relates to stem cell fate, we first note that this network is not static but instead encodes the essential topology of a complex dynamical system (BOX 3) in which transcriptional activation and inhibition may loosely be thought of as ‘forces’ that push and pull the cell in different genetic directions. Thus, the state of a cell is determined by its transcriptional (or, more generally, its molecular) expression profile, which in turn depends dynamically on the regulatory interactions that are encoded in its underlying molecular regulatory architecture.
Box 3. Dynamical systems, attractors and multi-stability.
The molecular state of a cell can be described by its state vector s(t) = [m1(t),m2(t),m3(t),…,mn(t)], in which mn(t) denotes the concentration of the ith molecular component at time t.The set of all possible molecular configurations is called the ‘state space’. A dynamical system is a mathematical description of how a system’s state vector changes over time based on the interactions between all the various components in the system (in the form of a set of coupled differential or difference equations, for example). Owing to the coupling between molecular components, the expression levels of the different components in a dynamical system generally change over time in a coordinated way, and this coordination restricts the trajectories that the system may take in state space over time. An attractor of a dynamical system is a minimal subset of state space A, such that all trajectories starting in the vicinity of A approach A eventually. Intuitively, attractors can be thought of as stable preferred states in which all the various interactions in the system are balanced and towards which the system is drawn over time. Attractors can be fixed points, corresponding to static stationary states, or more complex sets, corresponding to dynamic states such as limit cycles (oscillators) or strange (chaotic) attractors79. For a given attractor A, the subset of state space NA for which all trajectories starting in NA approach A for large time is known as the ‘basin of attraction’ of A. Some dynamical systems have many coexisting attractors, in which case the system is said to be ‘multi-stable’. The basins of attraction of the various attractors in a multi-stable system partition the state space into discrete pieces. As stationary attractors can intuitively be associated with the minima of an ‘energy-like’ function79, in the context of cellular differentiation this partitioning is sometimes referred to as the attractor landscape69 (see also FIG. 2).
Mathematical models can help to better understand the molecular basis of cell behaviour and can be approached at various different levels62–64. For example, coarse-grained models, such as Boolean networks, that assume that genes adopt a binary ‘ON’ or ‘OFF’ state and regulate each others’ expression through simple Boolean functions are useful in determining the collective behaviour of large complex regulatory networks65. By contrast, differential equation models, which focus in detail on smaller regulatory circuits in which additional information (such as mRNA and protein production and degradation rates) are known, are useful when examining the fine details of regulatory dynamics36. The most successful examples of integration of mathematical modelling with experimental approaches are from model organisms, such as yeast and bacteria64, which have generated a wealth of data. However, advances in experimental techniques are now increasingly facilitating the development of mathematical models of mammalian cell fate control by providing the required data. This has lead to an increasing interest in the application of techniques that have been developed in model organisms to mammalian cell biology66–72.
The central notions of this joint theoretical–experimental approach to cell fate go back to the 1940s, to the work of the physicist Max Delbrück73,74 and the developmental biologist Conrad Waddington75,76. Over 50 years ago Waddington presented his now famous ‘epigenetic landscape’ as a conceptual picture of development75,76. Waddington’s view was that development occurs similarly to a ball rolling down a sloping landscape containing multiple ‘hills’ and ‘valleys’: as development progresses, cells take different paths down this landscape and so adopt different fates, and uncontrolled differentiation does not occur because the hills act as barriers by separating the landscape into distinct valleys (cell types). So, in this view, differentiation is not terminal, but instead different cell states are maintained by epigenetic barriers that can be overcome given sufficient perturbation. In Waddington’s words “This ‘landscape’ presents, in the form of a visual model, a description of the general properties of a complicated developing system in which the course of events is controlled by many different processes that interact in such a way that they tend to balance each other.” (REF. 77). Although Waddington viewed this epigenetic landscape as a qualitative conceptualization of development, the idea that cell types may be related to ‘balanced states’ of an underlying regulatory system bears a striking resemblance to the modern mathematical notion of attractors of dynamical systems78,79.
Attractor landscapes
Although formal mathematical definitions are complicated78, broadly speaking an attractor can be thought of as a balanced state (or set of states) towards which a system will converge given sufficient time79. Consider, for example, a marble at rest at the bottom of a bowl. If perturbed away from the bottom of the bowl, the marble will track out a transient trajectory around the sides of the bowl, only to finally come to rest at the bottom again: the default resting position of the marble is at the bottom of the bowl, and this is the state towards which the marble is attracted to regardless of where it starts in the bowl. Consider now the more interesting case of a marble rolling around a smooth convoluted surface with many hills (local maxima) and valleys (local minima), as illustrated in FIG. 2. Now the marble will come to rest at the bottom of one of many possible valleys, with its final resting place depending on its starting position and the nature of the particular perturbation that displaced it from its initial state. In this case, each local minimum is an attractor of the marble’s dynamics, and we might refer to the surface as a whole as the attractor landscape.
Figure 2. Cellular reprogramming as navigation through a complex attractor landscape.

In a complex cellular attractor landscape there might be many coexisting stationary attractors (here represented as local minima), each of which might be associated with a unique molecular signature. In this view, cellular reprogramming corresponds to guiding the cell through the landscape from one local minimum to another (shown by the dotted arrows). As there might be many distinct paths between minima (both direct and through intermediary minima), reprogramming from one cell type to another might be achieved though numerous different routes5,108,120.
More formal descriptions of dynamical systems, attractors and attractor landscapes are given in BOX 3; however, this intuitive picture of attractors as local minima of a complex ‘energy-like’ landscape is conceptually informative, and the parallels with Waddington’s epigenetic landscape are clear. In the context of cellular differentiation, an attractor is an internal molecular state (or set of states) towards which the cell is drawn, in which all the molecular forces that are pushing and pulling the cell in different molecular directions are balanced. Thus, attractors correspond to stable molecular configurations and have accordingly been associated with different cell types65,80. A system that exhibits many coexisting attractor states is said to be a multi-stable system; the notion that different cell types may correspond to different stable states of an underlying multi-stable regulatory system was first suggested by Delbrück73,74. The notion that cell types might correspond more generally to attractors of ‘high-dimensional’ regulatory networks was first proposed by Kauffman65,80 and has been examined extensively in the theoretical published literature since the late 1960s36,65,80–83.
Despite this longstanding theoretical interest, direct experimental evidence that different cell types might correspond to attractors of multi-stable genetic regulatory networks has been provided only recently66,68,84–89. For example, in 2005 Huang and co-workers85 provided the first evidence that mammalian cell types might correspond to attractors of a high-dimensional dynamical system. To do so, they took advantage of the fact that human HL60 promyelocytic progenitor cells can be triggered to differentiate into neutrophils in vitro if they are stimulated with all-trans retinoic acid (ATRA) or dimethylsulphoxide (DMSO). By taking samples for microarray analysis at different time points during the differentiation process, they showed that ATRA and DMSO initially triggered different genetic responses, which, however, ultimately converged over time to a common stable pattern of gene expression. This ‘homing in’ is characteristic of an attracting state and suggests that the HL60 neutrophil state is an attractor of an underlying molecular regulatory network. Similarly, others have shown that if sub-optimal ATRA stimulation is removed before commitment is complete, HL60 cells do not differentiate into neutrophils but instead revert back to the promyelocytic state66. Stability in the face of weak perturbations is another hallmark of an attracting state, so this work indicates that the HL60 promyelocytic state is also an attractor of an underlying molecular regulatory network.
In the context of mammalian ES cell biology, although direct evidence for attracting states has not yet been provided, indirect evidence for a self-sustaining self-renewing state in mouse ES cells was recently provided by Ying and co-workers90. They showed that, if shielded from inductive stimuli through fibroblast growth factor receptor and extracellular signal-regulated kinase signalling and treated with a glycogen synthase kinase 3 inhibitor to restore viability, mouse ES cells can self-renew in the absence of additional maintenance factors such as leukaemia inhibitory factor (LIF). This indicates that the ES self-renewing state is a self-sustaining ‘ground state’ of the core transcriptional circuitry.
The notion that cell types correspond to attractors of underlying regulatory networks is appealing from a systems biology point of view, as attracting states do not depend solely on individual regulatory elements, but rather result from the collective behaviour of the cell’s molecular regulatory circuitry as a whole. However, in many circumstances cell phenotypes are not well defined, and there might be substantial variability between cells, even within a clonal population in a homogeneous environment91. Thus, in addition to a deterministic control by molecular regulatory circuits, it has long been suggested that cell fate specification also has a stochastic element92–95.
Stochasticity and stem cell fate
Gene expression is an inherently ‘noisy’ process96: owing to the stochasticity of molecular processes, such as transcription and translation (intrinsic noise), and the effect of environmental noise on these processes (extrinsic noise), gene and protein expression levels in a given cell are continuously fluctuating97. As molecular noise can markedly affect cell behaviour, cells have adapted a range of sophisticated mechanisms to control molecular noise98. For example, they use molecular mechanisms, such as the 26S proteasome, to buffer noise by targeting transcriptional pre-initiation complexes for degradation99,100. Furthermore, cells can use epigenetic regulatory agents, such as polycomb group repressors56, to restrict the transcriptional activation of developmental genes101. In addition, they use regulatory network motifs, such as negative feedback loops, to modulate the levels of noise102,103.
In the context of stem cell differentiation, there has been a longstanding interest in the role of stochasticity in determining cell fate92–95. For example, in the early 1960s McCulloch, Till and Siminovitch92 examined the distribution of stem cell-like colony forming units (CFUs) in the spleens of irradiated mice following the injection of a suspension of adult mouse bone marrow cells. They found that the proportion of CFUs per colony varied greatly from colony to colony and was consistent with a ‘birth’ and ‘death’ process in which cell fate decisions (that is, to differentiate or self-renew) were made stochastically. Similarly, in the 1980s Ogawa and co-workers93,95 studied pairs of cells derived from single haematopoietic progenitors (‘paired progenitors’). They showed that if isolated and allowed to form separate colonies in vitro, paired progenitors show remarkably variable and seemingly uncorrelated patterns of differentiation.
These classic papers suggest that, in addition to deterministic control by an underlying regulatory network, stem cell fate specification also has an intrinsically stochastic element. Furthermore, numerous reports have suggested that rather than being a destabilizing force to be minimized, molecular noise can have a positive role in determining cell fate104,105. A key observation in this regard is that molecular noise can give rise to robust heterogeneity at the cell population level96,104 by triggering stochastic transitions between coexisting attractor states84 (BOX 4). For example, it has been suggested that in microorganisms noise-driven heterogeneity in a clonal population allows adaptation during times of stress without the need for genetic mutations, by providing a means for individual cells to ‘explore’ different phenotypes in a dynamic manner104,105. This view is supported by the observation that in yeast the expression of proteins involved in responses to environmental changes are more noisy than those involved in protein synthesis106.
Box 4. A stochastic multi-stable switch.
Consider the simple motif in which two transcription factors activate their own expression and mutually repress each others’ expression (see the figure, part a). This type of feedback naturally gives rise to multi-stability86,126 and provides the cell with the ability to make all-or-none fate decisions in response to external cues. The following stochastic differential equations describe the expression levels of two transcription ) factors (x1 and x2 that are interacting in this way:
In these equations k1, k2 and k3 are the (normalized) rate constants at which transcription factors bind to promoters; K1 and K2 are (normalized) dissociation rate constants; b1 and b2 are (normalized) decay rate constants; σ1 σ2 and are constants determining the amplitude of noise in the system; and W denotes a Weiner process (Brownian motion). In this simple illustrative case we have assumed that each transcription factor binds cooperatively to its own promoter and to that of the other transcription factor as a homodimer (which is why x is raised to the power of two). In the absence of molecular noise (σ1=σ2=0) this model has many coexisting steady state attractors (for appropriate parameter regimes). In the presence of molecular noise (σ1, σ2>0), individual cells do not settle at a single attractor but instead stochastically switch between distinct states at a rate that depends on the amplitude of molecular noise. However, over time the joint probability density p(x1, x2) (that is, the probability of finding a cell with expression levels of (x1, x2)) settles to a stationary state, and a robust distribution of cell types is achieved. The figure (part b) shows the stationary probability distribution for a representative simulation of this system: red hot spots indicate preferred genetic configurations at which cells will accumulate, and blue indicates low probability configurations.
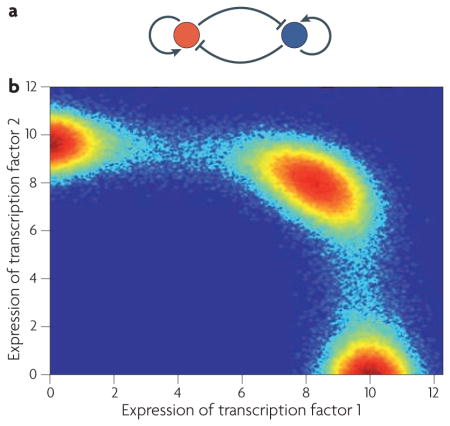
Evidence that a similar mechanism might give rise to heterogeneity in mammalian progenitor cell populations, allowing dynamic ‘priming’ of progenitors towards different lineages, has recently been provided by Chang and co-workers84. To do so, they studied heterogeneity in the expression of the stem cell surface marker SCA1 (stem cell antigen 1) in a clonal population of EML mouse multi-potent haematopoietic cells84. First, using flow cytometry they found that in EML cells SCA1 expression exhibits a characteristic bimodal distribution. Then, to probe the origin of this heterogeneity they used flow cytometry to isolate cells with the highest, middle and lowest SCA1 expression for further culture. Surprisingly, they found that over time all three selected fractions reconstituted the parental bimodal distribution. With the aid of mathematical analyses they identified discrete noise-driven transitions between two underlying and coexisting attracting states as one source of this universal reconstitution. This report is interesting, as it suggests that cells do not have a rigidly fixed identity but instead can transition stochastically between coexisting attracting states at a rate that depends on transcriptome-wide noise levels. However, crucially, at the population level the fraction of cells in the vicinity of each of the attracting states remains fixed in the long term (BOX 4). Thus, although cell identity might be somewhat indeterminate at the single cell level, the distribution of cell types at the population level is robust.
The notion that cell fate is controlled by the interplay between deterministic regulatory mechanisms and stochasticity is not new: a similar observation was made by McCulloch, Till and Siminovitch92, who observed that “individual cells within the population are not closely regulated” and that “it is the population as a whole that is regulated rather than individual cells.” However, the work of Chang and co-workers84 provides an elegant mechanism for this observation. Interestingly, another report (which was, in fact, published before the work of Chang and coworkers) has shown a remarkably similar phenomenon in mouse ES cells107. In this report, Chambers and co- workers107 used flow cytometry to profile NANOG expression in mouse ES cells using green fluorescent protein targeted to the Nanog locus. They found that, similarly to SCA1 expression in haematopoietic precursors, mouse ES cells also show variability in NANOG expression, which is undetectable in a fraction of OCT4-expressing cells. Importantly, they found that following cell sorting using flow cytometry, both NANOG-positive and NANOG-negative cell fractions had a heterogeneous distribution of NANOG expression over time and that ES cells lacking NANOG expression showed an increased propensity to differentiate. Their results suggest that NANOG expression levels fluctuate in mouse ES cells and that the NANOG-low expression phase might be a temporary ‘window of opportunity’, allowing dynamic priming of cellular commitment to differentiation.
Systems biology of cellular reprogramming
The reports discussed so far suggest that at the single cell level cell fates can exhibit a surprising degree of flexibility. In 2006 Yamanaka and co-workers5 made a remarkable discovery concerning the extent of this flexibility: they showed that following retroviral infection with just four transcription factors — OCT4, SOX2, KLF4 (Kruepper-like factor 4) and MYC — adult fibroblasts can be reprogrammed to a state that has many ES cell characteristics. These include an ES cell morphology, the ability to form teratomas (a type of tumour containing tissue from all three germ layers) following subcutaneous injection in nude mice and the ability to differentiate to all three germ layers in vitro. The cells were termed iPS cells on the basis of their similarity to ES cells.
Since these initial reports, the original reprogramming strategy of Yamanaka and co-workers has been refined by numerous groups108–118. In particular, reprogramming has been achieved using forced expression of various alternative reprogramming factors108,109 (including in the absence of forced MYC expression109–111) in a range of adult somatic cell types, such as fibroblasts5,6, hepatocytes112, gastric epithelial cells112, mesenchymal stem cells113 and neural stem cells114. Furthermore, improved selection criteria have allowed the derivation of more completely reprogrammed cells, which are similar to ES cells not only in morphology, differentiation capacity, response to cytokines such as LIF and the ability to form teratomas, but also in global genetic and epigenetic profiles115 and the ability to form viable chimeras following their injection into blastocysts116,117.
Taken together, these reprogramming reports indicate that somatic cell fate is not terminal, but instead that cellular integrity is preserved by reversible epigenetic barriers that can be overcome given the correct stimuli. These observations are in accordance with Waddington’s view of development and the idea of cell types as attractors. The fact that cellular reprogramming is a multistep process119 involving numerous (possibly stochastic120) transitions indicates that cellular reprogramming might correspond to navigation through a complex noisy attractor landscape (FIG. 2). Crucially, this landscape describes both the molecular characteristics of the various different cell types and the relationships between these different cell types — that is, how easy or difficult reprogramming between distinct cell types might be121.
As the processes involved in cellular reprogramming are highly complex, it is a considerable challenge to map this cell fate landscape. We view the generation of such a map as a long-term goal for stem cell systems biology that will require coordinated and sustained collaboration between scientists from a range of disciplines using both experimental and theoretical approaches. However, some first steps towards mapping this landscape might be taken immediately using current technologies. For example, the fact that reprogramming of various adult somatic cells to a self-renewing pluripotent state can be achieved by many different methods5,6,108,120 is consistent with, although not yet proof of, the existence of a core ES cell attractor. In particular, although Ying and co-workers90 have shown that the self-renewing state of an ES cell is self-sustaining if the cell is shielded from inductive signalling, the hallmark attractor characteristic of stability in the face of different weak perturbations has yet to be shown for the ES cell state. To confirm the presence of such an attractor, similar approaches to those taken in establishing the presence of attractors in the haematopoietic system could be adopted. For example, an informative experiment would be to use high-throughput techniques to measure temporal molecular expression patterns following the treatment of ES cells with suboptimal inductive stimuli (such as administering low-dose or short-period retinoic acid treatment, or applying suboptimal levels of key pluripotency factors that target and inhibit mRNA). This would help to determine whether there is an inductive point of no return before which perturbed cells revert back to the undifferentiated ES cell state when stimuli are removed and after which the undifferentiated ES cell state cannot be recovered simply by the removal of stimuli.
Conclusions
The published literature on stem cell systems biology and reprogramming indicates that cell fate might be controlled by a complex interplay between determinism and stochasticity. In the case of determinism, systems-level regulatory network dynamics define the molecular attracting states towards which the cell is drawn over time, and in the case of stochasticity systems-level molecular noise drives transitions between coexisting attractor states and ensures robustness at the population level.
However, current evidence for cellular attractors is limited to a few mammalian cell types. Additional experiments are needed to clarify how universal these initial observations are. Furthermore, direct evidence for molecular attractors is currently limited to the mRNA transcript level. As mRNA expression does not necessarily correlate with protein expression, additional studies are required to clarify how coordinated regulation at different molecular regulatory layers — including mRNA transcripts, proteins and protein complexes, histone modifications, RNA polymerase, signalling pathways and miRNAs — specifies cellular attractor states. Similarly, although there have been some reports detailing the temporal molecular dynamics of ES cell fate changes following perturbation122, our understanding of the systems-level molecular dynamics of cell fate specification, particularly at the single cell level, is still incomplete. For this reason, we anticipate that the further development and use of high-throughput single cell genetic, epigenetic and proteomic techniques106 will be necessary to elucidate the nature of cell to cell variability and to better dissect the role of molecular noise in determining cell fate. In addition, experimental advances will have to be continually integrated with computational models to construct an accurate quantitative understanding of the regulation of stem cell fate. In this regard, stem cell systems biology is an exciting field of research, as it is rich in both experimental and computational challenges and has the potential for genuinely collaborative research.
Recently there have been important and exciting advances in our understanding of stem cell fate specification and cellular reprogramming. However, we still know little about these intrinsically complex processes at the systems level. We hope that an integrated approach, in which experimental approaches provide the information that forms the theory and computational modelling refines experimental approaches, will help us to better understand the molecular basis of stem cell fate decision making and cellular reprogramming.
Acknowledgments
The authors thank J. Wang for supplying the NANOG interac-tome data used to create FIG. 1a and Y.-S. Ang for helping to compile the list of genes used to create the supplementary stem cell transcription network.
- Inner cell mass
Early cells in the embryo that generate all lineages of the mature organism but do not give rise to the placenta
- Blastocyst
The embryo before implantation, which contains at least two distinct cell types: the trophectoderm and the inner cell mass
- Feedback loop
A closed path in a network starting and ending at the same node and passing through intermediary nodes only once
- Feedforward loop
The union of two distinct paths in a network from a source node to a target node, passing through intermediary nodes only once
- Attractor
A stable balanced state of a dynamical system towards which nearby configurations are drawn over time. Attractors can be stationary states, limit cycles (oscillators) or even strange (chaotic)
- ChIP-on-chip
A high-throughput chromatin immunoprecipitation (ChIP) procedure that is used to identify binding sites for a specific transcription factor or other DNA-binding protein in the entire genome
- ChIP-seq
A procedure similar to ChIP-on-chip except that instead of hybridizing isolated DNA fragments bound by the protein of interest with a microarray, the fragments are amplified, size-selected and directly sequenced using massively parallel signature sequencing (MPSS)-based deep sequencing techniques
- ChIP-PET
A procedure that is similar to ChIP-on-chip and ChIP-seq. In this case isolated DNA from the ChIP portion of the experiment is digested into 18-nucleotide-long fragments that are concatenated, tagged and sequenced (known as paired-end ditags (PETs)). The sequences of the PETs are then reassembled and compared with the genome to identify actual binding sites
- Reverse engineering
Inferring regulatory interactions from high-throughput datasets using computational and statistical inference techniques
- Induced pluripotent stem (iPS) cell
A type of pluripotent stem cell that can be produced by various adult somatic cell types by forced expression of certain combinations of key embryonic stem cell-associated transcription factors
- Epigenetic modifier
A substance that causes a change in gene expression without changing DNA sequence
- Molecular noise
Stochastic fluctuations in molecular expression levels originating from the inherent the indeterminism of molecular processes and the unpredictable variability of the extracellular environment
- Multi-stable system
A dynamical system that supports the existence of two or more coexisting attractors for some region of parameter space
- 26S proteasome
Large multi-subunit protease complex that selectively degrades multiubiquitylated proteins. It contains a 20S particle that carries the catalytic activity and two regulatory 19S particles
Footnotes
DATABASES
UniProtKB: http://www.uniprot.org
NANOG | OCT4 | SOX2 |
FURTHER INFORMATION
The Black Family Stem Cell Institute homepage: http://www.blackfamilystemcell.org/BFSCIresearch.html
Integrated Stem Cell Molecular Interactions database: http://amp.pharm.mssm.edu/iscmid
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Contributor Information
Ben D. MacArthur, Email: ben.macarthur@mssm.edu.
Avi Ma’ayan, Email: avi.maayan@mssm.edu.
Ihor R. Lemischka, Email: ihor.lemischka@mssm.edu.
References
- 1.Gage F. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- 2.Wagers A, Weissman I. Plasticity of adult stem cells. Cell. 2004;116:639–648. doi: 10.1016/s0092-8674(04)00208-9. [DOI] [PubMed] [Google Scholar]
- 3.Pittenger M, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–161. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 4.Weissman IL. Translating stem and progenitor cell biology to the clinic: barriers and opportunities. Science. 2000;287:1442–1446. doi: 10.1126/science.287.5457.1442. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 7.Xie H, Ye M, Feng R, Graf T. Stepwise reprogramming of B cells into macrophages. Cell. 2004;117:663–676. doi: 10.1016/s0092-8674(04)00419-2. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanna J, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920–1923. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- 10.Loh Y, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nature Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 11.Boyer L, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Chu J, Shen X, Wang J, Orkin S. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Avilion AA, et al. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003;17:126–140. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chambers I, et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 15.Nichols J, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- 16.Mitsui K, et al. The homeoprotein nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113:631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364–368. doi: 10.1038/nature05284. This study derives a high-confidence protein–protein interaction network in ES cells centred around the transcription factor NANOG. [DOI] [PubMed] [Google Scholar]
- 18.Muller F-J, et al. Regulatory networks define phenotypic classes of human stem cell lines. Nature. 2008;455:401–405. doi: 10.1038/nature07213. This study uses innovative computational techniques to derive an extended network for stem cell pluripotency. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aderem A. Systems biology: its practice and challenges. Cell. 2005;121:511–513. doi: 10.1016/j.cell.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 20.Kitano H. Computational systems biology. Nature. 2002;420:206–210. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- 21.Kirschner MW. The meaning of systems biology. Cell. 2005;121:503–504. doi: 10.1016/j.cell.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- 23.Alon U. An introduction to systems biology: design principles of biological circuits. Chapman & Hall/CRC; Boca Raton: 2007. [Google Scholar]
- 24.Sontag E. Mathematical control theory. Springer; New York: 1998. [Google Scholar]
- 25.Wiggins S. Introduction to applied nonlinear dynamical systems and chaos. Springer; New York: 2003. [Google Scholar]
- 26.McQuarrie D, Allan D. Statistical mechanics. University Science Books; Sausalito: 2000. [Google Scholar]
- 27.Lee TI, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 28.Ihmels J, et al. Revealing modular organization in the yeast transcriptional network. Nature Genet. 2002;31:370–378. doi: 10.1038/ng941. [DOI] [PubMed] [Google Scholar]
- 29.Shen-Orr S, Milo R, Mangan S, Alon U. Network motifs in the transcriptional regulation network of Escherichia coli. Nature Genet. 2002;31:64–68. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- 30.Isalan M, et al. Evolvability and hierarchy in rewired bacterial gene networks. Nature. 2008;452:840–845. doi: 10.1038/nature06847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kidder BL, Yang J, Palmer S. Stat3 and c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS ONE. 2008;3:e3932. doi: 10.1371/journal.pone.0003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 33.Spooncer E, et al. Developmental fate determination and marker discovery in hematopoietic stem cell biology using proteomic fingerprinting. Mol Cell Proteomics. 2008;7:573–581. doi: 10.1074/mcp.M700292-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Hinsby AM, Olsen JV, Mann M. Tyrosine phosphoproteomics of fibroblast growth factor signaling: a role for insulin receptor substrate-4. J Biol Chem. 2004;279:46438–46447. doi: 10.1074/jbc.M404537200. [DOI] [PubMed] [Google Scholar]
- 35.Harary F. Graph theory. Westview; Boulder: 1994. [Google Scholar]
- 36.Tyson JJ, Chen K, Novak B. Network dynamics and cell physiology. Nature Rev Mol Cell Biol. 2001;2:908–916. doi: 10.1038/35103078. [DOI] [PubMed] [Google Scholar]
- 37.Ma’ayan A, et al. Formation of regulatory patterns during signal propagation in a mammalian cellular network. Science. 2005;309:1078–1083. doi: 10.1126/science.1108876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bromberg KD, Ma’ayan A, Neves SR, Iyengar R. Design logic of a cannabinoid receptor signaling network that triggers neurite outgrowth. Science. 2008;320:903–909. doi: 10.1126/science.1152662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rual JF, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 40.Barrios-Rodiles M, et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science. 2005;307:1621–1625. doi: 10.1126/science.1105776. [DOI] [PubMed] [Google Scholar]
- 41.Basso K, et al. Reverse engineering of regulatory networks in human B cells. Nature Genet. 2005;37:382–390. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- 42.Ma’ayan A. Network integration and graph analysis in mammalian molecular systems biology. IET Syst Biol. 2008;2:206–221. doi: 10.1049/iet-syb:20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bansal M, Belcastro V, Ambesi-Impiombato A, di Bernardo D. How to infer gene networks from expression profiles. Mol Syst Biol. 2007;3:78. doi: 10.1038/msb4100120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D’haeseleer P, Liang S, Somogyi R. Genetic network inference: from co-expression clustering to reverse engineering. Bioinformatics. 2000;16:707–726. doi: 10.1093/bioinformatics/16.8.707. [DOI] [PubMed] [Google Scholar]
- 45.Berger S, Posner J, Ma’ayan A. Genes2Networks: connecting lists of gene symbols using mammalian protein interactions databases. BMC Bioinformatics. 2007;8:372. doi: 10.1186/1471-2105-8-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang J, et al. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nature Cell Biol. 2008;10:353–360. doi: 10.1038/ncb1698. [DOI] [PubMed] [Google Scholar]
- 48.Singh SK, Kagalwala MN, Parker-Thornburg J, Adams H, Majumder S. REST maintains self-renewal and pluripotency of embryonic stem cells. Nature. 2008;453:223–227. doi: 10.1038/nature06863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cole M, Johnstone S, Newman J, Kagey M, Young R. Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev. 2008;22:746–755. doi: 10.1101/gad.1642408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X, et al. Yamanaka factors critically regulate the developmental signaling network in mouse embryonic stem cells. Cell Res. 2008;18:1177–1189. doi: 10.1038/cr.2008.309. [DOI] [PubMed] [Google Scholar]
- 51.Marson A, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mackay JP, Sunde M, Lowry JA, Crossley M, Matthews JM. Protein interactions: is seeing believing? Trends Biochem Sci. 2007;32:530–531. doi: 10.1016/j.tibs.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 53.Ji H, Vokes SA, Wong WH. A comparative analysis of genome-wide chromatin immunoprecipitation data for mammalian transcription factors. Nucleic Acids Res. 2006;34:e146. doi: 10.1093/nar/gkl803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ulitsky I, Shamir R. Identification of functional modules using network topology and high-throughput data. BMC Syst Biol. 2007;1:8. doi: 10.1186/1752-0509-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mathur D, et al. Analysis of the mouse embryonic stem cell regulatory networks obtained by ChIP-chip and ChIP-PET. Genome Biol. 2008;9:R126. doi: 10.1186/gb-2008-9-8-r126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boyer LA, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 57.Johnson R, et al. REST regulates distinct transcriptional networks in embryonic and neural stem cells. PLoS Biol. 2008;6:e256. doi: 10.1371/journal.pbio.0060256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu G, et al. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009;23:837–848. doi: 10.1101/gad.1769609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Venkatesan K, et al. An empirical framework for binary interactome mapping. Nature Methods. 2009;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paddison PJ, et al. A resource for large-scale RNA-interference-based screens in mammals. Nature. 2004;428:427–431. doi: 10.1038/nature02370. [DOI] [PubMed] [Google Scholar]
- 61.Ding L, et al. A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell. 2009;4:403–415. doi: 10.1016/j.stem.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 62.Mogilner A, Wollman R, Marshall WF. Quantitative modeling in cell biology: what is it good for? Dev Cell. 2006;11:279–287. doi: 10.1016/j.devcel.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 63.Wilkinson DJ. Stochastic modelling for quantitative description of heterogeneous biological systems. Nature Rev Genet. 2009;10:122–133. doi: 10.1038/nrg2509. [DOI] [PubMed] [Google Scholar]
- 64.Hasty J, McMillen D, Isaacs F, Collins JJ. Computational studies of gene regulatory networks: in numero molecular biology. Nature Rev Genet. 2001;2:268–279. doi: 10.1038/35066056. [DOI] [PubMed] [Google Scholar]
- 65.Kauffman SA. The origins of order: self-organization and selection in evolution. Oxford Univ. Press; 1993. [Google Scholar]
- 66.Huang A, Hu L, Kauffman S, Zhang W, Shmulevich I. Using cell fate attractors to uncover transcriptional regulation of HL60 neutrophil differentiation. BMC Syst Biol. 2009;3:20. doi: 10.1186/1752-0509-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bar-Yam Y, Harmon D, de Bivort B. Systems biology: attractors and democratic dynamics. Science. 2009;323:1016–1017. doi: 10.1126/science.1163225. [DOI] [PubMed] [Google Scholar]
- 68.Chang H, Oh P, Ingber D, Huang S. Multistable and multistep dynamics in neutrophil differentiation. BMC Cell Biol. 2006;7:11. doi: 10.1186/1471-2121-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang S, Ingber D. A non-genetic basis for cancer progression and metastasis: self-organizing attractors in cell regulatory networks. Breast Dis. 2007;26:27–54. doi: 10.3233/bd-2007-26104. [DOI] [PubMed] [Google Scholar]
- 70.Kramer BP, Fussenegger M. Hysteresis in a synthetic mammalian gene network. Proc Natl Acad Sci USA. 2005;102:9517–9522. doi: 10.1073/pnas.0500345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brock A, Chang H, Huang S. Non-genetic heterogeneity — a mutation-independent driving force for the somatic evolution of tumours. Nature Rev Genet. 2009;10:336–342. doi: 10.1038/nrg2556. [DOI] [PubMed] [Google Scholar]
- 72.Enver T, Pera M, Peterson C, Andrews PW. Stem cell states, fates, and the rules of attraction. Cell Stem Cell. 2009;4:387–397. doi: 10.1016/j.stem.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 73.Delbruck M. Unités biologiques douées de continuité génétique. Editions du Centre National de la Recherche Scientifique; Paris: 1949. pp. 33–35. [Google Scholar]
- 74.Thomas R. Laws for the dynamics of regulatory networks. Int J Dev Biol. 1998;42:479–485. [PubMed] [Google Scholar]
- 75.Waddington C. Organisers & genes. Cambridge Univ. Press; Cambridge, UK: 1940. [Google Scholar]
- 76.Waddington C. The strategy of the genes. George Allen & Unwin; London: 1957. [Google Scholar]
- 77.Waddington CH. In: The Development of Animal Behavior: a Reader. Bolhuis JJ, Hogan JA, editors. Vol. 22. Blackwell; Oxford: 1999. [Google Scholar]
- 78.Milnor J. On the concept of attractor. Commun Math Phys. 1985;99:177–195. [Google Scholar]
- 79.Strogatz SH. Nonlinear dynamics and chaos: with applications to physics, biology, chemistry, and engineering. Westview; Boulder: 2000. [Google Scholar]
- 80.Kauffman S. Homeostasis and differentiation in random genetic control networks. Nature. 1969;224:177–178. doi: 10.1038/224177a0. [DOI] [PubMed] [Google Scholar]
- 81.MacArthur BD, Please CP, Oreffo ROC. Stochasticity and the molecular mechanisms of induced pluripotency. PLoS ONE. 2008;3:e3086. doi: 10.1371/journal.pone.0003086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chickarmane V, Troein C, Nuber UA, Sauro HM, Peterson C. Transcriptional dynamics of the embryonic stem cell switch. PLoS Comput Biol. 2006;2:e123. doi: 10.1371/journal.pcbi.0020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cinquin O, Demongeot J. High-dimensional switches and the modelling of cellular differentiation. J Theor Biol. 2005;233:391–411. doi: 10.1016/j.jtbi.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 84.Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–547. doi: 10.1038/nature06965. This paper provides evidence of coexisting mammalian attractor states and switching between coexisting attractors at the single cell level owing to transcriptome-wide fluctuations in protein expression levels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang S, Eichler G, Bar-Yam Y, Ingber D. Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys Rev Lett. 2005;94:128701. doi: 10.1103/PhysRevLett.94.128701. This study provides the first experimental evidence that a mammalian cell type corresponds to a high-dimensional attractor of an underlying dynamical system. [DOI] [PubMed] [Google Scholar]
- 86.Xiong W, Ferrell JE. A positive-feedback-based bistable “memory module” that governs a cell fate decision. Nature. 2003;426:460–465. doi: 10.1038/nature02089. [DOI] [PubMed] [Google Scholar]
- 87.Becskei A, Séraphin B, Serrano L. Positive feedback in eukaryotic gene networks: cell differentiation by graded to binary response conversion. EMBO J. 2001;20:2528–2535. doi: 10.1093/emboj/20.10.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ferrell JE, Jr, Machleder EM. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science. 1998;280:895–898. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- 89.Graf T, Stadtfeld M. Heterogeneity of embryonic and adult stem cells. Cell Stem Cell. 2008;3:480–483. doi: 10.1016/j.stem.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 90.Ying QL, et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sigal A, et al. Variability and memory of protein levels in human cells. Nature. 2006;444:643–646. doi: 10.1038/nature05316. [DOI] [PubMed] [Google Scholar]
- 92.Till JE, McCulloch EA, Siminovitch L. A stochastic model of stem cell proliferation, based on the growth of spleen colony-forming cells. Proc Natl Acad Sci USA. 1964;51:29–36. doi: 10.1073/pnas.51.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Suda T, Suda J, Ogawa M. Disparate differentiation in mouse hemopoietic colonies derived from paired progenitors. Proc Natl Acad Sci USA. 1984;81:2520–2524. doi: 10.1073/pnas.81.8.2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ogawa M, Porter P, Nakahata T. Renewal and commitment to differentiation of hemopoietic stem cells (an interpretive review) Blood. 1983;61:823–829. [PubMed] [Google Scholar]
- 95.Suda T, Suda J, Ogawa M. Single-cell origin of mouse hemopoietic colonies expressing multiple lineages in variable combinations. Proc Natl Acad Sci USA. 1983;80:6689–6693. doi: 10.1073/pnas.80.21.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McAdams HH, Arkin A. Stochastic mechanisms in gene expression. Proc Natl Acad Sci USA. 1997;94:814–819. doi: 10.1073/pnas.94.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Swain PS, Elowitz MB, Siggia ED. Intrinsic and extrinsic contributions to stochasticity in gene expression. Proc Natl Acad Sci USA. 2002;99:12795–12800. doi: 10.1073/pnas.162041399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Arias AM, Hayward P. Filtering transcriptional noise during development: concepts and mechanisms. Nature Rev Genet. 2006;7:34–44. doi: 10.1038/nrg1750. [DOI] [PubMed] [Google Scholar]
- 99.Zwaka TP. Keeping the noise down in ES cells. Cell. 2006;127:1301–1302. doi: 10.1016/j.cell.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 100.Szutorisz H, Georgiou A, Tora L, Dillon N. The proteasome restricts permissive transcription at tissue-specific gene loci in embryonic stem cells. Cell. 2006;127:1375–1388. doi: 10.1016/j.cell.2006.10.045. [DOI] [PubMed] [Google Scholar]
- 101.Chi AS, Bernstein BE. Developmental biology: pluripotent chromatin state. Science. 2009;323:220–221. doi: 10.1126/science.1166261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Austin DW, et al. Gene network shaping of inherent noise spectra. Nature. 2006;439:608–611. doi: 10.1038/nature04194. [DOI] [PubMed] [Google Scholar]
- 103.Thattai M, van Oudenaarden A. Intrinsic noise in gene regulatory networks. Proc Natl Acad Sci USA. 2001;98:8614–8619. doi: 10.1073/pnas.151588598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nature Rev Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- 105.Thattai M, van Oudenaarden A. Stochastic gene expression in fluctuating environments. Genetics. 2004;167:523–530. doi: 10.1534/genetics.167.1.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Newman JRS, et al. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- 107.Chambers I, et al. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230–1234. doi: 10.1038/nature06403. This study shows that NANOG expression fluctuates in ES cells and that it provides a temporary predisposition towards cell differentiation. [DOI] [PubMed] [Google Scholar]
- 108.Feng B, et al. Reprogramming of fibroblasts into induced pluripotent stem cells with orphan nuclear receptor Esrrb. Nature Cell Biol. 2009;11:197–203. doi: 10.1038/ncb1827. [DOI] [PubMed] [Google Scholar]
- 109.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 110.Nakagawa M, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nature Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- 111.Wernig M, Meissner A, Cassady JP, Jaenisch R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell. 2008;2:10–12. doi: 10.1016/j.stem.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 112.Aoi T, et al. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science. 2008;321:699–702. doi: 10.1126/science.1154884. [DOI] [PubMed] [Google Scholar]
- 113.Park IH, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 114.Kim JB, et al. Oct4-induced pluripotency in adult neural stem cells. Cell. 2009;136:411–419. doi: 10.1016/j.cell.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 115.Maherali N, et al. directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 116.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 117.Wernig M, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- 118.Sridharan R, et al. Role of the murine reprogramming factors in the induction of pluripotency. Cell. 2009;136:364–377. doi: 10.1016/j.cell.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mikkelsen TS, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–582. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hemberger M, Dean W, Reik W. Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nature Rev Mol Cell Biol. 2009;10:526–537. doi: 10.1038/nrm2727. [DOI] [PubMed] [Google Scholar]
- 122.Ivanova N, et al. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442:533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- 123.Daheron L, et al. LIF/STAT3 signaling fails to maintain self-renewal of human embryonic stem cells. Stem Cells. 2004;22:770–778. doi: 10.1634/stemcells.22-5-770. [DOI] [PubMed] [Google Scholar]
- 124.Nishimoto M, Fukushima A, Okuda A, Muramatsu M. The gene for the embryonic stem cell coactivator UTF1 carries a regulatory element which selectively interacts with a complex composed of Oct-3/4 and Sox-2. Mol Cell Biol. 1999;19:5453–5465. doi: 10.1128/mcb.19.8.5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yuan H, Corbi N, Basilico C, Dailey L. Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes Dev. 1995;9:2635–2645. doi: 10.1101/gad.9.21.2635. [DOI] [PubMed] [Google Scholar]
- 126.Zhang P, et al. Negative cross-talk between hematopoietic regulators: GATA proteins repress PU.1. Proc Natl Acad Sci USA. 1999;96:8705–8710. doi: 10.1073/pnas.96.15.8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.van den Berg DLC, et al. Estrogen-related receptor β interacts with Oct4 To positively regulate Nanog gene expression. Mol Cell Biol. 2008;28:5986–5995. doi: 10.1128/MCB.00301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang X, Zhang J, Wang T, Esteban MA, Pei D. Esrrb activates Oct4 transcription and sustains self-renewal and pluripotency in embryonic stem cells. J Biol Chem. 2008;283:35825–35833. doi: 10.1074/jbc.M803481200. [DOI] [PubMed] [Google Scholar]