

Abstract
Spectroscopy of myosin and actin has entered a golden age. High-resolution crystal structures of isolated actin and myosin have been used to construct detailed models for the dynamic actomyosin interactions that move muscle. Improved protein mutagenesis and expression technologies have facilitated site-directed labeling with fluorescent and spin probes. Spectroscopic instrumentation has achieved impressive advances in sensitivity and resolution. Here we highlight the contributions of site-directed spectroscopic probes to understanding the structural dynamics of myosin II and its actin complexes in solution and muscle fibers. We emphasize studies that probe directly the movements of structural elements within the myosin catalytic and light-chain domains, and changes in the dynamics of both actin and myosin due to their alternating strong and weak interactions in the ATPase cycle. A moving picture emerges in which single biochemical states produce multiple structural states, and transitions between states of order and dynamic disorder power the actomyosin engine.
Keywords: EPR, spin label, fluorescence, luminescence, disorder, mobility
INTRODUCTION
Since this subject was last covered in Annual Reviews (73), two major advances outside the realm of spectroscopy have helped produce a renaissance in the spectroscopic study of muscle. First, high-resolution structures of the actin protomer (35) and the myosin head (58) were solved in the early 1990s (Figure 1a), and considerable further progress has been made in crystallography, coordinated with cryo-electron microscopy, of myosin and actin (29). This greatly increased the molecular detail and plausibility of models explaining how actin and myosin transform the chemical energy from ATP hydrolysis to the mechanical work of muscle contraction.
Figure 1.

(a) Crystal structures of actin monomer (35) and myosin head (S1) (58). (Top) Oriented as in rigor, muscle fiber axis vertical, showing catalytic domain (CD), light-chain domain (LCD), essential light chain (ELC, dark gray) and regulatory light chain (RLC, light gray). (Bottom) Rotated to show the ATP-binding pocket and the actin-binding cleft. Residues proposed to form the actomyosin interface are cyan on myosin, tan on actin. (b) Proposed structural changes within S1 during muscle contraction based on crystal structures (19, 57) and on EPR data showing that a disorder-to-order transition occurs when myosin isomerizes from weak to strong binding (9). (c) Variable angle between CD [all shown bound to actin in the rigor orientation (red)] and LCD from crystal structures of S1 from scallop (32), chicken smooth muscle (19), and chicken skeletal muscle (58).
Figure 1b illustrates some of the principal hypotheses that developed from these advances. The best-characterized actin-myosin complex is the post-powerstroke rigor state, in which ADP-bound myosin binds strongly to actin at a ~45° angle. It was proposed that the actin-binding cleft is closed in this complex, while the ATP-binding pocket is open. This complex is short-lived, since ATP binds quickly and dissociates myosin from actin, when the nucleotide pocket closes and the actin-binding cleft opens. According to this model, ATP hydrolysis induces a large rotation of the light-chain domain (LCD), acting as a lever arm. A weakly bound pre-powerstroke complex is formed, characterized by dynamic disorder. In the proposed force-generating powerstroke, the lever arm rotates, coupled to closing of the cleft, opening of the pocket, and rigid ordering of the strongly bound myosin head. Figure 1b illustrates three key areas in which crystallographic results are ambiguous. (a) In myosin crystallography, which relies on the use of nucleotide analogs to trap stable complexes that mimic intermediates in the ATPase cycle, the correlation between structural state and biochemical state (defined by the bound nucleotide analog) is not consistent (Figure 1c) (19, 25, 27), probably because crystal packing perturbs the distribution between structural states, compared with solution, in which multiple structural states can populate a single biochemical state (50). (b) The key structures to be determined are complexes of actin and myosin, but there are no crystal structures containing both proteins. (c) Some of the key structural transitions involve dynamically disordered states, which cannot be defined by static crystal structures (20, 37, 75, 76). Spectroscopic methods have the potential to overcome these shortcomings and provide the means for direct testing and refining of these models.
In order for spectroscopic methods to test mechanisms in molecular detail, they must be site-directed—certain locations within the actin-myosin complex must be probed specifically. This has been made possible by a second major advance: technology for site-directed mutagenesis and expression of mutant muscle proteins, which now makes it possible to label virtually any residue by site-directed labeling (SDL). As discussed below, for myosin light chains this has been achieved in Escherichia coli (66), but it has not been achieved for skeletal muscle myosin heavy chain. Therefore, most SDL of myosin II heavy chain has been done either in Dictyostelium discoideum [starting from the native myosin gene (70)] or in insect (Spodoptera frugiperda) cells [for smooth muscle myosin fragments (91)]. Similarly, SDL of actin has been done mainly on yeast actin (38), although recent progress has been reported in expression of muscle actin in insect cells (2). SDL usually entails first producing a “Cys-lite” version of the protein, in which all the reactive Cys have been replaced by a nonreactive amino acid. A specific Cys labeling site is then introduced by mutagenesis, followed by labeling with a Cys-reactive probe (38, 70). Alternatively, spectroscopic signals can be introduced by the use of genetically encoded probes (72, 94) or nucleotide analogs (48). All these labeling methods are applicable to actin-myosin complexes, and some can be used in active muscle fibers. SDL has allowed researchers to probe previously unexplored domains of muscle proteins, and to test and refine new models suggested by crystal structures.
In spectroscopy, there have also been significant technological advances, such as time-resolved luminescence resonance energy transfer (LRET) (42, 83, 84), single-molecule fluorescence (92), total internal reflection fluorescence (TIRF) detection (26), time-domain electron paramagnetic resonance (EPR) (36), multifrequency EPR (50), and electrostatic potential measured by EPR (71b). These breakthroughs have allowed spectroscopy to test previously untestable molecular models of muscle structure and function. The present review surveys the recent use of spectroscopic probes to study myosin and actin structure and dynamics, focusing on site-specific probes that are applicable not only to isolated proteins in solution, but also to protein complexes and muscle fibers. To keep this review relevant to muscle, this review focuses on myosin II, the isoform found in muscle. Although primary emphasis is placed on probes of myosin, we also highlight some exciting recent probes of actin structural dynamics.
There have been recent reviews on site-directed spin labeling (33), distance measurement by EPR (34, 71), single-molecule fluorescence (63), and resonance energy transfer (RET) (64, 68) that discuss these spectroscopic methods in greater detail than this review. There are also more detailed reviews on actomyosin dynamics (75, 93), actomyosin interactions (86, 87), muscle mechanics (43), and muscle regulation (23) that have focused on spectroscopic data.
LUMINESCENCE AND ELECTRON PARAMAGNETIC RESONANCE
This review focuses on optical emission (luminescence) and nitroxide EPR, because these are the only techniques that have the combination of sensitivity and resolution needed to analyze purified proteins as well as large complexes and muscle fibers. These methods are capable of measuring both probe orientation (due to anisotropic interactions of the probe with polarized light or magnetic field) and interprobe distances in the 2–10 nm range (due to dipole-dipole interactions) with high resolution, quantitate both static and dynamic disorder of these distances and angles, and detect solvent accessibility. Because these techniques differ in the type of probe used and the physical parameters observed, they are complementary. EPR has the advantage that most spin labels are smaller than most optical labels and thus usually cause smaller steric perturbations. Another advantage is that a single nitroxide probe and a single EPR spectrometer can be used to detect rotational dynamics in the nanosecond (by conventional EPR) and microsecond (by saturation transfer EPR) time ranges, whereas optical spectroscopy typically requires two different probes and two different spectrometers, fluorescence and phosphorescence (73). EPR also has superior resolution in both orientation and probe-probe distance and can thus provide more quantitative and definitive information about the presence of disorder and multiple structural states. On the other hand, because the energy of an optical photon is 105 times that of a microwave photon, fluorescence is much more sensitive. Whereas EPR typically requires nanomoles of probe, fluorescence can be measured even at the single-molecule level, which can remove some sources of heterogeneity within an ensemble and thus compensate for the relatively low spectroscopic resolution of fluorescence (63). When both techniques are performed at micromolar concentrations, fluorescence data can be acquired much more rapidly, even within the millisecond timescale of biochemical kinetics (46, 90).
It is particularly in the measurement of interprobe distances that EPR and luminescence reveal their complementary natures. Conventional, continuous wave EPR is sensitive to distances from 0.8 to 2.5 nm, almost exactly complementing the range of 2 to 10 nm that is accessible to RET. The recently introduced pulsed EPR technique called DEER (double-electron-electron resonance), is sensitive to distances from 1.5 to 6 nm (24, 36), allowing EPR to take the place of RET in many cases. On the other hand, DEER requires frozen samples, whereas RET does not. Thus when constructing Cys mutants for labeling, it is not predictable whether EPR or luminescence will prove most useful or valid, and the wisest course usually is to do both.
MYOSIN LIGHT-CHAIN DOMAIN
Global LCD Dynamics
The lever arm model suggests that the LCD behaves as a rigid body that amplifies and transmits ATP-driven conformational changes in the myosin catalytic domain (CD) to the thick filament core (58). This hypothesis predicts a large force-generating rotation of the LCD relative to the CD (Figure 1b), and there have been many attempts to test this model using orientation-sensitive probes by EPR or luminescence. The first direct evidence in support of this model in contracting muscle was obtained from a study in which spin-labeled regulatory light chain (RLC) was exchanged for endogenous RLC in skinned scallop muscle fibers, and then analyzed with the high orientational resolution of EPR (4). These fibers were perfused with solutions to induce the three physiological states of muscle, and revealed two populations of LCD angles that varied only in their mole fraction (Figure 2).
Figure 2.

Distributions of regulatory light chain (RLC)-bound probe angle (relative to fiber axis) detected by EPR in muscle fibers (4). The total distribution (black; mean indicated by black arrowhead) is a linear combination of pre- (red, 74° mean) and post-powerstroke (green, 38° mean) distributions.
In rigor (no ATP, heads strongly bound to actin), a 38° population is predominant, with a much broader orientational distribution than observed previously for the CD (74), suggesting that there is some flexibility between the CD and LCD. In relaxation (ATP, no Ca2+), 38° and 74° angles were equally populated, and this distribution shifted toward the 38° angle in contraction (ATP + Ca2+) by 17%. In other words, both pre- and post-powerstroke structural states are populated in both relaxation and contraction, but only 17% of the heads change orientation upon activation of muscle, consistent with the 20% of heads previously found to be strongly attached to actin in active muscle, using probes on the CD (17). This 36° rotation is only about half that predicted from the most popular models, indicating that the actual lever arm swing in contraction is less than that suggested by crystal structures (Figure 1). Without the high resolution of EPR, even this 36° rotation would not be resolved, because the mean rotation between contraction and relaxation is only 3° (Figure 2), explaining why fluorescence polarization is insensitive to this rotation (discussed below). Transient EPR following the photolysis of caged ATP shows that this LCD rotation lags behind the initial phase of force generation (41). This observation suggests that the powerstroke has two roughly equal components—a disorder-to-order transition of the CD followed by a lever arm rotation (Figure 1).
The quantitative resolution by EPR of the pre- and post-powerstroke structural states of myosin in active muscle (4), coupled with simultaneous measurement of muscle force, enabled analysis of the coupling between thermodynamics and structural mechanics within the muscle filament lattice (5). This work showed that inorganic phosphate decreases active isometric muscle force by decreasing the average force per strongly attached myosin head, without changing the number of strongly attached heads as classically assumed. Thus force is limited by the free energy of the weak-to-strong transition within the coupled ensemble of actomyosin interactions (6).
LCD orientation was also measured from the polarized emission of a fluorescent probe on RLC (18, 30, 65). To determine accurately the orientation of the LCD, a bifunctional rhodamine dye was reacted with a series of Cys pairs engineered in a Cys-lite RLC construct (18). This labeled RLC was reconstituted into skinned rabbit muscle fibers, and fluorescence polarization was then measured as a function of physiological state. The mean tilt of the LCD changed by only a few degrees from rigor to relaxation, and the change from relaxation to contraction was not significant (18). As explained above, this result is consistent with that obtained using EPR (4), which had the necessary resolution to detect the small population of actively rotating heads. Subsequent studies employing rapid length steps did show significant LCD rotation that correlated with force, supporting the lever arm model (31). By studying several probes attached at different orientations, researchers found that both tilting and twisting of the LCD occurs upon rapid length changes in rigor (18, 31).
Fluorescence polarization was used to detect LCD orientation at the single-molecule level, with the potential of circumventing some sources of ensemble heterogeneity. In smooth muscle myosin, the RLC adopts two orientational states on actin during force generation (80), attributed to a disordered weak-binding state followed by an ordered strong-binding state, consistent with the disorder-to-order model of force generation (Figure 1b). In a single-molecule study of labeled RLC on skeletal myosin in rigor, using bifunctional rhodamine and TIRF polarization microscopy, a more heterogeneous orientational distribution was observed for the two-headed heavy meromyosin (HMM) than for the single-headed S1, suggesting a flexible connection between the CD and LCD (55).
Clear resolution by fluorescence of transitions between two distinct myosin head orientations on actin was finally obtained by combining time-resolution, single-molecule detection, and myosin V, a processive motor in which most heads bind to actin even in the presence of ATP (26b). This study resolved two interchanging orientations of the LCD (with bifunctional rhodamine attached to a calmodulin light chain) during active sliding on actin. These two LCD angles were tilted by about 42° (Figure 3)(26b) in remarkable agreement with the 36° tilt resolved previously by EPR in contracting muscle (Figure 3) (4). Owing to the cos2θ dependence of the observed signals, it is equally likely that the tilt is on the order of 70° in both cases..
Figure 3.

LCD probe angular distributions from EPR on contracting scallop muscle fibers (black) (4) and single-molecule fluorescence polarization on Myosin V (red) (26b).
The above methods measure the orientational distribution of the LCD but do not reveal whether disorder is static or dynamic. Microsecond rotational dynamics of the LCD has been measured by phosphorescence (56) and ST-EPR (61), using RLC-bound probes. Both studies showed microsecond rotational motions in relaxation, slower and more restricted motions in contraction, and much more restricted motions in rigor. Thus the LCD undergoes a disorder-to-order transition as it transitions from relaxation to contraction to rigor, as observed for the CD (9). However, the LCD is (a) less dynamic (more ordered) than the CD in relaxation and (b) more dynamic (less ordered) than the CD in rigor, adding to the evidence of a flexible linkage between the CD and LCD.
While changes in probe orientation and rotational motion dominated early work in testing the lever arm rotation model, distance measurement by RET between the CD and LCD has become more important in recent years. Initial LRET studies were limited to native labeling sites, which were not strategically placed to detect this predicted interdomain rotation (14, 51, 83, 85). Real progress required SDL. A breakthrough was achieved by fusing green and blue fluorescent proteins (GFP and BFP) to the N and C termini of D. discoideum CD (S1dC) (72). Fluorescence resonance energy transfer (FRET) showed these fluorescence proteins moved apart 15 to 18 Å when ATP or a posthydrolysis analog (ADP.AlF4 or ADP.V) is bound, but not when ADP or a prehydrolysis analog (ATPγS) is bound, supporting the proposal that the lever arm (attached to the C terminus) rotates, producing a severely bent myosin head, only in the posthydrolysis (pre-powerstroke) state (Figure 1b). However, this construct did not even contain a significant portion of the lever arm, so this experiment did not provide a quantitative test of the model.
RET studies discussed thus far could not match the orientational resolution of EPR, which resolved two distinct LCD states (4). Equivalent resolution with FRET was finally achieved by combining time-resolved fluorescence detection with SDL of Cys-lite myosin (Figure 4). A labeling site, C250, was engineered in the CD of Cys-lite D. discoideum myosin II full-length S1 that was optimally placed to detect movement of the LCD (70). FRET was measured to this site, labeled with tetramethylrhodamine (acceptor), from Oregon Green 488 (donor) on RLC (C114 or C116). Large interdomain movements were detected. Nucleotides decreased the distances between probes, indicating more acute angles, as predicted. However, time resolution revealed that, as was discovered previously with EPR in muscle fibers (4), multiple structural states were populated in a single biochemical state. Each of the rigor (A.M) and apo (M) biochemical states is populated predominantly by a single structural state, but actin perturbs the structure of apo-S1, making it even straighter. With the addition of ATP, three structural states are populated, but the major one is still postrigor. With the addition of the posthydrolysis analog ADP.V, the predominant state is the compact pre-powerstroke state as predicted (19), but two other structural states are also populated, including an intermediate structure not previously observed. Thus these FRET measurements confirm some of the predictions from crystallography, but they describe a heterogeneous structural distribution in solution, even in a well-defined biochemical state, and they suggest that at least one myosin structural state has not been revealed by crystallography.
Figure 4.

Position of the light chain domain relative to the catalytic domain (CD) measured by time-resolved fluorescence resonance energy transfer (FRET) within Dictyostelium discoideum S1. (Top) Four structural states resolved by FRET, with structural models constructed as described in Reference 70, by adding the converter domain and lever arm of the indicated PDBs to the D. discoideum CD. The structural states illustrate agreement with the FRET-measured interprobe distances. (Bottom) Table showing the distribution of these structural states within each of the four biochemical states. Fractions for M, M + ATP, and M.ADP.V are calculated from data provided in Reference 70 by normalizing to these three structural states; A.M data and mole fractions are from David Kast (personal communication).
Starting from a similar Cys-lite construct of smooth muscle myosin S1, LRET from a chelated Tb3+ donor (attached to C210, essentially the same site labeled in Reference 70) to an acceptor on RLC (84) confirmed that a posthydrolysis nucleotide analog (in this case ADP.AlF4) makes S1 more compact and found that actin makes S1.ADP slightly more compact, consistent with previous electron microscopic results.
LRET, using Tb3+ chelates bound to single-Cys RLC mutants, was used to measure distances between LCDs on the two heads of myosin in scallop muscle fibers (42) and in skeletal muscle HMM (15). In the muscle fiber study, the head-head separation was greatest in relaxation, least in rigor, and intermediate in contraction. Using four different labeling sites on RLC, it was found that the relative tilt of the two LCDs decreases by 30° in the force-generating weak-to-strong transition (42). The HMM study concluded that the two heads of myosin bind to actin in rigor in a strained configuration that is distinct from that of single-headed binding (15).
Flexibility Within the LCD
Spectroscopic measurements with probes on both essential light chain (ELC) and RLC have been compared to determine whether the LCD acts as a rigid body (lever arm). ST-EPR (8) and time-resolved phosphorescence anisotropy (TPA) (13) in myosin filaments showed that the microsecond rotational mobility of ELC is only slightly greater than that of RLC and substantially less than that of the CD. These results indicate that most of the flexibility within S1 is between the CD and LCD, not between the ELC and RLC. Similarly, RET between ELC and RLC probes showed little or no effect of nucleotides (51). These results are consistent with the proposition that the LCD acts as a semirigid lever arm.
The fluorescence polarization of bifunctional rhodamine, previously applied to the RLC as discussed above, was recently extended to the ELC in skinned rabbit psoas fibers (37). The data obtained in rigor, when combined with previous RLC data (12), could be reconciled with the nucleotide-free myosin crystal structure (Figure 5a) only by introducing both a 45° tilt (β) and a 25° twist (ϕ) between the ELC and RLC (Figure 5b). In rigor, the ELC had a narrower orientational distribution than the RLC, consistent with its proximity to the rigid actin bond. In relaxation, the RLC was more ordered, consistent with its proximity to the thick filament backbone. In contraction, the ELC orientational distribution was slightly more ordered than in relaxation, consistent with previous observations of RLC orientation by fluorescence (65) and EPR (4). These results, together with the ELC/RLC probe results discussed above, indicate that the principal joint of flexibility (compliance) within the myosin head is between the CD and LCD, but there is also some flexibility within the LCD (Figure 5).
Figure 5.

Conventional model for the rigor acto-S1 complex (a) compared with revised structural model (b), based on fluorescence polarization data using bifunctional rhodamine. Adapted with permission from Reference 37.
RLC Structure and Dynamics
Phosphorylation of S19 on RLC activates the actin-activated ATPase of smooth muscle myosin, inducing contraction. Less dramatic activation occurs in cardiac and skeletal muscle. The structural basis of this regulatory mechanism remains unknown, primarily because there is no crystal structure of the N-terminal 24 amino acids of any RLC, presumably because this segment is disordered and/or susceptible to proteolysis. This structural problem has been solved by site-directed spectroscopy. In a pioneering study, FRET was measured between probes attached to an engineered Cys at position 2 on skeletal RLC and three other sites within the LCD (66, 82). Although these results were not enough to construct a structural model, the measured distances later proved useful as constraints for modeling.
A more complete structural analysis was later achieved by site-directed spin labeling (49). Single-Cys mutagenesis was performed throughout smooth muscle RLC expressed in E. coli, and then EPR experiments were performed on spin-labeled RLC bound to intact and functional HMM[AU: Introduced above.] to determine side chain dynamics and solvent accessibility as affected by phosphorylation at S19. A significant phophorylation effect was only observed in the N-terminal 25 amino acids, showing that this region can properly be designated the phosphorylation domain. Cys-scanning SDL was performed on this domain, and the pattern of oxygen accessibility along the sequence was analyzed by EPR (Figure 6a). In the absence of phosphorylation, little or no periodicity was observed, suggesting a lack of secondary structural order in this region. However, phosphorylation induced a strong helical pattern in the first 17 residues, while increasing accessibility throughout the first 24 residues. The results support a model in which this disorder-to-order transition within the phosphorylation domain results in decreased head-head interactions, activating myosin in smooth muscle (49). Molecular dynamics simulations confirmed this result and produced the first atomic-resolution structural models of the phosphorylation domain in the presence and absence of phosphorylation (20) (Figure 6b). In these models, phosphorylation stabilizes a salt bridge between pS19 and R16, which in turn stabilizes the N-terminal helix. In silico mutation of this arganine to alanine or glutamate destabilizes this helix (21). Although the structural mechanism of communication between the phosphorylation domain and CD is not yet known, a fluorescence quenching study with acrylodan demonstrated that the phosphorylation domain is sensitive to the nucleotide state of myosin (47). Because this sensitivity depends on phosphorylation, it is likely that the more ordered form of the N-terminal helix reverses inhibitory head-head or head-tail interactions (49). Thermodynamic analysis of simulations revealed that phosphorylation of S19 in RLC is accompanied by a large, unfavorable decrease in entropy, which is offset by a slightly larger, favorable decrease in enthalpy (21). The small size of the total free energy change (6 kcal mol-1) helps ensure that this phosphorylation-dependent disorder-to-order transition is a highly reversible regulatory switch.
Figure 6.

Effect of phosphorylation on smooth muscle myosin regulatory light chain (RLC) structure. (a) EPR from site-directed spin labeling; the sequence dependence of accessibility changes dramatically, implying a transition from disorder to helical order (49). (b) Structural model. Blue, myosin heavy chain. Red, RLC (25–168) from crystal structure (PDB ID 1SEM). Purple, RLC (1–24), based on FRET (66), EPR (49), and molecular dynamics simulation (20, 21), showing that phosphorylation induces a disorder-to-order transition in the N-terminal domain of RLC.
MYOSIN CATALYTIC DOMAIN
Nucleotide Pocket
Based on X-ray crystal structures, it has been hypothesized that the nucleotide-binding pocket closes in weak-binding states and opens in strong-binding states (Figure 1b). FRET measurement between W512- and MANT-labeled nucleotide analogues was used to test and refine this hypothesis. When MANT-ADP is bound (strong binding), FRET efficiency was 13 ± 1.3% compared to 35.4 ± 5.5% for MANT-ATP (weak binding), corresponding to an increase of 6.7 Å in probe separation, suggesting that the nucleotide pocket is more open in the strong-binding state (89). Solvent accessibility measurements were consistent with these results. More recently, FRET was measured from an engineered W344 (upper 50 kDa domain) to MANT nucleotides (60). Again, MANT-ADP produced less FRET than MANT-ATP did, indicating that the two sites become closer in the weak (ATP) to strong (ADP) transition by 8.5 Å. This provides compelling evidence of a nucleotide-dependent structural change within the upper 50-kDa domain, becoming more compact in the weak-to-strong transition.
Nucleotide spin labels have provided a direct probe of opening and closing of the nucleotide pocket. An EPR study (48) using spin-labeled nucleotides and analogues [SL-ADP (Figure 7), SL-AMPPNP, SL-ADP•BeF3, or SL-ADP•AlF4] showed that the spin label is restricted (Figure 7a) in all cases when myosin is free in solution or weakly bound to actin, indicating a closed pocket, as predicted by crystal structures (Figure 1b). However, when SL-ADP was bound to myosin, the formation of a strongly bound complex (A.M.D) with actin revealed a new spectral component implying greatly increased mobility (Figure 7a) and an open nucleotide pocket, which had been predicted (Figure 1a) but never observed. Unexpectedly, it was observed that both the open and closed structural states of the pocket are populated (Figure 7a), indicating that there are two distinct structural states in the A.M.D biochemical state (Figure 7a). EPR was used to measure the Keq value for this transition. The thermodynamics of this structural equilibrium was determined from a van’t Hoff plot (Figure 7b), revealing that the large positive (unfavorable) enthalpy change in the pocket-opening reaction is offset almost precisely by a favorable entropy increase (order-to-disorder transition), thus balancing the reaction.
Figure 7.

Structure and thermodynamics of the nucleotide pocket deduced from EPR (48). (a) Low-field portion of EPR spectrum of SL-ADP bound to myosin in rabbit psoas muscle (orange), deconvoluted into its two components (blue and red). (b) van’t Hoff plot of Keq for opening of the pocket, determined from EPR.
Actin-Binding Cleft
One hypothesis to arise from the first actomyosin model (57) with support from more recent models (28, 78) is that the actin-binding cleft between upper and lower 50-kDa domains closes upon rigor binding to actin (Figure 1b). Several spectroscopic studies have been carried out to test this hypothesis in solution. Starting with a Trp-lite construct of the smooth myosin CD, Trp was engineered at position 425 in the actin-binding cleft (90). Solvent exposure of W425 was decreased by actin binding and increased by ATP, with the same kinetics as pyrene-actin fluorescence, suggesting that cleft closure coincides with the weak-to-strong actomyosin transition. Subsequently, a pair of Cys residues (537 and 416), one on each side of the cleft, was introduced into a Cys-lite mutant of D. discoideum myosin II CD (S1dC) (16). Pyrene fluorescence increased upon ATP binding and decreased upon actin binding. Since the opposite behavior had been expected, it was suggested that the pyrene probes are too large to fit inside the closed cleft. When spin labels were attached to the same sites, and spin-spin distances were measured by both continuous wave EPR and DEER, the results were more consistent with the conventional models (22). A recent EPR study on S1dC employed five different pairs of spin-labeling sites (Figure 8) and thus provided a more comprehensive picture (36). The data were consistent with actin-induced cleft closure, but most labeling sites gave evidence of two conformations of the cleft. These two structural states (open and closed clefts) were populated in each of the biochemical states (prehydrolysis, posthydrolysis, and rigor), with the closed state most highly populated in rigor. Thus actin binding is not required for cleft closure, nor is actin dissociation required for cleft opening. These results suggest that the conformational distribution of the cleft in solution is more complex and dynamic than in the crystal. This conclusion is consistent with previous studies of probe dynamics and accessibility (38).
Figure 8.
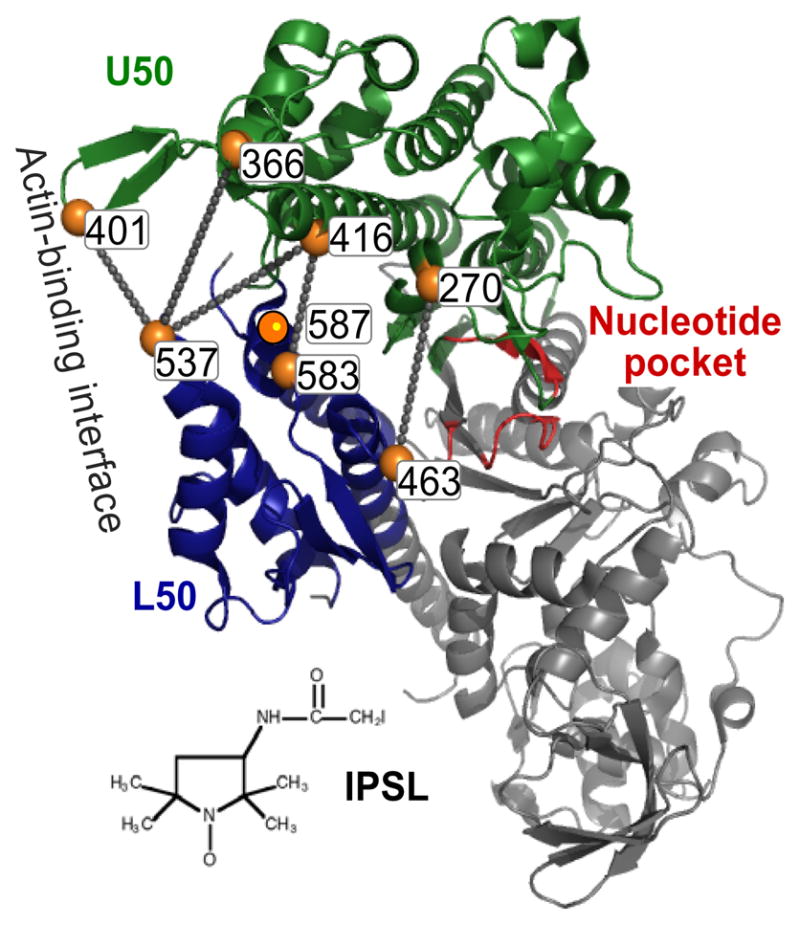
Pairs of spin-labeled Cys residues used to probe cleft closure in S1dC (36).
Force-Generating Region
Early spectroscopic studies of myosin showed that ATP binding enhances Trp fluorescence (81), and there is a further enhancement upon ATP hydrolysis (3). Spectral and structural analysis of Trp residues in the CD of skeletal myosin suggested that these changes arise primarily from W510 (59) [W512 in smooth muscle (89) and W501 in D. discoideum (7)], which is located on the rigid relay loop (44), just N-terminal from the relay helix that directly links the nucleotide-binding site to the SH1-SH2 helix and the converter domain and thus to the LCD. In particular, the relay loop and helix are coupled directly to the nucleotide-binding pocket through switch 2, which responds directly to both nucleotide binding and hydrolysis, so the fluorescence of this conserved Trp has proven to be a powerful tool in the study of myosin’s transient ATPase kinetics (45, 77). However, Trp fluorescence intensity does not provide well-defined structural information, as can be obtained from EPR or FRET.
Crystal structures suggest that the powerstroke, a transition from bent-to-straight structural states (Figure 9a), is driven by a similar bent-to-straight transition in the relay helix (Figure 9b). The reversal of this step, the recovery stroke, should be observable in isolated myosin upon the hydrolysis of ATP or with the binding of posthydrolysis nucleotide analogs such as ADP.V or ADP.AlF4. This was tested in S1dC, by measuring the distance between two probes at engineered Cys within the CD, one at the N terminus of the relay helix (C498) and another at 515 in the upper 50-kDa domain. Both DEER and FRET showed the predicted result: Addition of ATP, ADP.V, or ADP.AlF4 decreased the interprobe distance, demonstrating that the relay helix bends during the recovery stroke. Unexpectedly, both bent (pre-powerstroke) and straight (post-powerstroke) structural states (Figure 9) were populated simultaneously in a single biochemical state, trapped by a prehydrolysis analog (ADP.BeF3). This was confirmed by transient FRET after rapid ATP addition, which showed that the bent structural state is populated before ATP is hydrolyzed. This direct structural observation is consistent with previous kinetic measurements of Trp fluorescence in S1dC (45, 46).
Figure 9.

(a) Reversal of powerstroke (recovery stroke) detected by EPR (4). (b) Bending of relay helix in myosin recovery stroke, predicted by crystal structures and detected by DEER and FRET (R. Agafanov & Y. Nesmelov, personal communication).
The SH1-SH2 helix was among the first sites labeled covalently with probes on myosin, because SH1 (Cys 707) is the most reactive Cys residue and can therefore be labeled specifically with a wide range of thiol reagents. This is a sensitive region, so most probes result in partial inhibition of ATPase activity. The mobility of an iodoacetamide spin label (IASL), attached to SH1 in rabbit skeletal myosin, is sensitive to ATP binding and hydrolysis; its EPR spectra resolve three distinct structural states of this region (reviewed in References 73 and 75). This resolution is even greater when the microwave frequency is increased by a factor of 10 from the conventional 9.4 GHz (X-band) to 94 GHz (W band). When EPR spectra at both frequencies were recorded for IASL-labeled S1, it was possible not only to resolve the three structural states, but also to define the spin label’s amplitude and rate of motion in each state, giving detailed insight into the environment of the SH1-SH2 helix, which is so central to the force-generating function of myosin (50). Spectra obtained with a series of nucleotides permitted assignment of these structural states to crystal structures.
To extend this analysis to D. discoideum, which lacks SH1, a single-Cys mutant (T688C) was engineered to have Cys at the equivalent site (1). Spectra were identical for the two myosins in the apo and ADP-bound states, in which single (but distinct) structural states (probe mobilities) were observed. However, with bound ADP and phosphate analogs, (a) both proteins exhibit two resolved structural states (pre-powerstroke and post-powerstroke) in a single biochemical state (defined by the bound nucleotide), (b) in the two myosins, these structural states are essentially identical but are occupied to different extents as a function of the biochemical state. It is clear that myosin structural and biochemical states do not have a one-to-one correspondence, and that D. discoideum myosin II differs from muscle myosin in the coupling between biochemical and structural states. As in the case of the nucleotide spin labels, the quantitative resolution of EPR permitted the thermodynamic analysis of the powerstroke structural transition, revealing an increase in entropy (50), consistent with the hypothesis that the powerstroke is a disorder-to-order transition in which enthalpic and entropic changes are both large and compensatory.
Global CD Dynamics
Cross-linking Cys 707 (SH1) and Cys 697 (SH2) in the CD of myosin weakens the actin-myosin bond, producing a biochemical state with properties expected for the posthydrolysis complex A.M.ADP.P (reviewed in Reference 76). When these two thiols were cross-linked with a bifunctional spin label (BSL), this weak-binding state was trapped and the spin label was completely immobilized (Figure 10), reflecting the global orientation and dynamics of the myosin CD (76). Conventional EPR of BSL-S1 on oriented actin showed that the CD is partially disordered, as previously observed for ATP-induced weak binding. However, ST-EPR showed that this disorder is slow on the microsecond timescale, as previously observed for strong-binding states, suggesting that the trapped state is a “missing link” between weak and strong binding.
Figure 10.
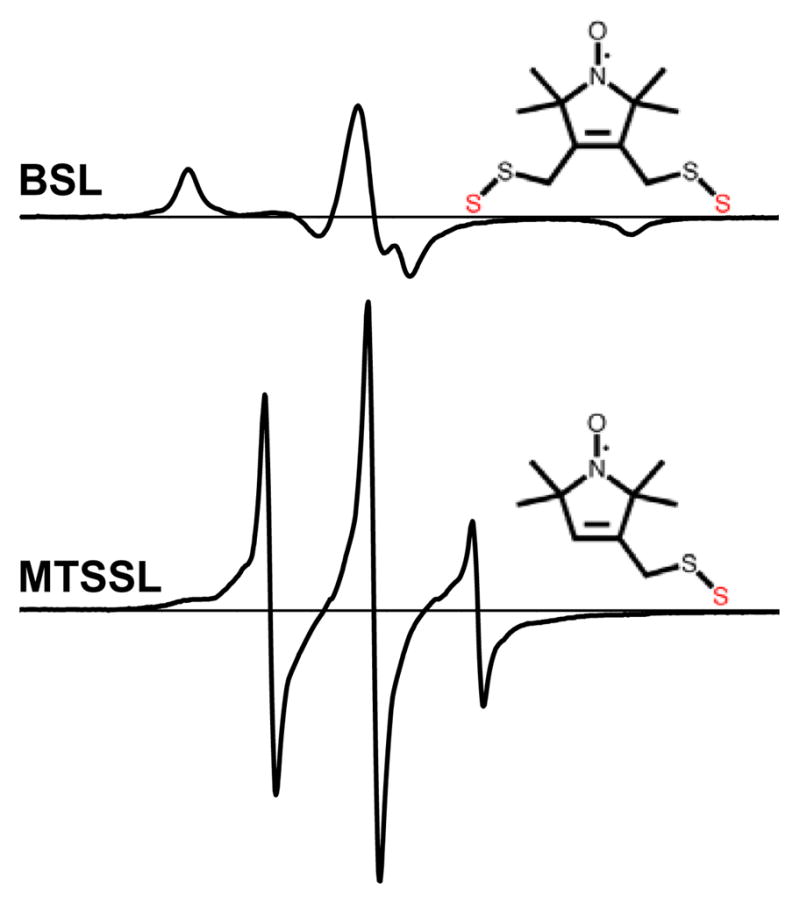
EPR spectra of myosin S1 labeled with a bifunctional methanethiosulfonate spin label (BSL), which is strongly immobilized, compared with the monofunctional methanethiosulfonate spin label (MTSSL), which undergoes almost complete nanosecond rotational mobility (76).
Results on myosin-bound spectroscopic probes, discussed above, were used to construct the model in Figure 11, showing that force and actin movement are generated in two steps, first a disorder-to-order transition in the CD and then rotation of the LCD (76). Of course, these and other transitions in Figure 11 are probably not strictly sequential, since in several biochemical states evidence is described above for a structural equilibrium among structural states.
Figure 11.

Updated model for the coupling of actomyosin ATP hydrolysis to force and movement, summarizing key results discussed in this review. Only actin-bound states are shown. Red and green signify weak (pre-force) and strong (force-generating) complexes, curved arrows signify orientational disorder, and upward steps indicate stages in the cycle where force and movement are likely to be imparted to actin. A, actin; M, myosin; T, ATP; D, ADP; P, inorganic phosphate. Text under each state indicates distinguishing properties of the catalytic domain (76). Prime symbol (′) indicates a second structural state corresponding to the same biochemical state (defined by the active-site ligand). Although these primed and unprimed states are shown in a sequence, they are presumably interconverting in solution or muscle fibers.
ACTIN DYNAMICS
Spectroscopy has shown that the changes in structural dynamics caused by the formation of the actin-myosin complex and the weak-to-strong transition are not limited to myosin. Indeed, one of the most widely exploited muscle probes is pyrene iodoacetamide attached to C374 on actin, which is strongly quenched by strong, but not weak, myosin binding (39). Previous spectroscopic studies have shown that the unbound actin filament exhibits intermonomer torsional and bending motion, as well as internal dynamics within the actin monomer (reviewed in Reference 75). Both optical (mainly TPA) and EPR (mainly ST-EPR) experiments showed that strong myosin binding constrains the dynamics of the actin filament at substoichiometric levels, indicating that the effects are propagated cooperatively along the filament. These observations suggest that as myosin isomerizes from the weak to strong binding state, in a disorder-to-order transition, the actin filament also undergoes a transition from dynamic disorder to order. There is a correlation between inhibited actomyosin interactions due to actin modification (e.g., covalent modification, isoform differences, or mutation) and alterations in actin filament structural dynamics (75).
How does actin structural dynamics change in the transition between weak and strong binding? This was analyzed by performing TPA experiments in the absence and presence of saturating ATP, in the brief steady state before ATP is depleted (54). In the absence of ATP, the final anisotropy rises nonlinearly with myosin binding, indicating that each myosin cooperatively restricts the dynamics of about three actin protomers (Figure 12a). At saturation, weakly bound myosin has almost as much effect as strongly bound myosin, but a much smaller effect is seen at lower binding levels. Strikingly, the dependence is precisely linear—the cooperativity is completely lost. Thus (a) actin undergoes a partial ordering transition upon weak binding and a more profound one upon strong binding (Figure 12b), and (b) in the weak-to-strong transition both myosin and actin undergo disorder-to-order transitions. The functional significance of this finding was recently underscored by the finding that oxidative stress inhibits muscle contraction by perturbing actin dynamics in the weak complex but not in the strong complex, thus decreasing the magnitude of the weak-to-strong transition (52).
Figure 12.
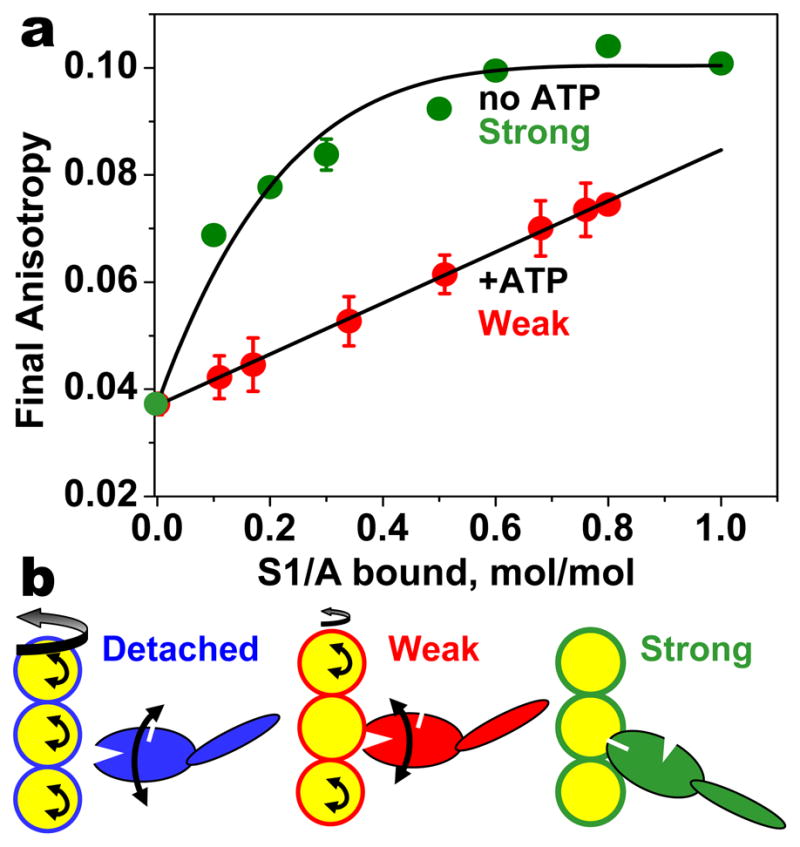
(a) The effect of S1 on TPA of actin in the absence (strong binding) and the presence (weak binding) of saturating ATP (54). (b) Both myosin and actin undergo disorder-to-order transitions upon attachment and upon the weak-to-strong transition.
Further support for actin’s active role in the actomyosin interaction came from a single-molecule FRET study, which measured structural changes within the actin protomer, using rhodamine on N41 as donor and IC5 on C374 as acceptor (40). Actin switches between two states, defined by low- and high-FRET efficiencies, on a timescale of seconds. Active interaction with myosin favors the high-FRET conformation, but cross-linked actin (which does not interact actively with myosin) favors the low-FRET conformation.
Recent studies have probed actin’s structural response to myosin in skinned muscle fibers. Actin was labeled with either rhodamine-phalloidin or Alexa-ATP, and polarization showed a myosin-dependent biphasic response to an ATP pulse (from photolysis of caged ATP), indicating that actin rotates during contraction (10). A similar approach was used to detect orientational signals simultaneously from actin (labeled on phalloidin), myosin (labeled on RLC), and Alexa-ADP (bound to the active site of myosin) (69). Following caged ATP photolysis, the initial phases of RLC rotation (presumably due to cross-bridge detachment), actin rotation, and ADP release (which is presumably simultaneous with ATP binding) were essentially simultaneous, consistent with the model that actin is responding to ATP-induced dissociation of myosin. Another study measured the effects of weak and strong myosin binding on the orientation and dynamics of actin in muscle (11). Actin monomers were labeled either in the nucleotide-binding pocket or on one of five actin residues and then incorporated into muscle fibers. Strongly and weakly bound myosin produced opposite effects on probe orientation, confirming similar conclusions from solution studies (54) (Figure 12) and indicating that actin is an active participant in muscle contraction.
ACTOMYOSIN INTERFACE
There are no high-resolution structures of actin-myosin cocrystals; however, several models have been constructed for these complexes by docking crystal structures into electron microscopy images (28, 57, 67, 79). Although these actomyosin structural models are only for the strongly bound complexes and they do not include dynamics of the interaction, they provide testable hypothesis for spectroscopic measurements in solution.
Before the development of SDL, spectroscopic studies on the actomyosin complex were limited to placing probes on residues located outside of the proposed interface (reviewed in Reference 75). A recent study used yeast actin and D. discoideum myosin to introduce single Cys mutations into four labeling sites; located in the proposed weak-binding and strong-binding interfaces of each protein, and both fluorescent and spin labels were attached to each (Figure 13) (38). While formation of the rigor complex resulted in decreased probe mobility at all four sites, both myosin sites and one of the actin sites showed remarkably high probe mobility even after complex formation. Complex formation decreased solvent accessibility for both actin-bound probes but increased it for the myosin-bound probes. These results are not consistent with a simple model in which there are discrete weak and strong interfaces with only the strong interface forming under strong-binding conditions, nor are they consistent with a model in which surface residues become rigid and inaccessible upon complex formation. In fact, the actin-myosin interface remains dynamic, especially on the surface of myosin, even under rigor binding conditions.
Figure 13.
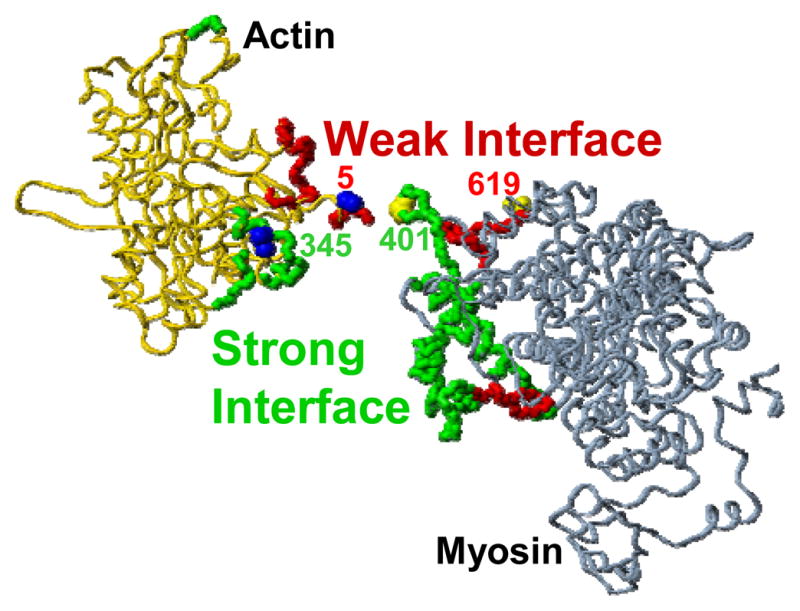
Proposed actin-myosin interface: Yeast actin (left) and S1dC (right) are shown in their rigor orientation (filament axis vertical) but separated horizontally to reveal the proposed interface involved in strong binding (green) and weak binding (red). The four labeling sites used in Reference 38 are indicated.
RET has been used to measure distances between labels on actin and myosin, with the objective of testing models for the actin-myosin complex. In a study on smooth muscle myosin, two distances within the rigor complex were consistent with most models, but no effect of ADP was observed, indicating that the known effect of ADP on the LCD orientation of actin-bound smooth muscle S1 is not detectable with the chosen probe in the CD (88). Further studies of this kind, with several pairs of probes in well-defined sites, coupled with computational analysis incorporating data from crystallography, electron microscopy, and FRET or DEER are needed to provide more rigorous tests of actin-myosin complex structures (62).
CONCLUSIONS AND PERSPECTIVES
Structural dynamics, disorder, and conformational heterogeneity are essential features of the actomyosin machine, especially involving myosin II, which is a low-duty-cycle motor. Therefore, crystal structures serve as the building blocks (models for structural states) that the spectroscopist uses to determine the actual mechanisms (i.e., the coupling between biochemical and structural states in solution and in muscle). EPR and luminescence have the required properties—sensitivity and resolution—to lead this effort, and the two approaches are complementary. Spectroscopic resolution, in both frequency and time, has allowed these methods to demonstrate that a single biochemical state of myosin or actin is populated by two or more structural states. Recent results show that both myosin and actin undergo coupled weak-to-strong (disorder-to-order) transitions that are crucial to muscle contraction and regulation. Future advances will continue to depend on advances in SDL, computational simulation, and spectroscopic instrumentation, correlating structural changes in multiple domains with functional measurements of force, movement, and biochemical kinetics. These advances will lead to more direct tests of complex molecular models, connecting the “separate” parts of actomyosin in space and time, and defining which of these relationships and events are most crucial for the function of muscle contraction.
SUMMARY POINTS.
The discovery of high-resolution structures for myosin and actin along with the development of expression systems for these proteins has helped to produce a renaissance in the spectroscopic study of muscle. Crystal structures are the building blocks (structural states) that the spectroscopist uses to determine the actual mechanisms (i.e., the coupling between biochemical and structural states).
Unlike crystallography, spectroscopy can resolve structural features of (a) proteins and protein complexes that are not crystallizable and (b) partially disordered regions of proteins.
EPR and luminescence are complementary methods and are most effectively used in tandem.
Although luminescence (fluorescence) is much more sensitive, EPR has smaller probes and more intrinsic resolution.
The weak-to-strong transition in myosin, which generates force on actin, occurs in at least two structural steps, first a dynamic disorder-to-order transition of the catalytic domain and then a tilting of the LCD.
Spectroscopic resolution—in both frequency and time—allows both EPR and fluorescence to show that a single biochemical state of myosin or actin is populated by two or more structural states.
Quantitation of structural states’ populations gives insight into thermodynamics as well as structure.
Myosin and actin both undergo coupled weak-to-strong (disorder-to-order) transitions that are crucial to muscle contraction and its regulation.
Dynamics and disorder are critical aspects of myosin and actin structure and function.
FUTURE ISSUES.
To assess the relationships of actomyosin dynamics to biochemical kinetics, site-directed spectroscopy should be measured in the transient phase of the ATPase reaction. This will require further development of transient spectroscopic methods.
To assess the coordination among structural elements of the actomyosin machine, simultaneous spectroscopic measurements should be made with probes on two or more sites.
To assess the functional relevance of detected structural changes within actomyosin, spectroscopic measurements should be done in the presence and absence of known functional mutations.
This field will continue to be driven by advancements in technology: multifrequency EPR, high-throughput time-resolved spectroscopy, single-molecule detection of fluorescence, and solid-state NMR.
Because myosin II is a low-duty-cycle motor, insight into its most important structural transition—the transition from weak binding (dynamic disorder) to strong binding (order)—requires more measurements capable of resolving weak (brief) interactions and dynamic disorder.
The recent development of in vivo labeling techniques is likely to have a profound impact on this field in the next decade.
An increasing number of spectroscopic studies will continue to be done in relationship to muscle disorders. This will depend to some extent on biological technology (e.g., generation of transgenic animals and expression systems) and spectroscopic developments that increase sensitivity for studying small and unstable samples.
Acknowledgments
We thank Octavian Cornea for expert assistance in preparation of the manuscript. We obtained helpful comments from many colleagues, but we especially thank Josh Baker, Roger Cooke, Nariman Naber, Malcolm Irving, Yuri Nesmelov, and Ewa Prochniewicz. This work was supported by NIH grants AR32961 and AG26160 to DDT. DK was supported by NIH training grant AR07612.
TERMS/DEFINITIONS LIST
- Actin binding cleft
gap between upper and lower 50-kDa domains of myosin that may close to allow strong binding to actin
- Anisotropy (r)
luminescence polarization signal used to measure rotational dynamics in the time scale of excited-state lifetime
- CD
catalytic domain
- ELC
essential light chain
- Electron paramagnetic resonance (EPR)
spectroscopic detection of microwave absorption by paramagnetic molecules in a magnetic field, providing high-resolution information about the probes environment, orientation, and motion
- Fluorescence
a type of the luminescence in which light is emitted from the singlet excited state, which typically has a lifetime in the nanosecond range
- FRET
fluorescence resonance energy transfer
- LCD
light-chain domain
- Lever arm hypothesis
a proposed mechanism for muscle contraction in which the LCD of myosin rotates to produce the “powerstroke”
- LRET
luminescence resonance energy transfer
- Luminescence
emission of light from an electronic excited state, including fluorescence and phosphorescence
- Phosphorescence
a type of the luminescence in which light is emitted from the triplet excited state, which typically has a lifetime in the millisecond range
- Resonance energy transfer (RET)
transfer of excited state energy from a luminescent donor to an acceptor, with a rate proportional to r−6
- RLC
regulatory light chain
- S1dC
catalytic domain of Dictyostelium discoideum myosin II
- Saturation transfer EPR (ST-EPR)
EPR done under saturating conditions that make the spectra sensitive to microsecond rotational motions
- SDL
site-directed labeling
- Spin label
paramagnetic probe based on nitroxide group
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any biases that might be perceived as affecting the objectivity of this review.
Contributor Information
David D. Thomas, Email: ddt@umn.edu.
David Kast, Email: kast0040@umn.edu.
Vicci L. Korman, Email: korma002@umn.edu.
LITERATURE CITED
- 1.Agafonov RV, Titus MA, Nesmelov YE, Thomas DD. Muscle and nonmuscle myosins probed by a spin label at equivalent sites in the force-generating domain. Proc Natl Acad Sci USA. 2008;105:13397–402. doi: 10.1073/pnas.0801342105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anthony Akkari P, Nowak KJ, Beckman K, Walker KR, Schachat F, Laing NG. Production of human skeletal alpha-actin proteins by the baculovirus expression system. Biochem Biophys Res Commun. 2003;307:74–79. doi: 10.1016/s0006-291x(03)01133-1. [DOI] [PubMed] [Google Scholar]
- 3.Bagshaw CR, Eccleston JF, Eckstein F, Goody RS, Gutfreund H, Trentham DR. The magnesium ion-dependent adenosine triphosphatase of myosin. Two-step processes of adenosine triphosphate association and adenosine diphosphate dissociation. Biochem J. 1974;141:351–64. doi: 10.1042/bj1410351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker JE, Brust-Mascher I, Ramachandran S, LaConte LE, Thomas DD. A large and distinct rotation of the myosin light chain domain occurs upon muscle contraction. Proc Natl Acad Sci USA. 1998;95:2944–49. doi: 10.1073/pnas.95.6.2944. EPR study, first experimental evidence demonstrating lever arm rotation in contraction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker JE, LaConte LE, Brust-Mascher II, Thomas DD. Mechanochemical coupling in spin-labeled, active, isometric muscle. Biophys J. 1999;77:2657–64. doi: 10.1016/S0006-3495(99)77100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker JE, Thomas DD. A thermodynamic muscle model and a chemical basis for A.V. Hill’s muscle equation. J Muscle Res Cell Motil. 2000;21:335–44. doi: 10.1023/a:1005615925390. [DOI] [PubMed] [Google Scholar]
- 7.Batra R, Manstein DJ. Functional characterisation of Dictyostelium myosin II with conserved tryptophanyl residue 501 mutated to tyrosine. Biol Chem. 1999;380:1017–23. doi: 10.1515/BC.1999.126. [DOI] [PubMed] [Google Scholar]
- 8.Baumann BA, Hambly BD, Hideg K, Fajer PG. The regulatory domain of the myosin head behaves as a rigid lever. Biochemistry. 2001;40:7868–73. doi: 10.1021/bi002731h. [DOI] [PubMed] [Google Scholar]
- 9.Berger CL, Thomas DD. Rotational dynamics of actin-bound intermediates of the myosin adenosine triphosphatase cycle in myofibrils. Biophys J. 1994;67:250–61. doi: 10.1016/S0006-3495(94)80476-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borejdo J, Shepard A, Dumka D, Akopova I, Talent J, et al. Changes in orientation of actin during contraction of muscle. Biophys J. 2004;86:2308–17. doi: 10.1016/S0006-3495(04)74288-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borovikov YS, Dedova IV, dos Remedios CG, Vikhoreva NN, Vikhorev PG, et al. Fluorescence depolarization of actin filaments in reconstructed myofibers: the effect of S1 or pPDM-S1 on movements of distinct areas of actin. Biophys J. 2004;86:3020–9. doi: 10.1016/S0006-3495(04)74351-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brack AS, Brandmeier BD, Ferguson RE, Criddle S, Dale RE, Irving M. Bifunctional rhodamine probes of myosin regulatory light chain orientation in relaxed skeletal muscle fibers. Biophys J. 2004;86:2329–41. doi: 10.1016/S0006-3495(04)74290-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown LJ, Klonis N, Sawyer WH, Fajer PG, Hambly BD. Independent movement of the regulatory and catalytic domains of myosin heads revealed by phosphorescence anisotropy. Biochemistry. 2001;40:8283–91. doi: 10.1021/bi010566f. [DOI] [PubMed] [Google Scholar]
- 14.Burmeister Getz E, Cooke R, Selvin PR. Luminescence resonance energy transfer measurements in myosin. Biophys J. 1998;74:2451–58. doi: 10.1016/s0006-3495(98)77953-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chakrabarty T, Xiao M, Cooke R, Selvin PR. Holding two heads together: stability of the myosin II rod measured by resonance energy transfer between the heads. Proc Natl Acad Sci USA. 2002;99:6011–6. doi: 10.1073/pnas.082024299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conibear PB, Bagshaw CR, Fajer PG, Kovacs M, Malnasi-Csizmadia A. Myosin cleft movement and its coupling to actomyosin dissociation. Nat Struct Biol. 2003;10:831–5. doi: 10.1038/nsb986. [DOI] [PubMed] [Google Scholar]
- 17.Cooke R, Crowder MS, Thomas DD. Orientation of spin labels attached to cross-bridges in contracting muscle fibres. Nature. 1982;300:776–8. doi: 10.1038/300776a0. [DOI] [PubMed] [Google Scholar]
- 18.Corrie JE, Brandmeier BD, Ferguson RE, Trentham DR, Kendrick-Jones J, et al. Dynamic measurement of myosin light-chain-domain tilt and twist in muscle contraction. Nature. 1999;400:425–30. doi: 10.1038/22704. [DOI] [PubMed] [Google Scholar]
- 19.Dominguez R, Freyzon Y, Trybus KM, Cohen C. Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the prepower stroke state. Cell. 1998;94:559–71. doi: 10.1016/s0092-8674(00)81598-6. [DOI] [PubMed] [Google Scholar]
- 20.Espinoza-Fonseca LM, Kast D, Thomas DD. Molecular dynamics simulations reveal a disorder-to-order transition upon phosphorylation of smooth muscle myosin. Biophys J. 2007;93:2083–90. doi: 10.1529/biophysj.106.095802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Espinoza-Fonseca LM, Kast D, Thomas DD. Thermodynamic and structural basis of phosphorylation-induced disorder-to-order transition in the regulatory light chain of smooth muscle myosin. J Am Chem Soc. 2008;130:12208–9. doi: 10.1021/ja803143g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fajer MI, Li H, Yang W, Fajer PG. Mapping electron paramagnetic resonance spin label conformations by the simulated scaling method. J Am Chem Soc. 2007;129:13840–6. doi: 10.1021/ja071404v. [DOI] [PubMed] [Google Scholar]
- 23.Fajer P. Conformational switching in muscle. Adv Exp Med Biol. 2004;547:61–80. doi: 10.1007/978-1-4419-8861-4_6. [DOI] [PubMed] [Google Scholar]
- 24.Fajer PG, Gyimesi M, Málnási-Csizmadia A, Bagshaw CR, Sen KI, Song L. Myosin cleft closure by double electron-electron resonance and dipolar EPR. J Phys Condens Matter. 2007;19:285208, 10. [Google Scholar]
- 25.Fisher AJ, Smith CA, Thoden JB, Smith R, Sutoh K, et al. X-ray structures of the myosin motor domain of Dictyostelium discoideum complexed with MgADP.BeFx and MgADP.AlF4. Biochemistry. 1995;34:8960–72. doi: 10.1021/bi00028a004. [DOI] [PubMed] [Google Scholar]
- 26.Forkey JN, Quinlan ME, Goldman YE. Measurement of single macromolecule orientation by total internal reflection fluorescence polarization microscopy. Biophys J. 2005;89:1261–71. doi: 10.1529/biophysj.104.053470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b.Forkey JN, Quinlan ME, Shaw MA, Corrie JE, Goldman YE. Three-dimensional structural dynamics of myosin V by single-molecule fluorescence polarization. Nature. 2003;422:399–404. doi: 10.1038/nature01529. Single-molecule polarized TIRF resolves myosin LC domain tilting transitions. [DOI] [PubMed] [Google Scholar]
- 27.Holmes KC. Muscle contraction. Novartis Found Symp. 1998;213:76–92. [PubMed] [Google Scholar]
- 28.Holmes KC, Angert I, Kull FJ, Jahn W, Schroder RR. Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature. 2003;425:423–7. doi: 10.1038/nature02005. [DOI] [PubMed] [Google Scholar]
- 29.Holmes KC, Schroder RR, Sweeney HL, Houdusse A. The structure of the rigor complex and its implications for the power stroke. Philos Trans R Soc London B Biol Sci. 2004;359:1819–28. doi: 10.1098/rstb.2004.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hopkins SC, Sabido-David C, Corrie JE, Irving M, Goldman YE. Fluorescence polarization transients from rhodamine isomers on the myosin regulatory light chain in skeletal muscle fibers. Biophys J. 1998;74:3093–110. doi: 10.1016/S0006-3495(98)78016-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hopkins SC, Sabido-David C, Van Der Heide UA, Ferguson RE, Brandmeier BD, et al. Orientation changes of the myosin light chain domain during filament sliding in active and rigor muscle. J Mol Biol. 2002;318:1275–91. doi: 10.1016/s0022-2836(02)00189-4. [DOI] [PubMed] [Google Scholar]
- 32.Houdusse A, Szent-Gyorgyi AG, Cohen C. Three conformational states of scallop myosin S1. Proc Natl Acad Sci USA. 2000;97:11238–43. doi: 10.1073/pnas.200376897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat Struct Biol. 2000;7:735–9. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 34.Jeschke G. Distance measurements in the nanometer range by pulse EPR. Chemphyschem. 2002;3:927–32. doi: 10.1002/1439-7641(20021115)3:11<927::AID-CPHC927>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 35.Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC. Atomic structure of the actin:DNase I complex. Nature. 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- 36.Klein JC, Burr AR, Svensson B, Kennedy DJ, Allingham J, et al. Actin-binding cleft closure in myosin II probed by site-directed spin labeling and pulsed EPR. Proc Natl Acad Sci USA. 2008;105:12867–72. doi: 10.1073/pnas.0802286105. DEER with multiple spin label pairs, used to detect structural changes in the myosin actin-binding cleft. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knowles AC, Ferguson RE, Brandmeier BD, Sun YB, Trentham DR, Irving M. Orientation of the essential light chain region of myosin in relaxed, active and rigor muscle. Biophys J. 2008;95:3882–91. doi: 10.1529/biophysj.108.131508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korman VL, Anderson SE, Prochniewicz E, Titus MA, Thomas DD. Structural dynamics of the actin-myosin interface by site-directed spectroscopy. J Mol Biol. 2006;356:1107–17. doi: 10.1016/j.jmb.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 39.Kouyama T, Mihashi K. Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur J Biochem. 1981;114:33–8. [PubMed] [Google Scholar]
- 40.Kozuka J, Yokota H, Arai Y, Ishii Y, Yanagida T. Dynamic polymorphism of single actin molecules in the actin filament. Nat Chem Biol. 2006;2:83–6. doi: 10.1038/nchembio763. Single-molecule FRET within actin reveals two structural states within the actin protomer that are in slow equilibrium and determine active interaction with myosin. [DOI] [PubMed] [Google Scholar]
- 41.LaConte LE, Baker JE, Thomas DD. Transient kinetics and mechanics of myosin’s force-generating rotation in muscle: resolution of millisecond rotational transitions in the spin-labeled myosin light-chain domain. Biochemistry. 2003;42:9797–803. doi: 10.1021/bi034288r. [DOI] [PubMed] [Google Scholar]
- 42.Lidke DS, Thomas DD. Coordination of the two heads of myosin during muscle contraction. Proc Natl Acad Sci USA. 2002;99:14801–6. doi: 10.1073/pnas.232161999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lombardi V, Piazzesi G, Reconditi M, Linari M, Lucii L, et al. X-ray diffraction studies of the contractile mechanism in single muscle fibres. Philos Trans R Soc London B Biol Sci. 2004;359:1883–93. doi: 10.1098/rstb.2004.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malnasi-Csizmadia A, Kovacs M, Woolley RJ, Botchway SW, Bagshaw CR. The dynamics of the relay loop tryptophan residue in the Dictyostelium myosin motor domain and the origin of spectroscopic signals. J Biol Chem. 2001;276:19483–90. doi: 10.1074/jbc.M010886200. [DOI] [PubMed] [Google Scholar]
- 45.Malnasi-Csizmadia A, Pearson DS, Kovacs M, Woolley RJ, Geeves MA, Bagshaw CR. Kinetic resolution of a conformational transition and the ATP hydrolysis step using relaxation methods with a Dictyostelium myosin II mutant containing a single tryptophan residue. Biochemistry. 2001;40:12727–37. doi: 10.1021/bi010963q. [DOI] [PubMed] [Google Scholar]
- 46.Malnasi-Csizmadia A, Woolley RJ, Bagshaw CR. Resolution of conformational states of Dictyostelium myosin II motor domain using tryptophan (W501) mutants: implications for the open-closed transition identified by crystallography. Biochemistry. 2000;39:16135–46. doi: 10.1021/bi001125j. [DOI] [PubMed] [Google Scholar]
- 47.Mazhari SM, Selser CT, Cremo CR. Novel sensors of the regulatory switch on the regulatory light chain of smooth muscle myosin. J Biol Chem. 2004;279:39905–14. doi: 10.1074/jbc.M407062200. [DOI] [PubMed] [Google Scholar]
- 48.Naber N, Purcell TJ, Pate E, Cooke R. Dynamics of the nucleotide pocket of myosin measured by spin-labeled nucleotides. Biophys J. 2007;92:172–84. doi: 10.1529/biophysj.106.090035. Spin-labeled nucleotide analogs demonstrate that open and closed states of the myosin nucleotide pocket are populated in actin-bound myosin.ADP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nelson WD, Blakely SE, Nesmelov YE, Thomas DD. Site-directed spin labeling reveals a conformational switch in the phosphorylation domain of smooth muscle myosin. Proc Natl Acad Sci USA. 2005;102:4000–5. doi: 10.1073/pnas.0401664102. Site-directed spin labeling shows that phosphorylation induces a disorder-to-order transition in the N-terminal domain of the RLC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nesmelov YE, Agafonov RV, Burr AR, Weber RT, Thomas DD. Structure and dynamics of the force-generating domain of myosin probed by multifrequency electron paramagnetic resonance. Biophys J. 2008;95:247–56. doi: 10.1529/biophysj.107.124305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palm T, Sale K, Brown L, Li H, Hambly B, Fajer PG. Intradomain distances in the regulatory domain of the myosin head in prepower and postpower stroke states: fluorescence energy transfer. Biochemistry. 1999;38:13026–34. doi: 10.1021/bi991164z. [DOI] [PubMed] [Google Scholar]
- 52.Prochniewicz E, Spakowicz DJ, Thomas D. Changes in actin structural transitions associated with oxidative inhibition of muscle contraction. Biochemistry. 2008;47:11811–17. doi: 10.1021/bi801080x. [AU: Please update. If not in press by April 2009, then remove from list and insert into text.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deleted in proof
- 54.Prochniewicz E, Walseth TF, Thomas DD. Structural dynamics of actin during active interaction with myosin: different effects of weakly and strongly bound myosin heads. Biochemistry. 2004;43:10642–52. doi: 10.1021/bi049914e. Phosphorescence shows that weak and strong binding of myosin have profoundly different effects on actin filament dynamics. [DOI] [PubMed] [Google Scholar]
- 55.Quinlan ME, Forkey JN, Goldman YE. Orientation of the myosin light chain region by single molecule total internal reflection fluorescence polarization microscopy. Biophys J. 2005;89:1132–42. doi: 10.1529/biophysj.104.053496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramachandran S, Thomas DD. Rotational dynamics of the regulatory light chain in scallop muscle detected by time-resolved phosphorescence anisotropy. Biochemistry. 1999;38:9097–104. doi: 10.1021/bi9902945. [DOI] [PubMed] [Google Scholar]
- 57.Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, et al. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 58.Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, et al. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261:50–8. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 59.Reshetnyak YK, Andreev OA, Borejdo J, Toptygin DD, Brand L, Burstein EA. The identification of tryptophan residues responsible for ATP-induced increase in intrinsic fluorescence of myosin subfragment 1. J Biomol Struct Dyn. 2000;18:113–25. doi: 10.1080/07391102.2000.10506651. [DOI] [PubMed] [Google Scholar]
- 60.Robertson CI, Gaffney DP, 2nd, Chrin LR, Berger CL. Structural rearrangements in the active site of smooth-muscle myosin. Biophys J. 2005;89:1882–92. doi: 10.1529/biophysj.105.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roopnarine O, Szent-Gyorgyi AG, Thomas DD. Microsecond rotational dynamics of spin-labeled myosin regulatory light chain induced by relaxation and contraction of scallop muscle. Biochemistry. 1998;37:14428–36. doi: 10.1021/bi9808363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Root DD, Stewart S, Xu J. Dynamic docking of myosin and actin observed with resonance energy transfer. Biochemistry. 2002;41:1786–94. doi: 10.1021/bi015869o. [DOI] [PubMed] [Google Scholar]
- 63.Rosenberg SA, Quinlan ME, Forkey JN, Goldman YE. Rotational motions of macromolecules by single-molecule fluorescence microscopy. Acc Chem Res. 2005;38:583–93. doi: 10.1021/ar040137k. [DOI] [PubMed] [Google Scholar]
- 64.Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nat Methods. 2008;5:507–16. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sabido-David C, Hopkins SC, Saraswat LD, Lowey S, Goldman YE, Irving M. Orientation changes of fluorescent probes at five sites on the myosin regulatory light chain during contraction of single skeletal muscle fibres. J Mol Biol. 1998;279:387–402. doi: 10.1006/jmbi.1998.1771. [DOI] [PubMed] [Google Scholar]
- 66.Saraswat LD, Lowey S. Subunit interactions within an expressed regulatory domain of chicken skeletal myosin. Location of the NH2 terminus of the regulatory light chain by fluorescence resonance energy transfer. J Biol Chem. 1998;273:17671–9. doi: 10.1074/jbc.273.28.17671. [DOI] [PubMed] [Google Scholar]
- 67.Schroder RR, Manstein DJ, Jahn W, Holden H, Rayment I, et al. Three-dimensional atomic model of F-actin decorated with Dictyostelium myosin S1. Nature. 1993;364:171–4. doi: 10.1038/364171a0. [DOI] [PubMed] [Google Scholar]
- 68.Selvin PR. Principles and biophysical applications of lanthanide-based probes. Annu Rev Biophys Biomol Struct. 2002;31:275–302. doi: 10.1146/annurev.biophys.31.101101.140927. [DOI] [PubMed] [Google Scholar]
- 69.Shepard AA, Dumka D, Akopova I, Talent J, Borejdo J. Simultaneous measurement of rotations of myosin, actin and ADP in a contracting skeletal muscle fiber. J Muscle Res Cell Motil. 2004;25:549–57. doi: 10.1007/s10974-004-5073-6. [DOI] [PubMed] [Google Scholar]
- 70.Shih WM, Gryczynski Z, Lakowicz JR, Spudich JA. A FRET-based sensor reveals large ATP hydrolysis-induced conformational changes and three distinct states of the molecular motor myosin. Cell. 2000;102:683–94. doi: 10.1016/s0092-8674(00)00090-8. Frequency-domain FRET between CD and LCD of myosin S1 resolves multiple lever arm angles. [DOI] [PubMed] [Google Scholar]
- 71.Steinhoff HJ. Inter- and intramolecular distances determined by EPR spectroscopy and site-directed spin labeling reveal protein-protein and protein-oligonucleotide interaction. Biol Chem. 2004;385:913–20. doi: 10.1515/BC.2004.119. [DOI] [PubMed] [Google Scholar]
- 71b.Surek JT, Thomas DD. A paramagnetic molecular voltmeter. J Magn Reson. 2007;190:7–25. doi: 10.1016/j.jmr.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Suzuki Y, Yasunaga T, Ohkura R, Wakabayashi T, Sutoh K. Swing of the lever arm of a myosin motor at the isomerization and phosphate-release steps. Nature. 1998;396:380–3. doi: 10.1038/24640. [DOI] [PubMed] [Google Scholar]
- 73.Thomas DD. Spectroscopic probes of muscle cross-bridge rotation. Annu Rev Physiol. 1987;49:691–709. doi: 10.1146/annurev.ph.49.030187.003355. [DOI] [PubMed] [Google Scholar]
- 74.Thomas DD, Cooke R. Orientation of spin-labeled myosin heads in glycerinated muscle fibers. Biophys J. 1980;32:891–906. doi: 10.1016/S0006-3495(80)85024-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thomas DD, Prochniewicz E, Roopnarine O. Changes in actin and myosin structural dynamics due to their weak and strong interactions. Results Probl Cell Differ. 2002;36:7–19. doi: 10.1007/978-3-540-46558-4_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thompson AR, Naber N, Wilson C, Cooke R, Thomas DD. Structural dynamics of the Actomyosin complex probed by a bifunctional spin label that crosslinks SH1 and SH2. Biophys J. 2008;95:5238–46. doi: 10.1529/biophysj.108.138982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van Duffelen M, Chrin LR, Berger CL. Kinetics of structural changes in the relay loop and SH3 domain of myosin. Biochem Biophys Res Commun. 2005;329:563–72. doi: 10.1016/j.bbrc.2005.01.152. [DOI] [PubMed] [Google Scholar]
- 78.Volkmann N, Hanein D, Ouyang G, Trybus KM, DeRosier DJ, Lowey S. Evidence for cleft closure in actomyosin upon ADP release. Nat Struct Biol. 2000;7:1147–55. doi: 10.1038/82008. [DOI] [PubMed] [Google Scholar]
- 79.Volkmann N, Ouyang G, Trybus KM, DeRosier DJ, Lowey S, Hanein D. Myosin isoforms show unique conformations in the actin-bound state. Proc Natl Acad Sci USA. 2003;100:3227–32. doi: 10.1073/pnas.0536510100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Warshaw DM, Hayes E, Gaffney D, Lauzon AM, Wu J, et al. Myosin conformational states determined by single fluorophore polarization. Proc Natl Acad Sci USA. 1998;95:8034–9. doi: 10.1073/pnas.95.14.8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Werber MM, Szent-Gyorgyi AG, Fasman GD. Fluorescence studies on heavy meromyosin-substrate interaction. Biochemistry. 1972;11:2872–83. doi: 10.1021/bi00765a021. [DOI] [PubMed] [Google Scholar]
- 82.Wolff-Long VL, Tao T, Lowey S. Proximity relationships between engineered cysteine residues in chicken skeletal myosin regulatory light chain. A resonance energy transfer study. J Biol Chem. 1995;270:31111–8. doi: 10.1074/jbc.270.52.31111. [DOI] [PubMed] [Google Scholar]
- 83.Xiao M, Li H, Snyder GE, Cooke R, Yount RG, Selvin PR. Conformational changes between the active-site and regulatory light chain of myosin as determined by luminescence resonance energy transfer: the effect of nucleotides and actin. Proc Natl Acad Sci USA. 1998;95:15309–14. doi: 10.1073/pnas.95.26.15309. Developed LRET to detect lever arm motion in skeletal myosin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xiao M, Reifenberger JG, Wells AL, Baldacchino C, Chen LQ, et al. An actin-dependent conformational change in myosin. Nat Struct Biol. 2003;10:402–8. doi: 10.1038/nsb916. [DOI] [PubMed] [Google Scholar]
- 85.Xu J, Root DD. Domain motion between the regulatory light chain and the nucleotide site in skeletal myosin. J Struct Biol. 1998;123:150–61. doi: 10.1006/jsbi.1998.4023. [DOI] [PubMed] [Google Scholar]
- 86.Yanagida T. Muscle contraction mechanism based on actin filament rotation. Adv Exp Med Biol. 2007;592:359–67. doi: 10.1007/978-4-431-38453-3_30. [DOI] [PubMed] [Google Scholar]
- 87.Yengo CM, Berger CL. Fluorescence resonance energy transfer in acto-myosin complexes. Results Probl Cell Differ. 2002;36:21–30. doi: 10.1007/978-3-540-46558-4_3. [DOI] [PubMed] [Google Scholar]
- 88.Yengo CM, Chrin LR, Berger CL. Interaction of myosin LYS-553 with the C-terminus and DNase I-binding loop of actin examined by fluorescence resonance energy transfer. J Struct Biol. 2000;131:187–96. doi: 10.1006/jsbi.2000.4296. [DOI] [PubMed] [Google Scholar]
- 89.Yengo CM, Chrin LR, Rovner AS, Berger CL. Tryptophan 512 is sensitive to conformational changes in the rigid relay loop of smooth muscle myosin during the MgATPase cycle. J Biol Chem. 2000;275:25481–7. doi: 10.1074/jbc.M002910200. Engineered single tryptophan mutant in smooth muscle myosin. [DOI] [PubMed] [Google Scholar]
- 90.Yengo CM, De La Cruz EM, Chrin LR, Gaffney DP, 2nd, Berger CL. Actin-induced closure of the actin-binding cleft of smooth muscle myosin. J Biol Chem. 2002;277:24114–9. doi: 10.1074/jbc.M111253200. [DOI] [PubMed] [Google Scholar]
- 91.Yengo CM, Fagnant PM, Chrin L, Rovner AS, Berger CL. Smooth muscle myosin mutants containing a single tryptophan reveal molecular interactions at the actin-binding interface. Proc Natl Acad Sci USA. 1998;95:12944–9. doi: 10.1073/pnas.95.22.12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-overhand: single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–5. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- 93.Zeng W, Conibear PB, Dickens JL, Cowie RA, Wakelin S, et al. Dynamics of actomyosin interactions in relation to the cross-bridge cycle. Philos Trans R Soc London B Biol Sci. 2004;359:1843–55. doi: 10.1098/rstb.2004.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002;3:906–18. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]