

1 Introduction
The Working Group ‘Overall Blood Supply Strategy with Regard to Variant Creutzfeldt-Jakob Disease (vCJD)’ was formed in 2001 by request of the German Federal Ministry of Health in a letter dated January 26, 2001, consisting of staffs from the Paul-Ehrlich-Institut, the Robert Koch-Institut and the Federal Ministry of Health as well as external experts. On August 17, 2001 the report prepared by this Working Group was submitted to the Ministry of Health and published after a discussion in the National Advisory Committee ‘Blood’ (Arbeitskreis Blut) as a brochure and online [1]. After reports were published in the UK in 2004 on the transmission of the vCJD pathogen by blood transfusion, the Working Group became active again, preparing an update of the report on April 13, 2004 which was published in scientific journals [2, 3]. One of the conclusions of this report was: ‘The secondary route of infection by blood could largely be stopped as soon as a suitable screening test becomes available. No such test is currently available nor is its availability foreseeable. Providing resources for developing suitable test methods should therefore be treated with high priority.’ Early in 2008 the Working Group has started meeting again with slightly different members in order to assess progress made in the field of diagnostics, particularly with regard to the development of screening assays for blood donations. Another task has been to critically evaluate the requirements regarding an implementation of such test assays in the present basic framework.
The previous report of the Working Group has only been published a short time ago [2], therefore the present statement will not provide an extensive exploration of the whole set of vCJD problems again. Here we rather specifically discuss significant new aspects in the context of developing diagnostic assays. In order to illustrate the scope of the clinical difficulties, we give a brief survey on the epidemiology of vCJD and a presentation of how to clinically and diagnostically differentiate recently published borderline cases. Because the most recent development of assays has predominantly targeted the detection of pathological prion protein (PrP), new aspects of the pathophysiological impact of prions and their detectability are discussed.
Tests currently under development as well as recently published methods are presented in brief, without aiming for completeness in this treatise or for an elaborate in-depth assessment. The essential focus is rather on presenting and debating the general framework regarding the implementation of such assays. Relevant are the currently prevailing premises and the present regulation on the approval of the market access of screening tests for vCJD in Europe. Before the introduction of their wide-spread application in the screening of blood donors, the performance of the test assays must be thoroughly evaluated regarding their sensitivity and particularly their specificity. Preconditions of such evaluation studies in the context of implementing European regulations into the German legal system are discussed in detail. Ethical issues are particularly challenging, especially informing test persons / blood donors about a vCJD diagnosis that is still fatal, while a reliable confirmation test is lacking. At last, practical and technical requirements will be presented that must be considered on implementation.
Without putting the general aim of effectively screening of blood donors for vCJD into question, the present report of the Working Group points out considerable difficulties when implementing such assays under the prevailing preconditions. It should provide impetuses for the advancement of the handling of the assays by the regulatory bodies and for decision-making regarding the pressing ethical issues. These considerations might also be valuable for other diseases that will come into the focus in the future.
2 Present Epidemiological Data on vCJD, Clinical and Diagnostic Differentiation from Sporadic CJD
In the early 1920s human spongiform encephalopathies were described by H.G. Creutzfeldt [4] and A. Jakob [5]. In the 1930s it could be demonstrated that scrapie, a spongiform encephalopathy of sheep and goats, is a transmissible neurodegenerative disease. Histological investigations of the brain of animals and humans who had contracted transmissible spongiform encephalopathy (TSE), using routine staining methods like hematoxylin/eosin staining, showed spongious alterations, loss of nerve cells, and in some cases amyloid accumulations. The degree of the morphological changes can vary in different regions of the brain. Therefore the diagnostic standard procedure includes the identification of the pathogen PrPTSE in the brain.
2.1 Epidem io logy of v CJD
This form of the disease is associated etiologically and pathoge-netically with bovine spongiform encephalopathy (BSE). So far, more than 200 cases of vCJD have been reported world-wide. These patients are considerably younger than those affected by the sporadic form of CJD, and the course of the disease is prolonged (table 1). Psychiatric abnormalities are predominant (mostly in the shape of depression or psychosis) that can last for several months without any neurological signs. Later there are painful dysesthesia and walking ataxia while dementia appears only late in the course of the disease. In vCJD, the abnormal PrP can usually be also detected in the peripheral lymphatic tissues (appendix, tonsils, lymph nodes) (table 1). However, the abnormal PrP has also been found in the spleen and in muscle tissue in the sporadic form of CJD [6]. Transmissions of vCJD by blood transfusions were observed in the UK (detailed discussion in [2]). It can be considered as favorable that so far there have been no signs of an adaption of the pathogen after transmission via blood [7]. Table 2 gives a survey of the vCJD cases reported to date.
Table 1.
Typical characteristics of vCJD compared to sporadic CJD
| vCJD | sporadic CJD | |
|---|---|---|
| Mean age at death | 30 years | 65 years |
| Mean duration of the disease | 14 months | 6 months |
| Clinical symptoms at the onset of disease | dysesthesia, behavioral changes | memory disorder, ataxia |
| EEG | non-typical | 66% PSWCs |
| Detection of 14-3-3 protein in cerebrospinal fluid | 50% | 94% |
| Magnetic resonance tomography | hyperintensities in the thalamus, 'pulvinar sign' | hyperintensities in the basal ganglia |
| Codon 129 genotype | MM | MM, MV, VV |
| Lymphatic system | PrPTSE | no PrPTSE |
| Neuropathologic characteristics | 'florid plaques' | variable, no 'florid plaques' |
PSWCs = Periodic sharp and slow wave complexes.
Table 2.
Presently known vCJD cases, as of August 19, 2008 [8]
| Country | Total number of cases | Secondary transmission (blood) | No residence in UK > 6 months |
|---|---|---|---|
| UK | 166 | 3 (+1∗) | |
| France | 23 | 22 | |
| Republic of Ireland | 4 | 2 | |
| Italy | 1 | 1 | |
| USA | 3 | 1 | |
| Canada | 1 | ||
| Saudi Arabia | 1 | 1 | |
| Japan | 1 | 1 | |
| The Netherlands | 2 | 2 | |
| Portugal | 2 | 2 | |
| Spain | 4 |
4 |
|
| Total | 208 | 3 (+ 1 ∗) | 36 |
1 case of a probable transmission of vCJD, recipient has died from other causes.
Based on a model calculation [9], in the previous report [2] the Working Group has come to the conclusion that in Germany, even in worst-case scenarios, blood transfusions would not significantly promote the possible incidence of vCJD. Therefore, the report argued against an exclusion of transfusion recipients from donating blood. Mathematic modeling studies for the UK region [10] also assume that a perpetuation of vCJD is conceivable, but improbable. According to actual calculations, the group at Imperial College in London does not consider blood transfusions to have any significant influence on the epidemiology of vCJD [11].
2.2 Clinical and Pathological Classification, Phenotype Diversity of CJD
It is crucial to be able to differentiate vCJD which has evidentially been transmitted by blood from other forms of human TSE for which there are as yet no indications for a transmissibility by blood. To illustrate this difficulty induced by the diversity in phenotype, some cases will be discussed here in detail. Recently, Mead et al. [12] have reported a CJD in a 39-year-old female patient who first consulted a physician in January of 1999 because of visual disturbance. She then developed cognitive impairment, halting gait, motility disorder of the eye, dysarthria, stereotypical movements and myoclonia as well as finally progressive ataxia and dementia. 14 months after the onset of the disease the patient died. The codon 129 genotype was homozygous for valine. In this subtype (most often molecular VV2 subtype) ataxia is prominent while there are also signs of cognitive impairment, tonus abnormalities, and myoclonia. Typical characteristics are the lack of periodic sharp and slow wave complexes, as also happened in this patient, as well as the detection of the 14-3-3 proteins in the cerebrospinal fluid. Magnetic resonance tomography (MRT) is compatible with this subtype in which often a symmetrical affection of the basal ganglia without cortical involvement is found (unpublished data). A report in the journal New Scientist [13] cites the first author, Simon Mead, as follows: ‘The consensus is that it's more likely sporadic, but we can't conclude either way until further experiments are done.’ The overall course of disease is compatible with sporadic CJD, as observed in molecular VV2 subtype (compare table 3), regarding the development of clinical symptoms, the results of additional technical investigations, and the duration of the disease.
Table 3.
Phenotypical CJD characteristics
| Molecular subtype | Clinical features | Neuropathology | PrP immunohistochemistry | |
|---|---|---|---|---|
| Frequent | MM1/MV1 |
dementia, cortical anopsy, myoclonia, short duration of disease (approximately 4 months) |
pronounced damage of the occipital cortex, PrP accumulations of the synaptic type |
 |
| MV2 |
ataxia, dementia, extrapyramidal motion disorder, long duration of disease (approximately 18 months) |
focal damage of the cortex, amyloid (kuru) plaques, focal plaque-like PrP accumulations |
 |
|
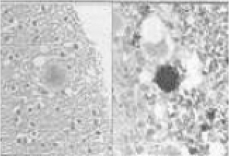
| VV2 | ataxia at onset, dementia at a late stage, average duration of disease (approximately 7 months) | pronounced damage of the subcortical structures as well as the brainstem, often spongiosis only located in the deeper cortical layers, plaque-like as well as perineuronal PrP accumulations | 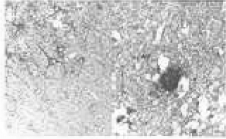 |
|
| Rare | MM2 thalamic (sFI) |
insomnia, dysautonomia, at a late stage ataxia and cognitive disturbance |
atrophy of the thalamus and the nucleus olivaris, lack of spongiosis possible |
 |
| MM2 cortical |
dementia for several months |
large confluent vacuoles with perivacuolar PrP accumulations |
 |
|
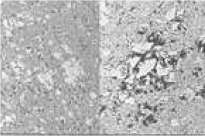
| VV1 | dementia at onset, later ataxia and extrapyramidal disorders | spongiosis, gliosis and loss of innervation of the cortical structures while omitting the brainstem and the cerebellum |  |
|
| vCJD | MM2b | psychiatric, dysaesthesia, ataxia, later dementia | spongiosis, gliosis and loss of innervation as well as 'florid' plaques | 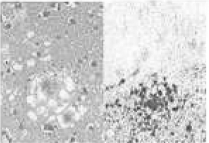 |
Gambetti et al. [14] have recently described a novel degenerative brain disease in humans that is associated with abnormal PrP. The disease occurred in patients of a mean age of 62 years and is clinically characterized by rapidly progredient cognitive disturbances as well as movement disorder. The clinical phase has a mean duration of 20 months. Brain tissue obtained by autopsy (10 patients) or by biopsy (1 patient) was investigated histologically and showed spongiform degeneration, astrogliosis and in some of the cases ‘micro-plaque’-like accumulations of the PrP and, after immunohistochemical staining, pronounced granular accumulations of the PrP. On the biochemical level there was comparatively little abnormal PrP that was less highly aggregated and the proteinase K(PK)-sensitive fraction of which was considerably larger than that in sporadic CJD. In addition, during enzymatic proteolysis the protein forms different protease-resistant cleavage products than those in the case of the well-known forms of CJD.
One year earlier a new subtype of CJD had been reported in Germany [15] that showed similarities to the above-mentioned cases described by Gambetti et al. [14]. The reports prompted a search for similar patients within the cohort of the German national reference center. So far, this search was unsuccessful. Regardless of the above-described cases that are difficult to classify, the molecular classification of sporadic CJD is based on the codon 129 genotype of the PrP gene (homozygous for methionine (MM) or valine (VV), or heterozygous (MV)) and on the biochemical properties of the abnormal PrP (PrPTSE type 1 or 2, differentiated by size and migration rate of the unglyco-sylized PrP band in the immunoblot). The molecular subtypes of the disease are accompanied by distinct clinical and neuropathological phenotypes of CJD (table 3).
MRT proves to be a diagnostic technique by which it is possible to differentiate individual CJD subtypes from each other and from vCJD, even before death. In addition, it is instrumental in identifying atypical courses of the disease at a very early stage (fig. 1a, 1b).
Fig. 1.

Patterns of lesions by MRT in sporadic CJD – differences in the molecular subtypes (weighting of diffusion). b Alterations in MRT in vCJD -increased signal in the posterior thalamus (FLAIR [fluid attenuated inversion recovery] imaging). The image has kindly been provided by Dr D. Collie,
2.3 Typing of PrPTSE
Cerebellar tissue of the patient that was homozygous for valine at codon 129 of the PRNP gene [12] was investigated by Western Blot in order to type PrPTSE. Differences to well-known PrPTSE types of sporadic CJD isoforms (fig. 2) became apparent. The relative proportion and electrophoretic migration pattern of the di-, mono- and unglycosylated bands of the PK-digested PrPTSE were more similar to the PrPTSE type found in vCJD. In contrast to vCJD-associated PrPTSE, the addition of EDTA (a chelating agent binding copper and zinc ions) to the PK digestion of the patient's PrPTSE caused a further slight reduction in the molecular weight of the three protein bands. From the results of their Western Blot investigations the authors concluded that the pathological PrP of the patient investigated represents a novel PrPTSE type that is different from PrPTSE types so far observed in sporadic CJD cases. Furthermore the three-dimensional structure differs from that displayed in vCJD-associated PrPTSE (from patients with MM at the PRNP codon 129). The authors classify the newly described PrPTSE type as ‘type 7’ in their classification system which is being developed over the past years. However, most experts prefer PrPTSE typing according to Parchi et al. [16]. According to this system, there are two basic PrPTSE types associated with sporadic CJD and one PrPTSE type associated with vCJD. Parchi et al. [16] were able to show that even minimal variations of the experimental conditions (e.g. the pH value) in PK digestion have considerable impact on the size of the resulting PrPTSE fragments and may possibly generate artificial PrPTSE types. In order to be able to assess the results by Mead et al. [12], it would therefore be helpful if the investigations could be reproduced using the method by Parchi et al. [16] under exactly defined pH and buffer conditions. On the one hand, by such investigations the slight difference in molecular weight could be additionally corroborated which is observed after EDTA treatment compared to the vCJD-associated PrPTSE type. On the other hand, this might also lead to a better understanding of the results shown in figure 4c in [12]. In this figure the PrPTSE of the patient after EDTA treatment and PK conspicuously resembles PrPTSE type 3 rather than the new type 7 – unlike the one shown in lane 2 of figure 4b in [12]. It is not possible to deduce from the published Western Blot data of a single case whether the newly described PrPTSE type is in any way connected with the BSE agent. Only transmission experiments in transgenic or normal mice might shed more light on a more precise characterization of the agent strain, allowing a continued sensitive epidemiological surveillance of the CJD trends.
Fig. 2.

PrPTSE types 1 and 2 in isoforms of sporadic CJD in Western Blot.
3 New Aspects of the Pathophysiology and the Diagnostics of Prions
In 1985 the PrP was identified as a gene product [17]. Since it is also found in healthy organisms, it is essential to be able to differentiate between the normal or cellular PrP, called PrPC, and the pathogenic infectious isoform, called PrPTSE in this report. Both forms of PrP, PrPC and PrPTSE, display the same composition of amino acids [18] and glycosylation pattern as well as the same lipid anchor because both proteins are coded by the same gene. The differences between PrPC and PrPTSE lie in their structural and physicochemical properties that are assumed to be responsible for infectivity. These properties permit the development of diagnostic assays for PrPTSE:
-
–
Isolated PrPC is water soluble, whereas PrPTSE cannot be dissolved (without losing its infectivity).
-
–
PrPC predominantly contains a-helices as its secondary structural element, whereas PrPTSE displays a high proportion of β-sheet structures [19, 20]. Conversion of PrPC to PrPTSE renders the PrP infectious.
-
–
PK treatment rapidly digests PrPC to oligopeptides, whereas in the case of PrPTSE PK treatment leads to the cleaving off of an N-terminal part of the PrP that is 68 amino acids in length. The cleavage site differs among the various prion strains and is located approximately between amino acid positions 87 and 107. The remaining molecule has a (strain-specific) molecular weight of around 27–30 kDa which is the reason why it is also called PrP27-30. For hours it resists further PK digestion which is true for almost every prion strain. Therefore, the presence or absence of pathological PK-resistant PrPTSE in tissue homogenates can be detected by Western Blot, for example. The PK concentration in these experiments must be high enough for a complete digestion of PrPC.
-
–
High molecular PrPTSE aggregates can also be detected in infected tissues, e.g. as amyloid plaques. In tissue homogenates aggregates can be separated from cellular, nonaggregated PrPC, i.e. by centrifugation, and be enriched in the sediment. Treatment with e.g. phosphotungstic acid can increase sedimentation [21].
Using immunohistochemistry, the misfolded PrPTSE is detected not only in a variety of plaques (kuru plaque, multicentric and florid plaques) but also in additional structures. The precondition for an immunohistochemical detection of PrPTSE is a denaturing pretreatment of the tissue sections samples (e.g. formic acid or guanidinium thiocyanate treatment) so that cellular PrPC can no longer be displayed. Additional treatment of the histological sections with PK can lead to an even more pronounced visibility of PrPTSE. The immunohistochemical methods are based on antibodies against the PrP, either in the form of polyclonal sera or of monoclonal antibodies (MABs); the latter are almost exclusively used today. As mentioned above, the composition of amino acids and the glycosylation pattern of PrPC and PrPTSE are identical, therefore no immunological or inflammatory reactions can be detected, either during the incubation stage or after clinical symptoms have developed. This explains the lack of serological detection methods like for other pathogens. Preferably knock-out mice that do not express PrP, so-called PrP0/0 mice, are used for the development of MABs. The best-known MABs are 3F4 [22] (reaction with human and hamster PrP), 13A5 [23] (reaction with hamster PrP) and 6H4 [24] (reaction with human, bovine, ovine, mouse and hamster PrP). These MABs bind to both PrPC and (denatured) PrPTSE so that a differentiation between cellular and pathogenic isoforms of the PrP becomes possible only after using additional diagnostic methods, e.g. PK digestion of PrPC as used for the Western Blot. So far, only few MABs have been described that selectively recognize PrPTSE due to the reaction with a conformational epitope. One of these is MAB 15B3 [24], a MAB of the IgM class. Above all, this MAB is used for precipitation and concentration of PrPTSE, while no additional properties of this MAB have yet been described. In the meantime, further conformation-specific MABs have been described [25,26,27]. Several MABs that were described to be PrPTSE-specific apparently react with aggregated infectious and noninfectious PrP [28]. Binding of PrPTSE can be achieved by means of MABs coupled to ‘microbeads‘; apparently MAB specificity does not play a role here [29].
In addition to detecting PrPs by antibody-binding reactions, the (differences in the) glycosylation pattern can be used for discriminating prion strains according to their lectin-binding properties [30]. A PK treatment is required for such a differentiation because the binding to lectin in sporadic CJD is mainly detectable in the non-cleaved PrP while in vCJD the PK-treated PrP also shows a strong reaction.
The correlation of infectivity with an immunohistochemical detection of PrPTSE in animal experiments (generally using in-tracerebral application of brain homogenate or homogenates of other tissues such as spleen) has led to the conclusion that immunohistochemical proof of PrPTSE in fact confirms a TSE disease. Differentiation of the cellular (nonpathogenic) isoform PrPC from the (pathogenic) isoform PrPTSE is crucial since currently this represents the only diagnostic marker. It must be kept in mind that, in the case of a TSE disease, PrPTSE must be specifically identified in a background of PrPC present in a concentration that is several magnitudes higher. Several methods that are described below are under development which should allow differentiating of PrPC from PrPTSE for diagnostic purposes.
So far, the established methods for diagnosing a TSE disease like CJD or vCJD are based on either immunohistopathological investigations or Western Blotting. In order to be able to identify PrPTSE by these methods, denaturing and/or proteolytic cleavage of PrPC are required. The presently widely used conformation-dependent immunoassay (CDI) [21] represents the only method to specifically identify PrPTSE even in a background of high concentrations of PrPC. With this method even PK-sensitive PrPTSE could be detected in the brain of patients with sporadic CJD [31]. However, these methods are restricted to post-mortem diagnostics or to tissue biopsies.
Resistance of pathogenic PrPTSE against PK digestion has long been known not to be absolute but rather dependent on the prion strain, too. For example, strain DROWSY (DY) is sensitive to PK digestion, in contrast to other strains including strain HYPER (HY) [32]. DY and HY are prion strains that cause transmissible encephalopathy of the mink (TME) and that have been further passaged in hamsters. As has been observed in strain DY, infectivity of PrPTSE is not associated with PK resistance of the PrP. Therefore some authors also use the terms sPrPTSE (PK-sensitive PrPTSE) and rPrPTSE (PK-resistant PrPTSE). Investigations of brain samples from patients with CJD have shown that more than 80% of total PrPTSE is PK-resistant [31]. Additional investigations in mice have confirmed an infectivity that is associated with PK-sensitive PrPTSE [33]. These investigations have used prion strains in which only small amounts of PrPTSE could be demonstrated by immunohistochemistry. Here a high infectivity titer was found (more than 107 infectious units/ g of brain homogenate), but no PK-resistant PrPTSE. However, it must be taken into consideration that the sensitivity of the Western Blot was comparatively low: the detection limit was 25 μg PrPTSE/g of wet weight.
In case PrPTSE isolated from brain homogenate is purified by differential centrifugation, and the supernatant is investigated for infectivity after high-speed centrifugation (220,000 × g for 30 min), only a minimal amount (if any) of PrPTSE can be demonstrated by Western Blot, but high infectivity titers. In this case also, infectivity seems to be associated mainly with PK-sensitive PrPTSE (an at least 200- to 1,000-fold higher infectivity titer than expected from the biochemical signal (Western Blot) has been described [34]). It is noteworthy that the low PrPTSE signal in Western Blot could be precipitated quantitatively with phosphotungstic acid, together with the whole amount of infectivity of the sample. This precipitation step can also be usefully applied in precipitating PK-resistant PrPTSE [21, 35]. It remains to be clarified to what extent this PrP preparation, present in the high-speed centrifugation supernatant, consists (only) of low molecular PrPTSE aggregates showing a very high specific infectivity. For example, it was reported that oligomers consisting of 14–28 PrPTSE molecules exhibit the highest infectivity. The molecular weight of these oligomers ranges between 300 and 600 kDa, and their size is between 17 and 27 nm. Oligomers consisting of as little as 5 PrPTSE molecules or less do not seem to be infectious [36].
Endogenous infectivity in the blood of experimentally infected animals can be demonstrated not only in the clinical but already in the preclinical stage of a TSE disease [37,38,39,40,41]. At least part of this infectivity is PK-sensitive [42]. An incubation of the endogenous infectivity in plasma with different concentrations of PK (100 and 500 μg) reduces the infectivity titer from 20.6 in the control without PK down to 8.4 and 5.2 infectious units/ml, respectively. However, it must be stated that the PK concentrations investigated were insufficient for a complete digestion of plasma proteins. Therefore, it cannot be excluded that a PK concentration sufficiently high for a complete proteolysis of plasma proteins would also have completely digested the infectivity in the plasma samples.
As mentioned above, to some extent infectivity can be PK-sen-sitive. Therefore, it must be pointed out that diagnostic assays using PK digestion for differentiation might miss part of the infectious PrPTSE and might thus prevent correct diagnostic conclusions.
4 Development of Diagnostic Assays
In statements regarding measures to prevent the transmission of vCJD via blood and blood products, various countries have addressed the necessity of developing diagnostic assays for the identification of vCJD infectivity in the blood. For example, the Working Group ‘Overall Blood Supply Strategy with Regard to Variant Creutzfeldt-Jakob Disease (vCJD)’ in their statement dated April 13, 2006 [2] pointed out that the development of screening assays was one of the key issues in preventing possible transmissions of vCJD by blood products, and in initiating therapeutic measures – supposed they would be available – at an early stage of infection. If a direct identification of vCJD-in-fected blood donors by screening assays was to become possible already in the incubation period, testing of donations would be particularly superior to any indirect measures that lack specificity, such as the prophylactic exclusion of groups of donors with an elevated risk in their history.
The major difficulty in developing such assays is the very low concentration of PrPTSE that is to be expected, in a background of the physiological PrPC that is present at an approximately 10,000-fold surplus. The analytical sensitivity of screening assays that seem to be appropriate for the detection of PrPTSE in serum or plasma must be far lower than 1 pg/ml PrPTSE [2]. Assuming that in an infected brain one infectious unit corresponds to approximately 0.01–1.0 μg, the required sensitivity of the assay would have to correspond to approximately 1–100 nl of a 10% brain homogenate [43]. Additional requirements are high specificity, robustness as well as applicability in blood donation testing.
The UK National Institute for Biological Standards and Control (NIBSC) provides a plasma panel containing variable amounts of infectious and noninfectious material from the brain or spleen of patients with vCJD (NIBSC CJD Research Centre, Plasma Panel, Reference code NHPM0/0015: Normal Human Plasma spiked with vCJD Ctrl Brain/Spleen, 22 tubes, concentration range). This as well as other panels have already been used in a comparative assessment of tests under development. In 2007 Phil Minor has reported the results of an external quality assessment involving seven laboratories using eight test assays. Four out of eight assays were able to detect <100 nl of a 10% brain homogenate containing the vCJD agent while three other assays detected <300 nl [43]. At a hearing of the DG Enterprise (Directorate General Enterprise and Industry) on October 27, 2007 in Brussels a company announced that they were aiming to obtain the CE marking for a test assay still in 2008 [44].
In the context of an epidemiological investigation, the British health authorities stated on February 6, 2007 their intention to test 60,000 plasma samples [45]. Companies were requested to provide assays for these investigations. As a first step in selecting an assay for these investigations, the comparative assessment of a plasma panel was performed by an independent laboratory. While the comparative assessment has been finished, the epidemiological investigation has not yet started.
Because the development of diagnostic assays is progressing considerably, it has become necessary to clearly determine which specific requirements should be made on a test able to detect vCJD in blood and which materials could be provided to characterize such an assay.
Currently various diagnostic assays are being developed, using different approaches. Such methods are based e.g. on binding to specific ligands, on the application of antibodies, on the detection or induction of conformation changes as well as on PCR-like protein amplification. Some of these assays will be briefly presented in the following.
4.1 Conformation-Dependent Immunoassay (CDI)
Detection of PrPTSE by CDI (originating from S. Prusiner's laboratory) uses the approach of ‘epitope exposure’ [21, 31]: defined epitopes that are at first inaccessible in the native PrPTSE are exposed by denaturation, followed by detection using a Europium-labeled antibody. Last year, at the conference ‘Prion 2007’ in Edinburgh, J. Safar reported that this assay was able to detect PrPTSE in LDL (low density lipoprotein) fractions in plasma from patients with sporadic CJD. For 20 patients with sCJD a sensitivity of the assay of 90% was found, while specificity was 98 and 95% using control samples from 103 normal blood donors and from 20 donors with dementia of other etiology (like e.g. Alzheimer's disease), respectively.
4.2 Misfolded Protein Diagnostic (MPD) Assay
To specifically detect PrPTSE, this assay employs a pyrene-la-beled palindromic sequence of prion peptides. As their characteristic molecular structural element, pyrene and its derivatives possess a conjugated aromatic system of four fused benzene rings. They are used among other things as dyes and as molecular probes in fluorescence spectroscopy. A palindrome is a row of signs that, on reading forward as well as backward, result in the same sequence of signs. The peptide employed in the MPD assay undergoes a cascade of coil to β-sheet conversion in the presence of PrPTSE, accompanied by a measurable fluorescence shift of the dye. In a blinded study using the MPD assay it was possible to detect PrPTSE in the plasma of 5 patients with sporadic CJD while the assay gave negative results for the plasma from 5 normal donors [46].
4.3 Epitope Protection Assay
This assay is based on the modification of amino acids of the PrP by reaction with free radicals that are released by chemical reactions. This results in the destruction of specific epitopes on the surface that can no longer be recognized by antibodies. Because the pathogenic PrPTSE is present in an aggregated form, part of the protein is protected against reaction with free radicals. Epitopes are thus preserved that can then react with monoclonal antibodies which allows to detect PrPTSE after disintegration of the aggregates in the immunoassay. The assay employs magnetic beads loaded with antibodies and is supposed to be able to detect less than 10 fg recombinant PrPTSE in the sample.
4.4 PMCA-Based Detection of PrPTSE
In 2001 Soto and colleagues [47] have published the ‘Protein Misfolding Cyclic Amplification’ (PMCA) as a method to amplify pathologically aggregated PrP in vitro. PrPTSE, the disease-associated form of the PrP, is first incubated in a surplus of normal, nonaggregated PrPC. Seeded nucleus-induced polymerization integrates PrP into available PrPTSE aggregates, causing them to virtually ‘grow’. Using sonication, these enlarged PrPTSE aggregates are then fragmented into smaller parts which in turn can function again as ‘matrices’ or ‘nuclei’ in another PMCA cycle. It is unclear whether such reaction products are infectious; in case they are, special safety measures would be required.
Recently, Atarashi et al. [48, 49] have reported to have enhanced the PMCA technique resulting in the QUIC Assay which is supposed to allow a considerably more rapid and feasible detection of PrPTSE than conventional PMCA methods. By employing recombinant PrP as a substrate, incubating the samples at elevated temperatures and shaking (instead of sonication), the procedure is supposed to enable the detection of approximately 1 infectious unit in 24 h. The QUIC Assay was able to discriminate between scrapie-infected and uninfected hamsters using cerebrospinal fluid. The procedure has only been described for the hamster model so far.
4.5 Amyloid Seeding Assay (ASA)
Colby et al. [50] have developed a method to induce polymerization of recombinant PrP into amyloid fibrils by using PrPTSE aggregates as seeds. The term ‘amyloid’ designates protein accumulations of diverse origin that can be detected in the light microscope and that share the following characteristics: i) fi-brillary ultrastructure, ii) affinity to the dye Congo red, and iii) characteristic β-sheet structures. The PrP amyloid produced in the ASA is detected by binding to the dye thioflavin T (that shows a fluorescence shift after the binding). With the aid of ASA it was possible to detect PrPTSE in diluted brain homogenates from patients with sporadic CJD as well as in a variety of prion strains passaged in both mice and hamsters. Conservatively estimated, the detection limit for PrPTSE in the ASA is supposed to be <1 fg, and test results are obtained within 24 h.
4.6 Cell Panel Assay
Mahal et al. [51] described a cell culture assay with four cell lines that have varying susceptibility to infections with mouse-adapted prion strains RML, 22L, Me7 (scrapie-associated agents) and 301C (BSE-associated agent). The assay allows to detect and discriminate these prion strains within 2 weeks. As yet, the application of the Cell Panel Assay is relatively limited because the susceptibility of the cell lines is confined to murine prion strains.
4.7 Detection of Prions in Organotypic Brain Slice Cultures
Falsig et al. [52] used the so-called ‘POSCA’ (prion organotypic slice culture assay) for ex vivo replication and titration of the prions that are to be detected. Organotypic slices from the cerebellum of PrP-overexpressing tga20 mice were incubated in cultures for extended periods of time. 35 days after contact with prion-containing material (scrapieRML brain homogenate), mouse cerebellar slices had amplified PrPTSE >105-fold, as shown by Western Blot. Other prion strains (79A, 139A, Me7 (scrapie-associated agents) and 301C (BSE-associated agent)) also showed PrPTSE amplification using POSCA, but with variable efficiency. However, for mouse-adapted agents of the chronic wasting disease (CWD) as well as other defined scrapie strains, tga20-POSCA gave negative results. The range of application of the assay might be considerably expanded because organotypic brain slice cultures can be established from mice with the required genotypes as well as from a variety of different host species.
4.8 Atomic Dielectric Resonance Spectroscopy (ADRS)
Fagge et al. [53] have used ADRS to determine dielectric characteristics of sample material with the help of radio/micro waves in order to analyze blood samples from CJD and vCJD patients and identify characteristic ‘ADR signatures’ in the blood. In a preliminary study using unblinded samples from normal blood donors and from patients with vCJD, sCJD as well as other neurological diseases, initially group-specific ADRS properties were determined, and statistic analysis systems were trained to classify unknown samples. In a subsequent study employing these analysis systems it was possible to correctly classify ten blinded samples (four of them vCJD, three sCJD, three other neurological diseases). The investigations show that on the molecular level differences between the blood samples from the different groups of donors can be identified, but these differences cannot be specified. Moreover, the authors point out that it will be necessary to further verify their results in considerably larger studies.
5 Evaluation and Market Access of vCJD Screening Assays
5.1 Regulation of in vitro Diagnostic Medical Devices in Europe
It is one of the aims of the development of specific vCJD assays to identify potential carriers of vCJD already by blood screening on the occasion of blood donation and to eliminate their donation in time. Thus these assays would be applied similar to virus screening tests already widely in use by blood donation services, e.g. anti-HIV-1/-2 screening assays.
In Europe virus diagnostics for infections with HIV, HBV, HCV and HTLV, so-called high-risk products, are regulated considerably more stringently than other diagnostics, due to the in vitro diagnostic medical devices (IVDs) directive [54]. The reasons behind this classification are the severe consequences of false test results, for those concerned as well as for third parties, for example in the case of transmission of these viruses by blood transfusion to other persons due to false-negative test results of the blood donor. Regarding those IVDs not classified as high-risk products, the manufacturers are given as much of a free hand as possible to assess the state of the art on their own responsibility and to declare by CE marking the marketability of the products within the Common Market of Europe. In the case of high-risk products a thorough examination by an independent institution (Notified Body) is foreseen. Specific quality requirements, the Common Technical Specifications (CTS), must be met. In addition, before reaching the European market, the product batches are also counterchecked in an independent laboratory (batch release testing).
Regarding the risk potential, diagnostic assays for vCJD intended to be used in the blood service will have to take a position similar to anti-HIV-1/-2 screening assays: Release or exclusion of a blood donation is directly dependent on each test result. Any false-negative test results might have severe consequences for recipients of blood transfusions. Any reactive and potentially false-positive test results would require extensive investigations to clarify these results, as far as this was possible at all for vCJD diagnostic assays. Furthermore, reactive test results would represent a heavy psychological burden for the donor. It has to be mentioned that the IVD directive has been written at a time when neither the disease vCJD was known nor diagnostic assays for vCJD-infected individuals were conceivable.
A formal application to the European Commission is required in order to include new diagnostic assays into list A (high-risk products), as has been explained in detail in article 14 of the directive 98/79/EG. After some preliminary discussions, it was left to UK – being the member state most affected by vCJD – to file an application to include vCJD diagnostic assays into list A, annex 2, of the IVD directive. Such an application was filed in 2007 and is supported by several other member states, among them Germany and France. At a public hearing in Brussels dated October 26, 2007, such a classification of vCJD assays was also supported by the European IVD manufacturing industry (European Diagnostic Manufacturers Association; EDMA) and the European Medicines Agency (EMEA). As long as list A of the IVD directive has not been expanded – the application filed for this according to article 14 [54] can still take years until an official decision is made – vCJD assays can be CE-marked without special requirements and marketed in the Common Market, even for application in blood donation screening. This widely unregulated situation is very alarming.
On the occasion of the public hearing in Brussels it became also apparent that the development of CTS regarding vCJD assays will be urgently needed. A significant step would have to be experimental assessment of the assays using well-characterized samples with known vCJD infectivity. This endeavor appears to be quite difficult because such reference samples for vCJD are not available due to the low number of cases. In the meantime efforts have been made to collect the scant sample material and to systematically make it available (see below). It will be inevitable to also include other sample material, such as samples from TSE animal models, in an initial validation of diagnostic assays for the detection of vCJD in human blood. Anyway, a simple transfer of those CTS already established for current virus screening assays to the newly developed vCJD assays is not possible for several reasons. Another problem is the required testing of clinical samples in respect of sensitivity. For example, at least 400 different positive patient samples are mandatory for a sensitivity trial of the various virus screening assays, without missing any sample. Certainly this criterion is not applicable at present to vCJD assays because such sample numbers are not available.
Investigating the specificity of such assays would also be difficult. A minimal specificity of 99.5% is required in the CTS for virus screening assays. The specificity of blood screening assays has to be determined using samples from 5,000 blood donors. By definition, an investigation has a false-positive result if an initially reactive test is repeatedly reactive using the same sample and if comparable control or confirmation tests have negative results. Also there must be no further indication of a corresponding infection (e.g. clinical symptoms) for the person concerned. To implement a novel diagnostic assay without a control or confirmation test would imply that it would be impossible to differentiate false-positive from correct true-positive test results in a population that is not well characterized. Therefore, it is almost impossible at present to estimate the specificity of a vCJD assay. CTS further require the investigation of samples from hospitalized patients and samples with potentially cross-reacting components. Regarding vCJD assays, an investigation of individuals with neurological diseases for whom vCJD has been ruled out seems to be indicated. It has to be discussed further to what extent the etiological agents of other human TSE diseases may or even should cross-react.
In the UK a committee composed of international experts has been appointed at NIBSC (vCJD Repository Oversight Committee) that develops criteria for making available to manufacturers of diagnostic assays the samples registered and stored in the ‘vCJD repository’ of NIBSC. The Paul-Ehrlich-Institut is part of the Oversight Committee and has decided to add samples from vCJD-infected primates that were collected in the context of an EU research project at the Paul-Ehrlich-Institut to this vCJD repository at NIBSC. Thus IVD manufacturers are offered a contact point and a procedure to obtain appropriate samples that are provided under generally accepted conditions. NIBSC has already asked one manufacturer to make their diagnostic assay available for validation investigations [55]. Future CTS for vCJD assays will certainly have to also be coordinated with this policy.
Regardless of the obvious difficulties, the attempt must be made to define mandatory quality criteria also for vCJD assays, whether in the form of CTS or otherwise.
5.2 Legal and Ethical Aspects Regarding the Development and Application of Diagnostic Assays
Performance evaluation studies involving test persons and patients are necessary for an evaluation of novel IVDs. In the case of validating vCJD screening assays this would also include the testing of blood donors. Many difficult questions arise, not only in the context of these studies but also on implementing such assays into routine screening of blood donors. The set of problems under scrutiny here is particularly complex. The particular situation in Germany including the basic legal aspects and ethical implications (sources of information regarding the regulations and legislation are listed there) is discussed in detail in ‘Legal and Ethical Aspects Regarding the Development and Application of Screening Assays to Detect vCJD Infections’ (see online supplemental material at www.karger.com/doi/10.1159/000188082).
In summary the following should be noted:
-
a)
Performance evaluations of IVDs are aimed at obtaining CE certification and thus marketability of such medicinal devices in the EU. Their purpose is to determine the safety and qualification of the IVDs for the intended application.
-
b)
In order to ensure health protection for the population, in accordance with section 5 paragraph 1 Transfusion Act (Transfusionsgesetz; TFG), IVDs that allow the detection of grave and epidemiologically significant infectious diseases have to be employed to determine the eligibility of a person to donate blood. This applies under the precondition that IVDs are technically safe and suitable and that sufficient safety cannot be reached by other measures (e.g. donor exclusion). However, in view of the presently known epidemiological situation of vCJD in Germany, in view of only four confirmed transmissions by blood transfusion in the UK and in view of the difficulties described above regarding performance evaluation, the implementation of vCJD diagnostic assays appears to be inadvisable at present.
-
c)
The performance evaluation study of an IVD for the detection of vCJD infections regularly requires the informed consent of the participant, see § 24 paragraph 1 Medical Devices Act (Medizinproduktegesetz; MPG). The only conceivable exception would be the investigation of factually anonymized samples from those individuals who are not at the same time donors in terms of the TFG, who are not suspected to carry any other disease and whose blood samples were taken for other reasons than performance evaluation studies.
-
d)
Already in the case of a ‘justifiable suspicion’ of a donor to be infected with vCJD, the physician responsible is obligated to eliminate the donation, to trace and inform as well as examine any recipients and to inform the authorities responsible, unless there is a reasonable assumption of its probably being a false-positive result (see also section 19 paragraph 1 sentence 1 and sentences 6–8 as well as section 3 TFG). In view of the largely unknown epidemiology of the vCJD infection, the latter assumption cannot be justified by statistical considerations alone. Rather it has to use as definitive guidance the sensitivity and specificity (to be supported by performance evaluation studies) as well as technical feasibility of the assays to be developed for detecting a vCJD infection. On the condition that there was a vCJD assay available, the fact alone that as yet there was no assay for confirmation or exclusion of a reactive vCJD screening test result, would be no sufficient reason to exclude a ‘justifiable suspicion’ for a vCJD infection in terms of the TFG, section19 paragraph 1 sentence 1.
-
e)
In investigations carried out in the context of or on the occasion of blood donation, employing an IVD to detect a vCJD infection, a positive result implies the duty of the physician responsible to inform and counsel the donor on this fact, provided that it must be assumed that the IVD has to be regarded as able to ‘confirm’ a vCJD infection (see also TFG, section 19 paragraph 1 sentence 4). In this regard it firstly depends on the respective state of development of the IVD whether it can be regarded as suitable for allowing a confirmed diagnosis of a vCJD infection, even at this early stage during performance evaluation studies. According to the current state of scientific knowledge, this cannot yet be answered affirmatively in view of the vCJD diagnostic assays under development.
-
f)
In case a positive test result is disclosed, the person concerned must receive counseling and advice by a physician, see also TFG, section 19 paragraph 1 sentence 5. But it must be taken into account that currently no diagnostic method is established to confirm a positive screening test result ante mortem and no therapy to treat or cure the terminal vCJD disease.
-
g)
On request, participants in a performance evaluation study regarding an IVD for detecting vCJD must be informed about the test results, see sections 19 and 34 Federal Data Protection Law (Bundesdatenschutzgesetz; BDSG). In this connection they must be informed before and after participation that a positive test result does not mean in every case that they are infected with vCJD and, even if they should actually be infected, that no statement can be made regarding the question of whether the disease will become symptomatic. Furthermore they should be offered counsel and help.
-
h)
The application of screening assays for detecting vCJD infections may have a negative effect on the general willingness to donate blood, but this fact is no adequate reason to interfere with performance evaluation studies or with the implementation of an IVD to detect vCJD infections in blood donations, at any rate unless such an infection of blood donors and in blood products can be excluded by other diagnostic methods.
-
i)
Before performance evaluation studies of an IVD can be started, ethics boards must be involved either because of section 24 paragraph 1 in conjunction with section 20 paragraphs 7 and 8 of the MPG and/or because of legal guidelines for physicians (see section 15 of the Medical Association's professional code of conduct).
6 General Conditions and Problems Expected to Arise on Implementation of vCJD Screening Assays
6.1 Sensitivity
Due to the scarcity of human samples, the stage of development of the diagnostic assays so far does not allow any statement on their sensitivity regarding detection of vCJD in the preclinical stage. A negative result does not rule out an infection with vCJD.
Considering the epidemiological situation in Germany which is presently characterized by a prevalence and incidence too low to be measured, it is impossible in the current situation to quantify the increase of safety that is to be gained by screening for vCJD in terms of avoiding transfusion-transmitted vCJD.
6.2 Specificity
Up to now available investigations using very small sample numbers indicate an unsatisfactory specificity. With all due caution one has to assume a rate of false-positive results of approximately 2%. Only by extensive studies using samples from blood donors will it be possible to establish a test-immanent specificity of the various assays. The medico-legal difficulties involved have been discussed above under section 5.
On behalf of a maximal safety for transfusion recipients, all donations showing reactive screening results would have to be destroyed and actually, as a consequence in the absence of confirmation tests, donors would have to be excluded from further blood donation. Moreover, to be consistent, a look-back procedure would have to be initiated covering the entire donor history as far back as 1980, and recipients of blood products as well as the plasma fractionation industry would have to be notified. In Germany in 2006 4,595,484 homologous whole blood donations and 1,731,921 plasma and thrombocyte apheresis donations were collected [56]. On average, whole-blood donors donate every 6 months and apheresis donors once every month [2]. Based on an assumed rate of 2% false-positive results, at least in the first year after implementation of the test, approximately 92,000 whole-blood donors and up to 35,000 apheresis donors would have to be informed and excluded from further donations. Because repeat donors tend to donate regularly for years, the effort regarding look-back measures would be extraordinary. The same applies to apheresis donors who donate very frequently and to plasma donors whose plasma for fractionation is part of a plasma pool used for the manufacture of various plasma products.
6.3 Ethical Problems
Based on the actual epidemiological situation in Germany, it can be assumed that in fact almost none of the reactive results would indicate a real preclinical vCJD. If donors as well as recipients of blood products produced earlier were informed about a very vague suspicion concerning a fatal disease that can neither be ultimately ruled out nor treated, a considerable psychological strain for those concerned would be caused that might have conceivable respective consequences, potentially culminating in suicide.
Those concerned will ask a number of questions:
-
–
When did the infection happen?
-
–
How likely is my becoming ill?
-
–
When will the disease break out, what are its symptoms?
-
–
In case I was infectious, how can I transmit the agent?
-
–
Do I have to inform my partner?
-
–
Is a sexual transmission possible?
-
–
May I become pregnant, and how high is the risk of transmission to the fetus?
-
–
If my blood is infectious, what do I have to do if I suffer an injury?
Based on the current state of knowledge, one would give soothing answers, but this would be in stark contrast to the measures to be initiated for the sake of consistency.
Donors concerned (as well as recipients of blood components from these donors) would have to be advised to stop to donate blood, plasma, sperm, tissues or organs in the future. On the occasion of every medical, surgical and dental intervention they would have to inform about their existing potential infectivity. Medical institutions would have to implement considerably more measures than before to minimize the risk of these patients to transmit pathogenic prions. In the UK several hundred hemophiliacs had been treated with coagulation products that were possibly contaminated with the vCJD agent. Experiences from these persons in the UK showed that for example it had become very difficult for these patients to receive dental treatment.
Medical personnel with a reactive result in a vCJD test would have to stop performing certain parts of the job that are associated with a higher risk of injury (similar to those individuals infected with hepatitis B or hepatitis C).
Apart from family distress and depression, grave disadvantages must be taken into consideration, e.g. when contracting life insurance, vocational disability insurance or private health insurance, because of a diagnostic result which is highly likely to be false in diagnosing preclinical vCJD. Apart from this, on implementing such diagnostic assays, adequately trained medical and pastoral personnel will have to be available to carry out information and counseling of the donors concerned, which is required in accordance with the TFG.
One way of overcoming this set of problems would be, on account of currently unreliable data, to generally not inform individuals about a reactive result on performing a vCJD test. However, if a vCJD case should actually be diagnosed later on, or even another person be infected or have been infected in the past, accusations of violating due diligence are to be expected. On the other hand, the specifications of the TFG must be observed (see also section 5). A justifiable suspicion for an infection with vCJD based on the test results will initiate a look-back procedure in addition to destroying the actual donation. However, the donor has to be informed only after a confirmed diagnosis of a symptomatic disease or an infection, even though further donations will most probably become useless on account of their inherent albeit small risk. Furthermore, the donor is often suspended electronically and will be deferred without previous explanation, e.g. on the occasion of a computer-based mobile data entry. Under the present legal position in Germany, only if the donor actively requests an explanation of the reason for deferral, the blood service is allowed to extensively inform him/her about a justifiable suspicion.
In this situation, legally accepted definitions of both a justifiable suspicion and a confirmed diagnosis of vCJD must already be developed before performance evaluation and before, during and after implementation of a new diagnostic assay. These definitions are to be continuously updated according to the state of knowledge.
In order to establish definite rules for all concerned and to prevent subsequent criminal conviction, a consensus must be reached on the legal, political as well as social level regarding the resulting direct and indirect consequences and measures.
6.4 Restrictions on Supply
Based on an average frequency of donating whole blood of once every 6 months and of donating by apheresis of once every month [2] and also including the numbers mentioned under section 6.2, in 2006, approximately 2.3 million persons donated whole blood and 144,000 persons blood components or intermediates obtained by apheresis.
Assuming again the scenario of 2% false-positive results, in the first 12 months after test implementation, approximately 46,000 whole blood and 2,900 apheresis donations would be excluded from further blood donations. Thus the supply would be reduced by approximatley 92,000 erythrocyte concentrates and 23,000 l of plasma obtained from whole blood donations and approximately 35,000 blood components obtained by apheresis techniques. A further deterioration in supply is to be expected, especially regarding erythrocytes that at this stage have already begun to fail to meet demands.
Furthermore, it must be taken into consideration that 3 new donors have to be recruited within 12 months to replace 1 donor regularly donating whole blood [2]. To keep up the present supply, approximately 140,000 persons have to be newly motivated to donate blood.
In the context of the increasingly tight blood supply situation in Germany, a retraction of donors of the magnitude mentioned above would inevitably result in further bottleneck situations in supply, in the worst-case scenario leading to a collapsed service. This also applies in cases when an information of the donor is abandoned and just the respective donation is not released for use.
Deficits in plasma donations – and to a lesser extent in plasma for fractionation produced from whole blood – would lead to serious problems in supplying patients with immunoglobulins, coagulation factors, and other medical drugs produced from plasma. Whether imported plasma that was tested negative for vCJD became available for compensation is doubtful because donor retractions related to a test implementation would lead to a further shortage of plasma as a resource also on an international level.
6.5 Feasibility
Particularly for large blood services, it will only be possible to screen for prions if high-throughput automats are available. On account of the necessary release of blood components within 24 h or less after collection, the mere duration of the test should not be significantly longer than 4 h. It is impossible to delay the release for several days, especially for short-lived preparations like thrombocyte concentrates that, adapted for patients, have to be applied urgently or vitally important. Presently no suitable automated systems and corresponding reagents are available for these requirements. Until a minimum test specificity of 99.5% is reached for the assays (minimum requirement of the common technical specifications regarding infection serological assays), an implementation of an assay will be problematic as a result of the difficulties described in section 6.2. A specificity that is even higher would be preferable.
In addition, the legislator has to generate regulations permitting to test the feasibility and specificity of diagnostic assays available or under development under the conditions of a blood service, so that their viability can be assessed at all. Legal certainty must be established here about the fact that an information of the donors, as well as consequences for the release of blood donations or look-back procedures and ultimately the recall of donations, may be abandoned if test results are indeterminate. This implies to deliberately run the risk of the highly improbable case, which however cannot be completely ruled out, that a reactive result is indeed attributable to preclinical vCJD which may become conspicuous only after months or even years. On account of previously applied technology and technical equipment requirements, a longer period of transition is needed before nationwide screening can be implemented, because probably methods have to be implemented that have never been used routinely in the blood services. This transition period will also be necessary to arrange for the required investments, computer-based support and quality management/validation of the diagnostic assays.
6.6 Costs
At present no estimation can be made regarding supplementary costs for investments, reagents, and personnel required.
6.7 Quality Management
On account of the rareness of the disease, it is not possible at present to provide suitable samples for external quality assurance. There are no independent positive or negative controls. Thus, it is not possible to compare results of different tests. Accordingly, a validation by the user is only possible in terms of a performance evaluation. Therefore, the establishment of appropriate conditions should be aimed for. For example, every diagnostic assay to be implemented should be evaluated with an independent panel of human blood samples from patients with different TSE isoforms and other neurodegenerative diseases as well as from healthy controls.
6.8 Safety Considerations
According to the type of assay selected, safety precautions have to be taken that largely prevent hazards for the personnel. This applies especially to techniques which potentially generate pathological PrPs. In that case in every institution performing such tests a BSL-3 laboratory is required with the corresponding investment and maintenance costs.
6.9 Availability
On implementation of screening tests for prions in blood donations, it has to be ensured that manufacturing capacities of the companies in question are sufficiently large to guarantee an uninterrupted supply with test kits of consistent quality. The same is true for technical support. To interrupt the screening after it was implemented because of unavailable test kits would also be precarious for ethical reasons. Because different approaches will probably be used in the assays, a short-term switch-over to instruments and reagents from other manufacturers will be almost impossible.
7 Conclusion and Assessment
The disease vCJD is definitely fatal according to the current state of knowledge and is transmissible by blood transfusion. To prevent transmission of vCJD by blood transfusion and to protect transfusion recipients, the existence of a suitable screening assay for vCJD in blood donors would be very valuable. Still in 2006 the last report of the Working Group came to the following conclusion: ‘No such test is currently available nor is its availability foreseeable. Providing resources for developing suitable test methods should therefore be treated with high priority’ [2]. This situation has changed in the meantime.
The detection of the pathological PrPTSE has to be very sensitive and simultaneously specific in a background of physiological PrPC present in a concentration that is several magnitudes higher. The amino acid sequence of PrPC is identical to that of PrPTSE but is simply folded differently. Therefore, high demands are made on the methods. At present there are various far advanced assays under development, so that it is only a question of time when the first screening assays for vCJD will be offered and marketed. At present, in accordance with the applicable IVD law, it would be rather simple for the manufacturers to obtain the CE marking for such assays and thus make them marketable. A formal application to the European Commission is required in order to include new diagnostic assays into list A (‘high-risk products’). Such an application has been filed but might still take some time. This process should be emphatically supported.
The mandatory verification and validation of such assays is especially difficult for technical reasons because suitable vCJD-positive samples are very scarce. Therefore, it is suggested to support the creation of suitable sample panels. Such panels should predominantly contain samples from patients with various TSE isoforms and with dementia of other etiologies as well as from healthy controls.
Provided that they are technically safe and suitable, IVDs for the detection of infections causing severe diseases like vCJD have to be employed in assessing the fitness of a blood donor in accordance with the TFG. This emphasizes the necessity of a performance evaluation study for such novel assays regarding their suitability and safety. After successful admission into list A of the IVD guideline [54], the performance of the assay must be tested in test persons and patients. A precondition for the performance evaluation study of an IVD for the detection of vCJD infections in blood donors is the informed consent of the participants. Participants in a study are entitled at any time to learn their test results and would have to receive suitable counseling in case the test was reactive. The ethics boards involved are also called upon extensively in designing such studies. The main issue is that by a reactive test result a seemingly healthy person would be put under suspicion of the imminent outbreak of a fatal disease for which the latency is unknown and that cannot be treated at this point, while confirmation assays to clarify this suspected diagnosis are lacking at present.
Similar ethical problems would result on implementation of such assays for routine screening of blood donors. A positive test result would imply the obligation of the physician responsible to inform the donor about this fact and to give advice, in case the assay was assumed to be reliable and suitable to detect a vCJD infection. Already in the case of a ‘justifiable suspicion’ that a donor was infected with vCJD, the physician responsible is obligated to eliminate the donation, to trace and inform as well as examine any recipients, and to notify the competent authorities, unless there is a reasonable assumption of its probably being a false-positive result. In the context of this, but also regarding the necessary performance evaluation of the diagnostic assays, guidelines for the information of donors and recipients have to be developed. On the part of the blood services involved, there is an enormous desire for legal security.
In principle, the current development of screening assays for the detection of vCJD is welcome. However, before such assays can be marketed and put to routine use, there remain considerable problems. In view of the presently known epidemiological situation of vCJD in Germany, in view of only four confirmed transmissions by blood transfusion in the UK, and in view of the difficulties described above regarding performance evaluation, the implementation of vCJD diagnostic assays appears to be inadvisable at present.
Members of the Working Group
Michael Beekes, Robert Koch-Institut, Berlin
Johannes Blümel, Paul-Ehrlich-Institut, Langen
Reinhard Burger, Robert Koch-Institut, Berlin
Christian von Dewitz, State Office of Health and Social Affairs (LaGeSo), Berlin
Albrecht Gröner, CSL Behring, Marburg
Margarethe Heiden, Paul-Ehrlich-Institut, Langen
Hans Kretzschmar, Institute of Neuropathology and Prion Research, Ludwig-Maximilians-University Munich
Micha Nübling, Paul-Ehrlich-Institut, Langen
Uwe Schlenkrich, German Hemophilia Society (DHG), Hamburg
Volkmar Schottstedt, Blood Services of the German Red Cross West (DRK), Hagen
Rainer Seitz (central coordination), Paul-Ehrlich-Institut, Langen
Johanna Strobel, Federal Ministry of Health, Bonn
Hannelore Willkommen, NewLab BioQuality GmbH, Regulatory Affairs and Biological Safety Consulting, Erkrath
Carl-Heinz Wirsing von König, HELIOS Clinical Center, Krefeld Inga Zerr, National Reference Center for the Surveillance of TSE, University Clinical Center, Göttingen
The members of the Working Group were appointed by the German Federal Minister of Health.
References
- 1.Joint press release of the Paul-Ehrlich-Institut and the Robert Koch-Institut. Gesamtstrategie Blutversorgung im Angesicht der BSE-Krise. www.pei.de/cln_115/nn_157198/DE/infos/presse/pm/archiv/2001/14.html
- 2.Working Group ‘Overall Blood Supply with Regard to Variant Creutzfeldt-Jakob Disease (vCJD)’: Overall Blood Supply with Regard to Variant Creutzfeldt-Jakob Disease (vCJD) – Report of the Working Group Commissioned by the German Federal Ministry of Health, April 13, 2006 (Update of the Report from August 17, 2001) Transfus Med Hemother. 2006;33(suppl 2) [Google Scholar]
- 3.Seitz R, von Auer F, Blümel J, Burger R, Buschmann A, Dietz K, Heiden M, Hitzler WE, Klamm H, Kreil T, Kretzschmar H, Nübling M, Offergeld R, Pauli G, Schottstedt V, Volkers P, Zerr I. Impact of vCJD on blood supply. Biologicals. 2007;35:79–97. doi: 10.1016/j.biologicals.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Creutzfeldt HG. Über eine eigenartige herdförmige Erkrankung des Zentralnervensystems. Z Ges Neurol Psychiatr. 1920;57:1–18. [Google Scholar]
- 5.Jakob A. Über eigenartige Erkrankungen des Zentralnervensystems mit bemerkenswertem anatomischem Befund (Spastische Pseudosklerose – Encephalomyelopathie mit disseminierten Degenerationsherden) Dtsch Z Nervenheilk. 1921;70:132–146. [Google Scholar]
- 6.Glatzel M, Abela E, Maissen M, Aguzzi A. Ex-traneural pathogenic prion protein in sporadic Creutzfeldt-Jakob disease. N Engl J Med. 2003;349:1812–1820. doi: 10.1056/NEJMoa030351. [DOI] [PubMed] [Google Scholar]
- 7.Bishop MT, Ritchie DL, Will RG, Ironside JW, Head MW, Thomson V, Bruce M, Manson JC. No major change in vCJD agent strain after secondary transmission via blood transfusion. PLoS ONE. 2008;3:1–6. doi: 10.1371/journal.pone.0002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The National Creutzfeldt-Jakob Disease Surveillance Unit (NCJDSU): Variant Creutzfeldt-Jakob Disease. Current data. www.cjd.ed.ac.uk/vcjdworld.htm.
- 9.Dietz K, Raddatz G, Wallis J, Müller N, Zerr I, Duerr HP, Lefèvre H, Seifried E, Löwer J: Blood transfusion and spread of variant Creutzfeldt-Jakob disease. Emerg Infect Dis. 2007;13:89–96. doi: 10.3201/eid1301.060396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke P, Will RG, Ghani AC. Is there the potential for an epidemic of variant Creutzfeldt-Jakob disease via blood transfusion in the UK? J R Soc Interface. 2007;4:675–684. doi: 10.1098/rsif.2007.0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garske T: Presentation at the Paul-Ehrlich-Institut on May 28, 2008.
- 12.Mead S, Joiner S, Desbruslais M, Beck JA, O'Donoghue M, Lantos P, Wadsworth JDF, Collinge J. Creutzfeldt-Jakob disease, prion protein gene codon 129VV, and a novel PrPSc type in a young British woman. Arch Neurol. 2007;64:1780–1784. doi: 10.1001/archneur.64.12.1780. [DOI] [PubMed] [Google Scholar]
- 13.Coghlan A. Mysterious death reignites vCJD fears. NewScientist. 2008;2637:11. [Google Scholar]
- 14.Gambetti P, Dong Z, Yuan J, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008;63:697–708. doi: 10.1002/ana.21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krebs B, Bader B, Klehmet J, Grasbon-Frodl E, Oertel WH, Zerr I, Stricker S, Zschenderlein R, Kretzschmar HA. A novel subtype of CreutzfeldtJakob disease characterized by a small 6 kDa PrP fragment. Acta Neuropathol. 2007;114:195–199. doi: 10.1007/s00401-007-0242-5. [DOI] [PubMed] [Google Scholar]
- 16.Parchi P, Giese A; Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- 17.Oesch B, Westaway D, Wälchli M, McKinley MP, Kent SBH, Aebersold R, Barry RA, Tempst P, Teplow DB, Hood LE, Prusiner SB, Weissmann C. A cellular gene encodes scrapie PrP(27-30) protein. Cell. 1985;40:735–746. doi: 10.1016/0092-8674(85)90333-2. [DOI] [PubMed] [Google Scholar]
- 18.Basler K, Oesch B, Scott M, Westaway D, Wälchli M, Groth DF, McKinley MP, Prusiner SB, Weissmann C. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gen. Cell. 1986;46:417–428. doi: 10.1016/0092-8674(86)90662-8. [DOI] [PubMed] [Google Scholar]
- 19.Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP(27-30) in water by infrared spectroscopy. Biochemistry. 1991;30:7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 20.Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, Prusiner SB. Conversion of alpha-helices into beta-sheets features the formation of the scrapie prion proteins. Proc Natl Acac Sci U S A. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrPSc molecules with different conformations. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 22.Kascsak RJ, Rubenstein R, Merz PA, TonnaDeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987;61:3688–3693. doi: 10.1128/jvi.61.12.3688-3693.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barry RA, Prusiner SB. Monoclonal antibodies to the cellular and scrapie prion protein. J Infect Dis. 1986;154:518–521. doi: 10.1093/infdis/154.3.518. [DOI] [PubMed] [Google Scholar]
- 24.Korth C, Stierli B, Streit P, Moser M, Schaller O, Fischer R, Schulz-Schaeffer W, Kretzschmar H, Raeber A, Braun U, Ehrensperger F, Hornemann S, Gluckshuber R, Riek R, Billeter M, Wüthrich K, Oesch B. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature. 1997;390:74–77. doi: 10.1038/36337. [DOI] [PubMed] [Google Scholar]
- 25.Moroncini G, Mangieri M, Morbin M, Mazzoleni G, Ghetti B, Gabrielli A, Williamson RA, Giaccone G, Tagliavini F. Pathogenic prion protein is specifically recognized in situ by a novel PrP conformational antibody. Neurobiol Dis. 2006;23:717–724. doi: 10.1016/j.nbd.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Ulrih NP, Skrt M, Veranic P, Galvani V, Vranac T, Curin Serbec V. Oligomeric forms of peptide fragments PrP(214-226) in solution are preferentially recognized by PrPSc-specific antibody. Biochem Biophys Res Commun. 2006;344:1320–1326. doi: 10.1016/j.bbrc.2006.04.046. [DOI] [PubMed] [Google Scholar]
- 27.Salguero FJ, Diaz-San Segundo F, Brun A, Cano MJ, Torres JM. Comparison of three monoclonal antibodies for use in immunohistochemical detection of bovine spongiform encephalopathy proteaseresistant prion protein. J Vet Diagn Invest. 2006;18:106–109. doi: 10.1177/104063870601800117. [DOI] [PubMed] [Google Scholar]
- 28.Biasini E, Seegulam ME, Patti BN, Solforosi L, Medrano AZ, Christensen HM, Senatore A, Chiesa R, Williamson RA, Harris DA. Noninfectious aggregates of the prion protein react with several PrPSc-directed antibodies. J Neurochem. 2008;105:2190–2204. doi: 10.1111/j.1471-4159.2008.05306.x. [DOI] [PubMed] [Google Scholar]
- 29.Morel N, Simion S, Frobert Y, Volland H, Mourton-Gilles C, Negro A, Sorgato MC, Creminon C, Grassi J. Selective and efficient immunoprecipitation of the disease-associated form of the prion protein can be mediated by non-specific interactions between monoclonal antibodies and scrapie-associated fibrils. J Biochem Chem. 2004;279:30143–30149. doi: 10.1074/jbc.M403896200. [DOI] [PubMed] [Google Scholar]
- 30.Pan T, Li R, Wong B-S, Kang S-C, Ironside J, Sy MS. Novel antibody-lectin enzyme-linked immunosorbent assay that distinguishes prion proteins in sporadic and variant cases of Creutzfeldt-Jakob disease. J Clin Microbiol. 2005;43:1118–1126. doi: 10.1128/JCM.43.3.1118-1126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Safar JG, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H, Serban A, Vey M, Baron H, Giles K, Miller BL, DeArmond SJ. Diagnosis of human prion disease. Proc Natl Acad Si U S A. 2005;102:3501–3506. doi: 10.1073/pnas.0409651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol. 1992;66:2096–2101. doi: 10.1128/jvi.66.4.2096-2101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barron RM, Campbell SL, King D, Bellon A, Chapman KE, Williamson RA, Manson JC. High titers of transmissible spongiform encephalopathy infectivity associated with extremly low levels of PrPSc in vivo. J Biol Chem. 2007;282:35878–35886. doi: 10.1074/jbc.M704329200. [DOI] [PubMed] [Google Scholar]
- 34.Berardi VA, Cardone F, Valanzano A, Lu M, Pocchiari M. Preparation of soluble infectious samples from scrapie-infected brain: new tool to study the clearance of transmissible spongiform encephalopathy agents during plasma fractionation. Transfusion. 2006;46:652–658. doi: 10.1111/j.1537-2995.2006.00763.x. [DOI] [PubMed] [Google Scholar]
- 35.Wadsworth JDF, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J. Tissue distribution of protein resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet. 2001;358:171–180. doi: 10.1016/s0140-6736(01)05403-4. [DOI] [PubMed] [Google Scholar]
- 36.Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particle. Nature. 2005;437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown P, Rohwer RG, Dunstan BC, MacAuley C, Gajdusek DC, Drohan WN. The distribution of infectivity in blood components and plasma derivatives in experimental models of transmissible spongiform encephalopathy. Transfusion. 1998;38:810–816. doi: 10.1046/j.1537-2995.1998.38998408999.x. [DOI] [PubMed] [Google Scholar]
- 38.Brown P, Cervenáková L, McShane LM, Barber P, Rubenstein R, Drohan WN. Further studies of blood infectivity in an experimental model of transmissible spongiform encephalopathy, with an explanation of why blood components do not transmit Creutzfeldt-Jakob disease in humans. Transfusion. 1999;39:1169–1178. doi: 10.1046/j.1537-2995.1999.39111169.x. [DOI] [PubMed] [Google Scholar]
- 39.Cervenáková L, Yakovleva O, McKenzie C, Kolchinski S, McShane L, Drohan WN, Brown P. Similar levels of infectivity in the blood of mice infected with human-derived vCJD and GSS strains of transmissible spongiform encephalopathy. Transfusion. 2003;43:1687–1694. doi: 10.1046/j.0041-1132.2003.00586.x. [DOI] [PubMed] [Google Scholar]
- 40.Houston F, Foster JD, Chong A, Hunter N, Bostock CJ. Transmission of BSE by blood transfusion in sheep. Lancet. 2000;356:999–1000. doi: 10.1016/s0140-6736(00)02719-7. [DOI] [PubMed] [Google Scholar]
- 41.Hunter N, Foster J, Chong A, McCutcheon S, Parnham D, Eaton S, MacKenzie C, Houston F. Transmission of prion diseases by blood transfusion. J Gen Virol. 2002;83:2897–2905. doi: 10.1099/0022-1317-83-11-2897. [DOI] [PubMed] [Google Scholar]
- 42.Yakovleva O, Janiak A, McKenzie C, McShane L, Brown P, Cervenáková L. Effect of protease treatment on plasma infectivity in variant CreutzfeldtJakob disease mice. Transfusion. 2004;44:1700–1705. doi: 10.1111/j.0041-1132.2004.04198.x. [DOI] [PubMed] [Google Scholar]
- 43.Minor P: NIBSC, presentation at the workshop of the European Commission on 26 October 2007 in Brussels.
- 44.Uger M: Amorfix Life Sciences, presentation at the workshop of the European Commission on 26 October 2007 in Brussels.
- 45.Health Protection Agency: TED-Publication 30072-2007, February 6, 2007.
- 46.Pan T, Sethi J, Nelsen C, Rudolph A, Cervenáková L, Brown P, Orser CS. Detection of misfolded prion protein in blood with conformationally sensitive peptides. Transfusion. 2007;47:1418–1425. doi: 10.1111/j.1537-2995.2007.01284.x. [DOI] [PubMed] [Google Scholar]
- 47.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 48.Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA, Priola SA, Caughey B. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods. 2007;4:645–650. doi: 10.1038/nmeth1066. [DOI] [PubMed] [Google Scholar]
- 49.Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, Johnson LM, Onwubiko HA, Priola SA, Caughey B. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods. 2008;5:211–212. doi: 10.1038/nmeth0308-211. [DOI] [PubMed] [Google Scholar]
- 50.Colby DW, Zhang Q, Wang S, Groth D, Legname G, Riesner D, Prusiner SB. Prion detection by an amyloid seeding assay. Proc Natl Acad Sci U S A. 2007;104:20914–20919. doi: 10.1073/pnas.0710152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci U S A. 2007;104:20908–20913. doi: 10.1073/pnas.0710054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Falsig J, Julius C, Margalith I, Schwarz P, Heppner FL, Aguzzi A. A versatile prion replication assay in organotypic brain slices. Nat Neurosci. 2008;11:109–117. doi: 10.1038/nn2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fagge TJ, Barclay GR, Stove GC, Stove G, Robinson MJ, Head MW, Ironside JW, Turner ML. Application of atomic dielectric resonance spectroscopy for the screening of blood samples from patients with clinical variant and sporadic CJD. J Transl Med. 2007;5:41. doi: 10.1186/1479-5876-5-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Directive 98/79/EG of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices. Official Journal of the European Communities L 331/1 of December 7, 1998.
- 55.Amorfix: Press Release of August 19, 2008.
- 56.Haschberger B, Henseler O, Hesse J, Heiden M, Seitz R. Bericht zur Meldung nach ß 21 TFG für die Jahre 2005 und 2006. Bundesgesundheitsbl Gesundheitsforsch Gesundheitsschutz. 2008;51:353–372. doi: 10.1007/s00103-008-0546-1. [DOI] [PubMed] [Google Scholar]