

Abstract
Aim
Individuals with homozygous (ZZ) alpha-1-antitrypsin (α1AT) deficiency are at an increased risk for liver damage, cirrhosis and hepatocellular carcinoma (HCC). The transgenic PiZ mouse, expressing the human α1AT mutant Z gene, is a valuable model for this disease. We studied PiZ mice in order to identify and characterize mechanisms involved in the development of HCC.
Methods
Tumor incidence and histology were studied, gene expression levels were surveyed with microarrays, RNA quantified with quantitative real time polymerase chain reaction and protein levels determined with immunoblots and immunohistochemistry.
Results
By 16–19 months of age, approximately 69% of the PiZ mice had developed tumors. HCC was present with no evidence of benign adenomas as pre-cancerous lesions. Tumors showed abnormal mitochondria, variable levels of steatosis, globular inclusions of α1AT mutant Z protein and metastases. PiZ mice that subsequently developed liver tumors had higher serum levels of α1AT mutant Z protein than those that did not develop tumors. Cyclin D1, a cell cycle protein, was upregulated in PiZ livers without tumors compared to Wt. cFOS, a component of AP-1 that may be involved in transforming cells and MCAM, an adhesion molecule likely involved in tumorigenesis and metastases, were elevated in tumors compared with livers without tumors.
Conclusion
In the PiZ model, many of the histological characteristics of HCC recapitulated features seen in human HCC, whether from individuals with homozygous ZZ liver disease or from unrelated causes in individuals that were not homozygous ZZ. The accumulation of mutant Z protein altered the regulation of several genes driving proliferation and tumorigenesis.
Keywords: alpha-1-antitrypsin deficiency, cyclin D1, PiZ mice, hepatocellular carcinoma, MCAM, liver injury
INTRODUCTION
The liver is the primary source of synthesis and secretion of alpha-1-antitrypsin, a serine protease inhibitor important in protecting tissues from extracellular proteolytic activity. 1 Damage to the liver can occur in individuals homozygous for the mutant Z allele of α1AT, (PIZZ homozygotes in World Health Organization nomenclature). The mutant Z gene encodes for a single amino acid substitution that results in a mutant protein with lysine substituted for glutamate 342, which misfolds and accumulates in the endoplasmic reticulum (ER) of hepatocytes. 2 Individuals with ZZ mutations are at increased risk for liver damage, cirrhosis and HCC. 3–5
Several different transgenic mouse models produce human α1AT mutant Z protein and have been used to study the development of liver disease. 6, 7 Transgenic Z#2 mice, which are no longer available, were reported to develop HCC by 16–24 months of age. 7 However, a different transgenic line, the PiZ mouse, showed “liver necrosis and inflammation” in mice from 6 to 20 months of age, but no consistently identified HCC. 6 Livers of the PiZ mice have been shown to undergo inflammation, injury with compensatory proliferation, development of abnormal mitochondria and an increase in apoptosis and autophagy compared to wild type (Wt) mice. 8–11 Accumulation of α1AT mutant Z protein alters gene expression patterns in PiZ livers. A microarray analysis from transgenic mice induced to express α1AT mutant Z showed alterations in numerous pathways, including lipid metabolism, proteasomal degradation, inflammation, proliferation and cell death. 12 Expression of α1AT mutant Z activated specific proteins such as those within the apoptotic pathway, as well as, BAP31, involved in ER quality control and NF-κB. 8, 13
In this report, we observe that HCC occurs in a significant number of PiZ mice older than one year, with tumors occurring in 69% of mice aged 16–19 months. Individual tumors were characterized with respect to cytoarchitecture, steatohepatitis, α1AT expression, occurrence of globules of α1AT mutant Z protein that were periodic acid Schiff and diastase digestion resistant (PAS+ diastase digestion) and expression levels of a number of genes. Several alterations were identified in pathways that could contribute to the mechanisms of disease development in PiZ livers. Expression of cyclin D1 was upregulated in young and aging PiZ mice without tumors, compared to age-matched Wt mice, which is in agreement with previous models of compensatory cellular proliferation in response to injury.9 Tumors showed elevated levels of Ki67, consistent with proliferation, although not all were cyclin D1 dependent. Tumor tissues had increased expression levels of cFOS, and melanoma cell adhesion molecule (MCAM), proteins implicated in pathways leading to tumorigenesis and metastases.
METHODS
Materials
Antibody sources: cyclin D1, Ki67 (Lab Vision, Fremont, CA); STAT3, PSTAT3, Pan-Actin (Cell Signaling Technology, Danvers, MA); SOCS3 (American Research Products, Belmont, MA); cFOS, MCAM (Santa Cruz Biotechnology, Santa Cruz, CA). RT-PCR sources: random hexamers, Superscript II RT, (Invitrogen, Carlsbad, CA); Fast Start SYBR Green Mix (Roche Diagnostics, Indianapolis, IN) or Qscript cDNA Supermix, PerfeCTA SYBR green (Quanta Biosciences, Gaithersburg, MD). Primers: (Sigma-Genosys, St. Louis, MO). Sequences: Primer Bank.14 (http://pga.mgh.harvard.edu/primerbank), ID numbers: B-2-microglobulin, 31981890a1; cyclin D1, 6680868a1; c-FOS, 6753894a1; FGF-1, 6753850a1 (supplementary S3); HPRT, 7305155a1; MCAM, 12746444a1; SOCS3, 6671758a1. Other reagents: Sigma-Aldrich (St. Louis, MO), Fisher Scientific (Pittsburgh, PA), Promega (Madison, WI).
Animals
Studies were approved by the St. Louis University and Washington University animal studies committees using the instructions from “Guide for Care and Use of Laboratory Animals” (NIH, 86-23). Age- and sex-matched mice were used with C57Bl/6J as Wt controls and PiZ maintained on a C57Bl/6J background. Mice had food and water ad libitum and 12 hour dark-light cycles in a barrier facility. Surgical procedures have been described. 11
Histology, Immunohistochemistry, Electron Microscopy
Tissues were formalin-fixed, embedded, and processed with haematoxylin and eosin or PAS+diastase digestion. Tumors: livers were examined from 4 PiZ mice of each sex, monthly, from 1–12 and at 14,16,18,20, and 24 months of age, with additional data at 14–24 months. Wt mice were examined. A pathologist (E.B.) examined tumors for altered foci, adenoma and HCC. Immunohistochemistry (IHC) was done as before.10 Dark nuclei were considered positive. Morphometry excluded several small areas outside the tumors containing hepatocytes with globules and unusual cytoarchitectural features. In IHC for cyclin D1, followed by PAS+ diastase digestion, positive nuclei were yellow. No significant difference was seen in the percent of positive nuclei in tissue stained for cyclin D1 and glycoprotein versus cyclin D1 only. (P>0.4) Staining for PSTAT3 involved pre-treatment with DNaseI. 15 Photomicrographs were done as before, and adjusted uniformly for contrast, color and clarity using Adobe Photoshop. 11 Electron microscopy was as described. 16
Immunoblots & Serum ELISA
Proteins (50 μg) were separated by SDS-PAGE, immunoblotted and developed with chemiluminescence (ECL Plus, G.E. Health Sciences), with 2–3 repetitions. 9 Human α1AT was measured in PiZ serum, with goat anti-human α1AT, coating antibody, (Cappel, Aurora, Ohio) and rabbit anti-human α1AT, capture antibody (Boehringer Mannheim/Roche, Indianapolis, IN). 17
Quantitative real time polymerase chain reaction (qRT-PCR) and Microarrays
RNA was prepared using Trizol (Ambion, Austin, TX) and the RNeasy mini-kit (Qiagen, Valencia, CA). cDNA and PCR protocols were from PrimerBank. 14 cDNA quantification was as published. 9 β-2-microglobulin or HPRT were reference genes. The Chromo 4 machine (Bio-Rad, Hercules, CA) was used for qRT-PCR. Values are means ± S.E. of fold differences relative to Wt, 2–4 trials. Cancer Pathway Specific, Oligo GEArray Mouse Cancer Pathway Finder Microarrays, OMM033 were used (SABiosciences, Frederick, MD) with 3 male Wt mice and 3 PiZ nontumor (Z), older than 1 year. To determine common pathway alterations, tumor RNA was pooled into PiZ tumor 1 (ZT1) and PiZ tumor 2 (ZT2). Complementary RNA probes, labeled with biotin-16-UT were hybridized to the microarrays, developed with chemiluminescence, detected with a CCD camera (AlphaInnotech, Santa Clara, CA) and analyzed by GEArray Expression Analysis Suite (SABiosciences), using background subtraction and interquartile normalization (normalizes to mean intensity of signals in the 25%–75% of the array.) Affymetrix microarrays MOE430A (Affymetrix, Santa Clara, CA) were processed in the GeneChip facility of the Siteman Cancer Center of Washington University. Liver RNA was from 3 months, male Wt, n=4, PiZ, n=4. Data, normalized to β-2-microglobin, was analyzed using Affymetrix software. http://bioinformatics.wustl.edu. Microarray data was confirmed with qRT-PCR and/or protein determinations.
Data Analyses
Chemiluminescent signals were quantified with densitometry (ImageJ from NIH, //rsb.info.nih.gov/nih-image). Morphometric quantification used ImageJ, green grayscale images with the RGB split, a constant threshold range, background correction and analysis, as described. 18 Statistical significance was calculated by unpaired t tests using Sigma Plot Software. (SPSS, Chicago, IL.)
RESULTS
Tumor Incidence
HCC has not been consistently observed in the PiZ mouse model, although age related liver injuries have been noted, including a mild increase in fibrosis (although not cirrhosis). 11, 19 In this study, tumor formation was observed in older PiZ mice whereas, with the exception of sporadic cases, liver tumors were not seen in mice younger than 12 months and rarely in Wt mice. Prospective examination of PiZ mice varying in age from 1 to 24 months, revealed a high rate of tumor formation that correlated with advancing age, Fig. 1A. Overall, in mice aged 14 to 24 months, 68% (+/−) 4.3% of PiZ mice (n=116) showed tumors compared to 2.0% (+/−) 1.6% of Wt mice (n=87). Liver tumors were present in approximately 46% of the PiZ mice by 14–15 months of age. Tumor incidence increased significantly to 69% by 16–19 months and to 82% by 20–24 months of age, Fig. 1A. In the Wt mice of 14–15 months of age there were no tumors, the 16–19 months group had an HCC, Fig. 1B. and the 20–24 months group had an angiosarcoma, Fig. S1E. Tumor incidence did not show a significant difference between males and females. For example, in the 14–15 months age group, 43% (+/−)14% of PiZ females had tumors compared to 50% (+/−)14% of males; P=0.72, n= 14 each.
Figure 1.

Tumors from Wt and PiZ mice A. Incidence of liver tumors as a function of age. Black bars=PIZ; open bars=Wt; 14–15 months (n=28 PiZ; n=21 Wt); 16–19 months (n=49 PiZ; n=31 Wt, 1 tumor HCC); 20–24 months (n=39 PiZ; n=35 Wt, 1 tumor, angiosarcoma) *Significant difference from PiZ 20–24 months, P=0.002 and PiZ 16–19 months, P<0.05. **Significant difference between PiZ & Wt, P<0.001. B. H&E of Wt tumor, HCC in upper right, adjacent tissue approximately to the left of diagonal line (50X, X refers to original magnification) C. H&E of HCC in PiZ liver with floating trabeculae. (50X) D. Metastatic HCC in PiZ, liver with floating trabeculae, left panel (50X), middle panel and right panel, lung metastases (400X & 100X) E. Electron photomicrograph of HCC from PiZ mouse liver. Normal mitochondrion, m; mega-mitochondrion, M; crystalline inclusion, C; nucleus, N; fat droplet, F; globule, G. Lower left, bar= 500 nm F. Representative tumors and adjacent tissues from 4 mice: top row, Tissue A; second row, Tissue B; third row, Tissue C; bottom row, Tissue D. Left panel, top to bottom, IHC for α1AT, tumor areas stain more than adjacent areas. Right panel, top to bottom, PAS+ diastase digestion: red globules. (50X) G. Percent area occupied by globules in adjacent or tumor regions in each tissue from the right panel of F. Morphometrc quantification: five random microscope fields/tumor and five fields/adjacent region. Values are means (+/−) S.E. A,B,C,D = Tissues A – D. Gray bars=adjacent tissue; black bars =tumor tissue. *P<0.001
Tumor Morphology and Histology
A subset of the tumors preserved for histological analyses were measured. The Wt HCC tumor was approximately 7 mm in diameter and the average diameter of the PiZ tumors was 6.4 (+/−) 0.6 mm, calculated from 39 tumors sampled from 16 mice.
Histological classification of the tumors followed the criteria for rodent tumors published by the International Agency for Research on Cancer. 20 HCCs had variable histology, as illustrated in Fig. 1. Alterations in the normal lobular organization and cytoarchitrecture of the livers along with cytologic atypia are shown in Fig. 1B, Wt tumor and Fig. 1C, PiZ tumor E (also Fig. S1). In the Wt tumor, expansile clusters of neoplastic hepatocytes had “pushing borders” into the adjacent parenchyma, Fig. 1B. (tumor to right of line, adjacent region to the left.) Tumor E was a large (approximately 20 mm) HCC; several foci of lung metastases were found in this animal. The hepatic HCC showed “floating trabeculae”, architecture often characteristic of HCC (Fig. 1C), as well as focal hemorrhage, Fig. S1H. Both tumor E and the Wt tumor had tumor cells that were poorly differentiated, steatotic, dysplastic and/or showed hyperchromatic nuclei and mitotic figures, Fig. S1B–G. Figure 1D, left panel, shows tumor F, a metastatic HCC, with “floating trabeculae” in the liver, while the middle and right panels show two views of the metastases to the lung. The liver tumor had regions of “coagulative necrosis”, Fig S1I. Other examples of HCC in tumors from PiZ mice are shown in Figs. 1F and S1, J–M. Globules of α1AT mutant Z were present in many of the PiZ tumors, Figs. 1F and S1 and S2. The histological characteristics of HCC in the PiZ model recapitulates many of those described in HCC from both human homozygous ZZ liver and in HCC from other causes in non-ZZ human liver. 3, 21, 22
Livers from older PiZ mice also showed altered foci characterized by cellular alterations with clonal features and apparent growth within the liver; in humans, these are the equivalent of dysplastic nodules noted in cirrhosis. Globular accumulations of α1AT mutant Z were usually prominent throughout these altered foci. Fig. S2A–C, shows abnormal staining patterns in altered foci. Some foci were relatively small in diameter (usually < 2 mm), did not disturb the overall lobular cytoarchitecture and had little or no compression of adjacent tissue. An example of a tumor that could be classified as hyperplasia in a rodent (or a dysplastic nodule) is shown in Fig. S2 D–G. The overall hepatic lobular structure was intact in the tissue but there was evidence of hepatocellular damage with inflammation, an altered staining pattern, cytologic dysplasia and globules of α1AT mutant Z. It has been previously hypothesized that adenoma formation is a precursor to HCC in ZZ models, although this has not been described in humans in whom HCC arises in the setting of advanced fibrosis/cirrhosis. In the PiZ mouse model, more than 50 livers with HCC and more than 100 without HCC were examined and no cases of benign hepatocellular nodules consistent with hepatic adenomas were identified.
Our previous data showed that mitochondrial abnormalities, including changes consistent with the degeneration which occurs with membrane depolarization, as well as mitochondrial autophagy, were present in human α1AT mutant Z liver and in the PiZ mouse model. 10 To determine if these processes might still be ongoing in the HCC cells, we examined the ultrastructure of the mitochondria in six samples of HCC from six different PiZ mice. The results showed many mega-mitochondria with unusual, branching morphology and cytoplasmic crystalline inclusions, indicative of abnormal mitochondria (Fig 1E)
Expression of α1AT Mutant Z Protein and Tumor Formation
To evaluate the role of expression levels of α1ATZ on the subsequent development of liver tumors, serum levels were assayed, which is a previously validated, non-invasive method of estimating hepatic expression levels. 6, 17 In the young cohort of PiZ mice that subsequently developed liver tumors, serum levels of α1AT were significantly higher, with mean values of 621 ± 25 μg/ml, n=79 as compared to 518 ± 19 μg/ml, n=37 for the group that did not develop tumors (P<0.01). In the older PiZ mice, approximately 10–30% of the hepatocytes (n=3) showed staining for α1AT mutant Z with no clear pattern of zonal distribution. Overall, in representative tumors taken from 4 PiZ mice with HCC, Fig. 1F, left panel, α1AT mutant Z expression was strong throughout the tumors, whereas the adjacent regions had less staining that varied from approximately 15%, Tissue A, and 10%, Tissue B to 60%, Tissue C and 45%, Tissue D. With the IHC staining, globules of α1AT mutant Z showed the characteristic brown stain. 6 Globules were also visualized using PAS+ diastase digestion on the tissues (Fig. 1F, right panel). In hepatocytes with steatosis and no visible stained cytoplasm, globules were usually absent. Dense clusters of globules were seen in several areas within each tumor. The fractional area occupied by globules was estimated by morphometic analysis (Fig. 1G). Except for Tissue D, which had tiny globules in most of the hepatocytes of the tumor, the areas occupied by globules within each tumor and adjacent regions were not significantly different. Previous studies 8, 23 have shown that a significant proportion of the total α1AT mutant Z protein within the liver is not present within the globules. Therefore, taken together with these data, there is the suggestion that it is the non-globule pool of α1AT mutant Z protein within the hepatocyte which may be more highly related to the development of HCC.
Cyclin D1 Expression and Localization
Cyclin D1 expression was increased in PiZ males relative to age matched Wt males, as shown with microarray analysis. In young PiZ mice (3 months) there was a 4.1 fold increase in mRNA for cyclin D1, a protein expressed in the G1 phase of the cell cycle, and an increase of 3.5 fold in mice older than 1 year. Quantitative RT-PCR results confirmed the microarray analysis, showing that the expression levels of cyclin D1 were increased in young PiZ mice and older PiZ mice relative to age matched Wt mice, as seen in Figure 2A. Protein levels of cyclin D1 were low in Wt livers and elevated in livers from PiZ mice, both young and old, as shown in the typical immunoblot, Figure 2B.
Figure 2.

Cyclin D1 expression is elevated in PiZ mice. A. qRT-PCR for cyclin D1 in PiZ mice Values represent relative increases above the values for age-matched Wt mice and are calculated as means (+/−) S.E. bars: light gray Z,Y =PiZ, young ( n=4); dark gray Z,O=PiZ, older (n=3) Wt,Y= Wt, young (n=4); Wt,O=Wt, older (n=3). B. Typical immunoblot of cyclin D1. Actin: loading control. C. Quantification: fraction of nuclei positive for cyclin D1. Values are means (+/−) S.E. calculated using 10 microscope fields per specimen. (200X) Bars: unshaded Wt, O (2 Wt mice, specimens stained for cyclin D1)), dark grey Z, O. (2 PiZ mice, specimens stained for cyclin D1). *Significant difference P<0.001
The fraction of nuclei staining positively for cyclin D1 has been tabulated in Figure 2C. Older PiZ mice expressed significantly greater levels of cyclin D1 than older Wt mice. The numbers of positive nuclei were approximately 4.4 fold higher in the older PiZ mice, an increase similar to the increase in mRNA levels seen in Figure 2A. These results correlated with prior measurements of DNA synthesis in male mice, as assessed by BrdU incorporation, which showed an approximately 6.5 fold increase in PiZ livers over Wt. 9 This upregulated expression of cyclin D1 in PiZ hepatocytes and their increased DNA replication, likely predispose the mice to HCC. In one study on mice that overexpressed cyclin D1 within hepatocytes, mitotic figures increased and adenomatous nodules or HCC developed, probably from replicative mutations occurring in the dividing hepatocytes. 24 Overexpression of cyclin D1 can trigger mitotic spindle abnormalities and chromosomal changes. 25
Levels of Cyclin D1 and Ki67 in Tumors and Adjacent Tissue
Five tumors and adjacent regions (Tissues A–D & Wt HCC) were arbitrarily chosen for illustration of cyclin D1 and Ki67 staining. Fig. 3, upper row, shows the nuclear localization of cyclin D1 in individual tumors and adjacent tissue. In general, tissues A, D and Wt had low cyclin D1 in the tumor regions, indicating that these tumors were not likely to be cyclin D1 dependent, while tumors B and C express higher levels of cyclin D1. Ki67 nuclear labeling was used as a measure of cellular proliferation, as illustrated in Figure 3, lower row. As expected, with the exception of tissue D, there were significantly higher levels of proliferation in the tumors compared to non-tumor regions.
Figure 3.

Heterogeneity of expression of cyclin D1 and Ki67 in representative tissues, A through D and Wt, HCC. Tumors with HCC are on the right side of each photomicrograph; adjacent regions are on the left side. Dark line: approximate boundary between tumors and adjacent regions. Upper row, Cyclin D1 staining; bottom row, Ki67 staining (Original magnification 200X)
Levels of cyclin D1 mRNA and protein were quantified in tumor tissues. The amount of cyclin D1 was not elevated in pooled PiZ tumor tissue measured in the cancer pathway microarray, with fold differences relative to Wt of 0.6 for 3 pooled tumors (ZT1,O) and 1.2 for 3 other pooled tumors (ZT2,O). Similarly, qRT-PCR, Figure 4A, did not show a significant difference in levels of cyclin D1 in pooled PiZ tumors relative to Wt liver tissue. Quantification of the cyclin D1 levels for the five individual tumors shown in Figure 3, indicated that there was considerable variability in the levels of cyclin D1. In Figure 4B, the percent of nuclei staining positively for cyclin D1 in the particular tumors showed a considerable range and surrounding regions did not always have similar levels of cyclin D1 as the tumor. The labeling index was also determined for Ki67 in tumor and non-tumor regions, as shown in Figure 4C. With the exception of tissue D, Ki67 was significantly higher in the tumor regions than adjacent areas, indicating that tumors A–C and Wt were actively proliferating. There wasn’t a clear correlation between levels of cyclin D1 and Ki67. In some of the PiZ tissues, tumors and adjacent regions had cyclin D1 expression levels that were similar, but proliferation was higher in the tumors. There may be several explanations for this, for example, deregulation of cell cycle checkpoints and inhibitors have been shown to occur in HCC. 26 Overall, in tumors, detectable cyclin D1 expression may not correlate with measures of proliferation. 27, 28
Figure 4.

Quantification of cyclin D1 and Ki67 in tumors. A. qRT-PCR for cyclin D1 in tumor tissue as fold increase relative to Wt, non-tumor. Bars: black, ZT1, O= pooled tumors set 1, older, n=3 or ZT2, O= pooled tumor set 2, older, n=3; Wt, older (n=3). B. Fraction of total nuclei staining for cyclin D1 in tumors and adjacent regions. Counts were from 5 random microscope fields per tumor and 5 per adjacent region. Values are means (+/−) S.E. Bars: light gray, adjacent region; black, tumor region. *Significant difference from adjacent region, P=0.01. C. Fraction of the Ki67 positive nuclei in tumors and adjacent regions. Symbols as in B. *P≤ 0.006.
Globules, Cyclin D1 and Ki67
Liver tissues were doubly stained to illustrate the distribution of hepatocytes with α1AT mutant Z protein globules and those with either cyclin D1 or Ki67. Figure 5A shows a typical example of liver tissue without tumors, with relatively low levels of globules and a cyclin D1 positive hepatocyte without globules. Liver tumors varied in the number of hepatocytes with both globules and cyclin D1 or Ki67. Figure 5B shows a tumor with hepatocytes containing clusters of tiny globules and very low cyclin D1 levels and Figure 5C shows a tumor with a hepatocyte containing larger globules and cyclin D1 positive staining. In the tumor of Figure 5D, concentrated clusters of globules are in the upper region but there are also several cells that have globules and express Ki67. Although the tumors show proliferating hepatocytes with globules, it is not clear if the globules have any effect on proliferation, which is deregulated in tumors compared to PiZ livers without tumors.
Figure 5.
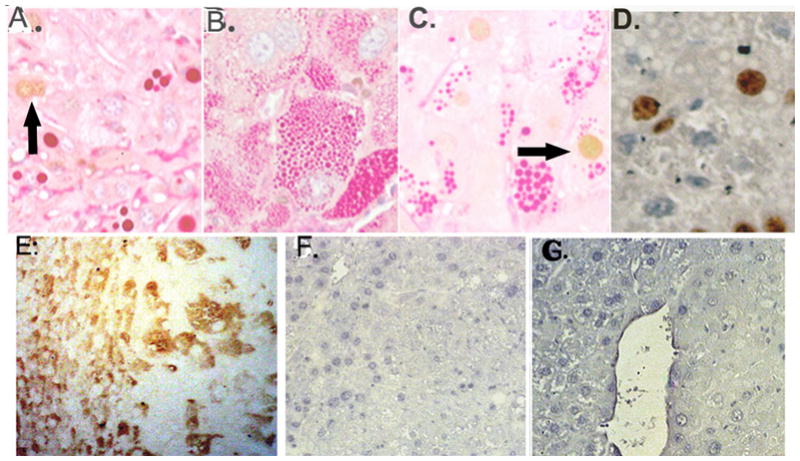
Tissue stained for cyclin D1, treated with PAS and diastase digested (no counterstain) A–C. A. Non-tumor tissue, globules, ~2 cyclin D1 positive nuclei, arrow B. Tissue D, tumor with tiny globules, no cyclin D1 staining shown C. Cyclin D1 in tumor hepatocyte with globules, arrow. D.(No PAS stain): Ki67 stain in tumor, globules appear as white spheres. (Original magnification 400X.) E. SOCS3, Tissue D, transitional region on left, tumor on right F. & G. PSTAT3 (enhanced DAB only), transitional left, tumor right.
Tumor Levels of SOCS3 and PSTAT3
Cyclin D1 transcription can be regulated by STAT3 (signal transducer and activator of transcription 3). 29 SOCS3, suppressor of cytokine signaling 3, is induced by STAT3 signaling but also functions as a negative regulator of STAT3 through its interactions with cytokine receptors. 29 One study found lowered SOCS3 levels and STAT3 activation in human HCC tissue compared to uninvolved, non-tumor regions. 30 Tissue D was stained for SOCS3 as shown in Figure 5E and greater SOCS3 staining was seen in the transitional region compared to the tumor region. In general, staining of SOCS3 protein was low overall in PiZ liver tissue. Staining for PSTAT3 on older PiZ tumors showed that nuclei stained very lightly in the adjacent tissue but levels of staining were lower in the tumors. This is shown in Figures 5F and 5G, transitional regions on the left, tumor regions on the right. (Nuclei from Wt liver don’t appear to stain while some nuclei in PiZ non-tumor tissues stain lightly. Data not shown.)
Quantification of Socs3, STAT3 and PSTAT3
In older PiZ mice Socs3 mRNA levels were lower than compared to age matched Wt mice, and in PiZ tumors the levels of Socs3 mRNA were similar to older Wt mice (Fig. 6A).
Figure 6.

Expression of Socs3 and STAT3 A. qRT-PCR. Levels of Socs3 mRNA measured in PiZ older or in pooled tumor sets. Values are fold increases relative to age matched Wt non-tumor. See Figures 2 and 4 for symbols and n-values. B. Typical immunoblots of PSTAT3 and STAT3 measured in Wt, O and Z, O. STAT3: 2 isoforms. Actin: loading control. # indomethacin treated, positive control. C. Quantification of typical immunoblot. Signals (densities) were calculated in ImageJ. No significant difference in STAT3, P> 0.05.
Prior work showed that STAT3 was not activated in young PiZ livers. 31 In the older mice, the lower Socs3 levels did not impact STAT3 activation, as only trace levels of PSTAT3 were detected in liver tissue from older Z mice, compared to the positive control, indomethacin treated mice (Fig. 6B). Total STAT3 levels were also determined in older Wt and older Z male mice (Fig 6B,C) and were comparable. While activation of STAT3 through IL6-JAK-STAT could increase levels of cyclin D1 transcription, no evidence for PSTAT3 was observed in PiZ livers. Other factors can be important in cyclin D1 regulation such as β catenin, AP-1 and NF-κB. 27, 32, 33
cFOS Localization in Tumors
cFOS, as part of AP-1, can regulate genes involved in tumor metastases and invasive growth. cFOS mRNA levels in older livers without tumors were not elevated in PiZ mice, (0.6) compared to Wt older mice. However, pooled tumor tissue ZT1, showed a 4.2 fold increase and ZT2, a 3.1 fold increase, using the cancer pathway microarray. Staining for cFOS was higher in the nuclei of hepatocytes from tumor tissue compared to adjacent tissue (Fig. 7A). Nuclear labeling was quantified, showing a range between 2 to 7 %, approximately, but was low in adjacent tissues, (Fig. 7B). Previously, in human HCC, higher expression levels of cFOS were observed in tumors than non-tumor tissue. 34 Enhanced expression of cFOS, as part of AP-1, and its targets has been noted in subtypes of HCC that may be derived from hepatic progenitor cells. 35
Figure 7.
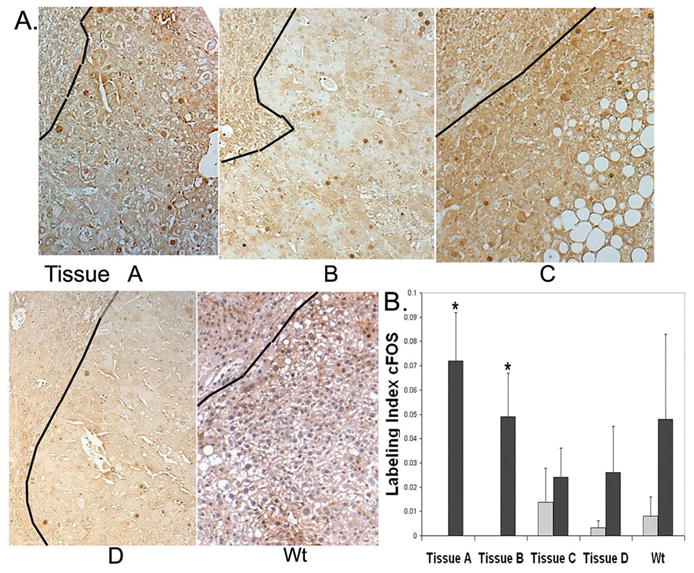
Levels of cFOS expression vary in tumor tissue. A. Nuclear staining for cFOS shown in tissues A–D and Wt. Each photomicrograph has approximate tumor area to right of black line, adjacent area to the left. B. Quantification: approximate labeling index of cFOS. Counting procedures and symbols as in Fig. 4B. *P<0.05.
MCAM Levels
We next examined MCAM, a cell adhesion molecule with elevated levels in PiZ tumors. Levels of MCAM mRNA in pooled tumor tissue from older PiZ mice were elevated by approximately 4.8 fold in ZT1 and 3.0 fold in ZT2, using the cancer pathway microarrays. In qRT-PCR assays, levels were elevated approximately 8–9 fold relative to age matched Wt (Fig. 8A). In PiZ non-tumor tissues, levels of MCAM were similar to Wt (0.8 fold). A typical immunoblot and a bar graph of the quantification of MCAM protein levels are illustrated in Figure 8B&C and indicate that the levels are significantly higher by approximately 4 fold in the tumor tissue from older PiZ mice as compared to age matched Wt or PiZ livers without tumors.
Figure 8.

Expression of MCAM A. qRT-PCR on MCAM mRNA, PiZ mice relative to Wt. Symbols as above. (n=3 for each category, Wt,O; PiZ,O; ZT1,O; ZT2,O) B. Typical immunoblot of MCAM liver homogenates from Wt, O; Z, O; and individual ZT, O. CB: Coomassie blue loading control. C. Quantification of typical immunoblot as in Figure 6. Symbols: gray= non-tumor (Wt, O & Z, O) black= ZT, O *Significant difference P= 0.014.
Discussion
Transgenic PiZ mice have been a valuable experimental model of alpha-1-antitrypsin deficiency liver disease. We found an increased propensity for HCC in PiZ mice, compared to age-matched Wt mice. Although dysplasia has been previously described in this model, this is a full description of HCC. These findings continue to show the utility of the PiZ mouse in recapitulating the injury found in human ZZ liver, as there is a well known increased risk for HCC in ZZ humans, especially in older adults. 3, 36 It is even suggested by previous data that this HCC risk in ZZ humans was not fully accounted for by cirrhosis, but rather could more directly involve the downstream consequences of a1AT mutant Z intracellular retention and accumulation. 3, 36 This is also consistent with our findings since mouse models of chronic liver disease, including the PiZ model, are rarely cirrhotic.
We have shown that young PiZ mice with higher serum levels of α1AT mutant Z were more likely to develop tumors with age than mice with lower levels. Previous work has shown that transgenic mice with higher copy numbers of the human α1AT mutant Z gene, had higher serum levels of α1AT than mice with lower levels of the gene. 6 PiZ mice, classified by high or low serum levels and treated with a chemical chaperone to improve the folding of α1AT mutant Z protein in the liver, responded with increased serum levels in proportion to initial levels. 17
Accumulation of the α1AT mutant Z protein results in caspase activation, autophagy, mitochondrial injury, and a low grade regenerative response. 8, 10, 31 Higher expression levels of α1AT mutant Z protein in the liver have been directly related to increased injury and enhanced proliferation and this could increase the probability of replicative mutations leading to clonal expansion and tumor formation. 9, 31 The approximately 4 fold higher cyclin D1 levels in PiZ mice versus Wt, as observed in this study, could facilitate cell cycle progression, since cyclin D1 forms a complex with the kinases cdk4 or cdk6, resulting in phosphorylation and inactivation of the retinoblastoma protein that inhibits cell cycle progression. 27 Additionally, elevated cyclin D1 expression can lead to aberrant mitotic spindles, centrosome amplification and aneuploidy, which could contribute to tumor formation. 25 Cyclin D1 may also contribute to tumorogenesis through its cdk-independent functions, involving its possible roles in regulating cellular metabolism and migration. 27 The enhanced cyclin D1 levels in PiZ mice indicate that regulatory pathways have become altered in the hepatocytes, and may drive tumorigenesis. In one mouse model of HCC involving inflammation, injury and increased proliferation, the IL-6 pathway was activated, resulting in the phosphorylation of STAT3, while ablation of IL-6 greatly reduced HCC incidence in the male mice. 37 However, our studies demonstrated that STAT3, a transcription factor that can increase cyclin D1 levels, was not phosphorylated in older PiZ mice, although SOCS3, a protein that can reduce STAT3 activation, had slightly lower mRNA levels in older PiZ mice than in Wt.
Tumors derived from a particular tissue type show alterations from the normal expression patterns in various molecular pathways and heterogeneity in the subsets of pathways that are deregulated. 38 We demonstrated that various hepatocellular tumors had altered expression of proteins involved in cancer pathways regulating proliferation, angiogenesis, invasive growth and metastasis. Proliferation (Ki67 labeling) was elevated, although it was not necessarily regulated by cyclin D1, as it was in the PiZ tumor free livers. cFOS, which together with certain other AP-1 family members forms dimers that are transcription factors, was significantly elevated in several of the tumors. The AP-1 dimer of cFOS with certain AP-1 members, can activate downstream targets such as VEGFD, a factor in angiogenesis; matrix metalloproteases that degrade the extracellular matrix and other targets that enhance invasive growth. 32 MCAM, a cell adhesion molecule expressed at low levels in endothelial cells, was elevated in PiZ tumors. MCAM can mediate some cell signaling and it is thought that this could lead to cytoskeletal re-arrangements.39 Its overexpression enhances tumorigenesis in certain cancers such as prostate cancer and antibodies to MCAM can inhibit angiogenesis. 40, 41 Overexpression of MCAM can promote tumor metastases in prostate cancers and melanoma. 41
Our study indicates that tumors had higher levels of α1AT mutant Z than adjacent tissue or tissue from animals without tumors. It is not clear whether the original tumor cells were derived from hepatocytes that had high expression levels or whether hepatocytes with high expression levels were more likely to provide “regenerative signals” to other cells. Levels of accumulation of globules did not clearly correlate with expression levels of α1AT mutant Z and likely represented long term and recently accumulated, polymerized mutant protein.
It is known that α-1-antitrypsin deficiency and other metabolic diseases can be risk factors for HCC. This study has identified several other genes that were altered in expression levels as the α1AT mutant Z protein accumulated and likely contributed to enhanced proliferation, tumor formation and metastases.
Supplementary Material
Acknowledgments
Supported by Alpha-1-Foundation (J.H.T), Cardinal Glennon Children’s Medical Center Pediatric Research Institute, Saint Louis University Liver Center, NIH grant DK-067489 (J.H.T.), Washington University Digestive Disease Research Core Center NIH PO3 DK52574, and NCI Cancer Center Support Grant #P30 CA91842 to Siteman Cancer Center of Washington University and Barnes Jewish Hospital for processing and analysis of the Affymetrix Gene Chip.
References
- 1.Huber R, Carrell RW. Implications of the three-dimensional structure of alpha 1-antitrypsin for structure and function of serpins. Biochemistry. 1989;28:8951–66. doi: 10.1021/bi00449a001. [DOI] [PubMed] [Google Scholar]
- 2.Teckman JH, Qu D, Perlmutter DH. Molecular pathogenesis of liver disease in alpha1-antitrypsin deficiency. Hepatology. 1996;24:1504–16. doi: 10.1002/hep.510240635. [DOI] [PubMed] [Google Scholar]
- 3.Rudnick DA, Perlmutter DH. Alpha-1-antitrypsin deficiency: a new paradigm for hepatocellular carcinoma in genetic liver disease. Hepatology. 2005;42:514–21. doi: 10.1002/hep.20815. [DOI] [PubMed] [Google Scholar]
- 4.Propst T, Propst A, Dietze O, Judmaier G, Braunsteiner H, Vogel W. Prevalence of hepatocellular carcinoma in alpha-1-antitrypsin deficiency. J Hepatol. 1994;21:1006–11. doi: 10.1016/s0168-8278(05)80609-9. [DOI] [PubMed] [Google Scholar]
- 5.Perlmutter DH. Pathogenesis of chronic liver injury and hepatocellular carcinoma in alpha-1-antitrypsin deficiency. Pediatr Res. 2006;60:233–8. doi: 10.1203/01.pdr.0000228350.61496.90. [DOI] [PubMed] [Google Scholar]
- 6.Carlson JA, Rogers BB, Sifers RN, et al. Accumulation of PiZ alpha 1-antitrypsin causes liver damage in transgenic mice. J Clin Invest. 1989;83:1183–90. doi: 10.1172/JCI113999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geller SA, Nichols WS, Kim S, et al. Hepatocarcinogenesis is the sequel to hepatitis in Z#2 alpha 1-antitrypsin transgenic mice: histopathological and DNA ploidy studies. Hepatology. 1994;19:389–97. [PubMed] [Google Scholar]
- 8.Lindblad D, Blomenkamp K, Teckman J. Alpha-1-antitrypsin mutant Z protein content in individual hepatocytes correlates with cell death in a mouse model. Hepatology. 2007;46:1228–35. doi: 10.1002/hep.21822. [DOI] [PubMed] [Google Scholar]
- 9.Rudnick DA, Liao Y, An JK, Muglia LJ, Perlmutter DH, Teckman JH. Analyses of hepatocellular proliferation in a mouse model of alpha-1-antitrypsin deficiency. Hepatology. 2004;39:1048–55. doi: 10.1002/hep.20118. [DOI] [PubMed] [Google Scholar]
- 10.Teckman JH, An JK, Blomenkamp K, Schmidt B, Perlmutter D. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am J Physiol Gastrointest Liver Physiol. 2004;286:G851–62. doi: 10.1152/ajpgi.00175.2003. [DOI] [PubMed] [Google Scholar]
- 11.Teckman JH, An JK, Loethen S, Perlmutter DH. Fasting in alpha1-antitrypsin deficient liver: constitutive [correction of consultative] activation of autophagy. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1156–65. doi: 10.1152/ajpgi.00041.2002. [DOI] [PubMed] [Google Scholar]
- 12.Hidvegi T, Mirnics K, Hale P, Ewing M, Beckett C, Perlmutter DH. Regulator of G Signaling 16 is a marker for the distinct endoplasmic reticulum stress state associated with aggregated mutant alpha1-antitrypsin Z in the classical form of alpha1-antitrypsin deficiency. J Biol Chem. 2007;282:27769–80. doi: 10.1074/jbc.M704330200. [DOI] [PubMed] [Google Scholar]
- 13.Hidvegi T, Schmidt BZ, Hale P, Perlmutter DH. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J Biol Chem. 2005;280:39002–15. doi: 10.1074/jbc.M508652200. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003;31:e154. doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Said J, Shintaku IP. Detection of estrogen receptors with monoclonal antibodies in paraffin sections. Am J Clin Pathol. 1988;90:120. doi: 10.1093/ajcp/90.1.120a. [DOI] [PubMed] [Google Scholar]
- 16.Teckman JH, Perlmutter DH. Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol. 2000;279:G961–74. doi: 10.1152/ajpgi.2000.279.5.G961. [DOI] [PubMed] [Google Scholar]
- 17.Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci U S A. 2000;97:1796–801. doi: 10.1073/pnas.97.4.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunter MG, Hurwitz S, Bellamy CO, Duffield JS. Quantitative morphometry of lupus nephritis: the significance of collagen, tubular space, and inflammatory infiltrate. Kidney Int. 2005;67:94–102. doi: 10.1111/j.1523-1755.2005.00059.x. [DOI] [PubMed] [Google Scholar]
- 19.Mencin A, Seki E, Osawa Y, et al. Alpha-1 antitrypsin Z protein (PiZ) increases hepatic fibrosis in a murine model of cholestasis. Hepatology. 2007;46:1443–52. doi: 10.1002/hep.21832. [DOI] [PubMed] [Google Scholar]
- 20.Mohr U. International Classification of Rodent Tumors Part I-The Rat 10. Digestive System. In: Mohr U, editor. IRAC Scientific Publications No. 122. International Agency for Research on Cancer, WHO; Lyon: 1997. [Google Scholar]
- 21.Eriksson S, Carlson J, Velez R. Risk of cirrhosis and primary liver cancer in alpha 1-antitrypsin deficiency. N Engl J Med. 1986;314:736–9. doi: 10.1056/NEJM198603203141202. [DOI] [PubMed] [Google Scholar]
- 22.Hadzic N, Quaglia A, Mieli-Vergani G. Hepatocellular carcinoma in a 12-year-old child with PiZZ alpha1-antitrypsin deficiency. Hepatology. 2006;43:194. doi: 10.1002/hep.21009. [DOI] [PubMed] [Google Scholar]
- 23.An JK, Blomenkamp K, Lindblad D, Teckman JH. Quantitative isolation of alphalAT mutant Z protein polymers from human and mouse livers and the effect of heat. Hepatology. 2005;41:160–7. doi: 10.1002/hep.20508. [DOI] [PubMed] [Google Scholar]
- 24.Deane NG, Parker MA, Aramandla R, et al. Hepatocellular carcinoma results from chronic cyclin D1 overexpression in transgenic mice. Cancer Res. 2001;61:5389–95. [PubMed] [Google Scholar]
- 25.Nelsen CJ, Kuriyama R, Hirsch B, et al. Short term cyclin D1 overexpression induces centrosome amplification, mitotic spindle abnormalities, and aneuploidy. J Biol Chem. 2005;280:768–76. doi: 10.1074/jbc.M407105200. [DOI] [PubMed] [Google Scholar]
- 26.Plentz RR, Park YN, Lechel A, et al. Telomere shortening and inactivation of cell cycle checkpoints characterize human hepatocarcinogenesis. Hepatology. 2007;45:968–76. doi: 10.1002/hep.21552. [DOI] [PubMed] [Google Scholar]
- 27.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology. 2004;145:5439–47. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]
- 28.Shoker BS, Jarvis C, Davies MP, Iqbal M, Sibson DR, Sloane JP. Immunodetectable cyclin D(1)is associated with oestrogen receptor but not Ki67 in normal, cancerous and precancerous breast lesions. Br J Cancer. 2001;84:1064–9. doi: 10.1054/bjoc.2001.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang S. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin Cancer Res. 2007;13:1362–6. doi: 10.1158/1078-0432.CCR-06-2313. [DOI] [PubMed] [Google Scholar]
- 30.Ogata H, Kobayashi T, Chinen T, et al. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology. 2006;131:179–93. doi: 10.1053/j.gastro.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 31.Rudnick DA, Shikapwashya O, Blomenkamp K, Teckman JH. Indomethacin increases liver damage in a murine model of liver injury from alpha-1-antitrypsin deficiency. Hepatology. 2006;44:976–82. doi: 10.1002/hep.21326. [DOI] [PubMed] [Google Scholar]
- 32.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–68. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 33.Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci. 2008;121:3853–7. doi: 10.1242/jcs.039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuen MF, Wu PC, Lai VC, Lau JY, Lai CL. Expression of c-Myc, c-Fos, and c-jun in hepatocellular carcinoma. Cancer. 2001;91:106–12. doi: 10.1002/1097-0142(20010101)91:1<106::aid-cncr14>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 35.Lee JS, Heo J, Libbrecht L, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410–6. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 36.Eriksson SG. Liver disease in alpha 1-antitrypsin deficiency. Aspects of incidence and prognosis. Scand J Gastroenterol. 1985;20:907–11. doi: 10.3109/00365528509088844. [DOI] [PubMed] [Google Scholar]
- 37.Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–4. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 38.Thorgeirsson SS, Lee JS, Grisham JW. Functional genomics of hepatocellular carcinoma. Hepatology. 2006;43:S145–50. doi: 10.1002/hep.21063. [DOI] [PubMed] [Google Scholar]
- 39.Anfosso F, Bardin N, Vivier E, Sabatier F, Sampol J, Dignat-George F. Outside-in signaling pathway linked to CD146 engagement in human endothelial cells. J Biol Chem. 2001;276:1564–9. doi: 10.1074/jbc.M007065200. [DOI] [PubMed] [Google Scholar]
- 40.Yan X, Lin Y, Yang D, et al. A novel anti-CD146 monoclonal antibody, AA98, inhibits angiogenesis and tumor growth. Blood. 2003;102:184–91. doi: 10.1182/blood-2002-04-1004. [DOI] [PubMed] [Google Scholar]
- 41.Wu GJ, Peng Q, Fu P, et al. Ectopical expression of human MUC18 increases metastasis of human prostate cancer cells. Gene. 2004;327:201–13. doi: 10.1016/j.gene.2003.11.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.