

Abstract
Addition of NaCN to isolated hepatocytes results in a marked and rapid decrease in cellular ATP content, and in the extrusion of a sizable amount of cellular Mg2+. This extrusion starts after a 10 min lag phase and reaches a maximum of 35 to 40 nmol Mg2+/mg protein within 60 min from the addition of CN−. A quantitatively similar Mg2+ extrusion is also observed following the addition of the mitochondrial uncoupler carbonyl cyanide p-trifluoromethoxy-phenylhydrazone (FCCP) but not that of the glycolysis inhibitor iodoacetate. The Mg2+ extrusion is completely inhibited by the removal of extracellular Na+ or the addition of imipramine, quinidine or glibenclamide, while it persists following the removal of extracellular Ca2+ or K+, or the addition of amiloride. An acidic extracellular pH or the removal of extracellular HCO3− inhibits the cyanide-induced Mg2+ extrusion by ≥80%. Taken together, these data suggest that the decrease in cellular ATP content removes a major Mg2+ complexing agent from the hepatocyte, and results in an extrusion of hepatic Mg2+ exclusively through a Na+-dependent exchange mechanism modulated by acidic changes in extracellular pH.
Keywords: Mg2+ homeostasis Na+/Mg2+ exchanger, chemical hypoxia, cyanide, FCCP, hepatocytes
Introduction
After potassium, magnesium (Mg2+) is the second most abundant cation within mammalian cells [1,2]. The majority of cellular Mg2+ is compartmentalized within nucleus, mitochondria and endo-sarco-plasmic reticulum [3], with concentrations ranging between 16 to 20mM in each of these compartments. Approximately 20% of total cellular Mg2+ content is in the cytoplasm, in the form of a complex with ATP (~4–5mM) and, to a lesser extent, other phosphonucleotides and cytosolic proteins [3]. As a result of this distribution, cytosolic free Mg2+ concentration is estimated to be ~0.7 mM Mg2+ or less than 4% of total cellular content [3]. While total and free cellular Mg2+ content do not change significantly under resting conditions, major fluxes of Mg2+ can cross the cell membrane in either direction as a result of hormonal or metabolic stimuli [reviewed in 4 and 5]. For the most part, these fluxes result in detectable changes in total cellular Mg2+ content whereas cytoplasm free [Mg2+] changes minimally, if at all [6].
The extrusion of Mg2+ across the cell membrane occurs via the distinct operation of a Na+-dependent and a Na+-independent mechanisms [7]. In the absence of functional cloning and more precise structural information, the Na+ dependent Mg2+ extrusion mechanism has been tentatively identified as a Na+/Mg2+ exchanger based upon its strict requirement for extracellular Na+ to operate [8] and its inhibition by non-specific Na+-transport inhibitors such as amiloride, quinidine or imipramine [see 7 as a review]. This exchanger is activated by the increase in cytosolic cAMP level that follows the stimulation of β-adrenergic, glucagon, or prostaglandin receptors as well as the addition of forskolin or cell-permeant cAMP analogs [5,9]. Under conditions in which the operation of this exchanger is inhibited, Mg2+ can still be extruded from the cell via a not well characterized Na+-independent mechanism [10] that utilizes various cations or anions [7] to favor Mg2+ efflux. The operation of these two distinct transport mechanisms in the hepatocyte membrane has been confirmed by our laboratory in purified liver plasma membrane vesicles [11]. Furthermore, our group has located the operation of the Na+-dependent and Na+-independent mechanism in the basolateral and the apical domain of the hepatocyte membrane, respectively [11], with only the Na+-dependent mechanism specifically requiring a cAMP-dependent phosphorylation to operate [12].
As more than 80% of cytoplasmic Mg2+ forms a complex with ATP, expectation is there that a marked decrease in ATP content will result in a proportional increase in cytoplasmic free [Mg2+]. Experiments performed in hepatocytes [13] and Jurkat cells [14], however, indicate that following addition of cyanide, cellular ATP decreases markedly and rapidly whereas cytosolic free Mg2+ increases only minimally [15] or not at all [14]. Therefore, possibility is there that cytosolic Mg2+ is either extruded from the cell or compartmentalized within organelles as a result of a change in cellular ion pattern. The decrease in ATP content can result in a decrease in the activity of the Na+/K+-ATPase [16] and in the activation of KATP channels [17, 18], a class of plasma membrane channels modulated by ATP and Mg2+. The release of K+ through this channel would counter-balance the entry of Na+ that occurs via Na+/H+ antiporter [19] and/or Na+/HCO3− cotransporter [20] following a marked cellular acidification as that observed under chemical hypoxia conditions [20].
Hence, the present study was undertaken: 1) to test the hypothesis that the lack of an increase in cytosolic free Mg2+ following a cyanide-induced decrease in cellular ATP level depends on an extrusion of Mg2+ from the cytoplasm into the extracellular space, and 2) to determine the relative role of the Na+-dependent and the Na+-independent transport mechanisms in mediating such an extrusion. The results reported here indicate that following cyanide administration Mg2+ is extruded from liver cells exclusively through a Na+-dependent mechanism highly responsive to acidic changes in extracellular pH.
Materials & Methods
Chemicals
All chemicals and reagents were from Sigma (St. Louis, MO). Collagenase type II was from Worthington (Freehold, NJ). Pinacidil and glibenclamide were from ICN Radiochemicals (Costa Mesa, CA).
Animals
Animal handling and experimental utilization conformed to the National Institutes of Health (NIH) guidelines for the care and use of laboratory animals, and the relative protocol was approved by Case Western Reserve University Institutional Animal Care and Use Committee (IACUC).
Cell Isolation
Male Sprague-Dawley rats (200 to 250g body weight) were anesthetized by i.p. injection of sature sodium pentobarbital solution (50 mg/Kg body weight). Following induction of deep anesthesia the abdomen was incised and the portal vein cannulated. Collagenase dispersed hepatocytes were isolated according to the procedure of Seglen as reported previously [21]. After isolation, cells were resuspended in a buffer containing (mM): 120 NaCl, 3 KCl, 1.2 KH2PO4, 1.2 MgCl2, 1.2 CaCl2, 10 glucose, 12 NaHCO3, 10 Hepes (pH 7.2), in the presence of O2:CO2 (95:5 v/v). Trypan Blue exclusion test indicated that the hepatocytes viability was 85±3% (n=9) immediately after isolation, and did not change significantly over the 4 hours of our experiment (84±5%, n=9).
Determination of Mg2+ Transport
Hepatocytes were washed twice in a medium similar to the one described above but devoid of Mg2+ (Mg2+ free buffer), and incubated therein, at the final concentration of 0.3–0.4 mg protein/ml, at 37°C, under continuous stirring and O2:CO2 flow (95:5 v/v). The amount of Mg2+ present as contaminant in the medium was measured by atomic absorbance spectrophotometry (AAS) in a Perkin Elmer 3100 and found to be less than 3μM. At selected time points, 0.7 ml aliquots of the incubation mixture were withdrawn in duplicates and rapidly sedimented in microcentrifuge tubes at 1200 rpm for 5 min. The supernatants were removed, and their Mg2+ content determined by AAS. The cell pellets were digested overnight in 0.7 ml of 10% HNO3. Following sedimentation of denatured protein at 14,000 rpm for 5 min in microfuge tubes, the Mg2+ content of the acid extracts was measured by AAS.
For the experiments in a Na+-free medium, NaCl and NaHCO3 were replaced with equimolar concentrations of choline chloride and KHCO3, respectively. Potassium hydroxide was used to attain pH 7.2. For the experiments in K+-free or Ca2+-free medium, these cations were simply removed from the incubation medium.
Determination of cytosolic free Mg2+ concentration
Changes in cytosolic free Mg2+ concentration ([Mg2+]i) were measured by Mag-Fura2 (Molecular Probe, Portland, OR) as described previously [6]. Hepatocytes were loaded with the cell permeant fluorescent ndicator Mag-Fura2-AM (10μM) for 20 min at room temperature to prevent entrapment of the dye within cellular organelles. Hepatocytes were then washed to remove excess dye and transferred into a thermostated cuvet (37°C), under continuous stirring, using a custom-built dual wavelength excitation fluorimeter (University of Pennsylvania Biomedical Instrumentation Group, Philadelphia, PA) equipped with narrow band width interference filters. The excitation wavelengths for the dual wavelength fluorimeter were 340 and 380 nm, respectively, whereas the emission wavelength was 510 nm. The Mg2+ concentration was calculated using the formula: [Mg2+]i = Kd * [(R-Rmin)/(Rmax − R)] * (Sf/Sb), where Kd = 1.2 mM [6,22]. Hepatocytes were incubated either in the presence or in the absence of extracellular Na+ using the buffer compositions described previously. Because of the unreliability in obtaining MagFura2 Rmax and Rmin in situ, calibration of changes in the fluorescence dual wavelength signal ratio was conducted using a cytosol-like buffer C containing (in mM) 130 KCl, 10 NaCl, 1.2 KH2PO4, 10 Hepes, and 0.5 MgCl2 at pH 7.2 and an amount of non-esterized MagFura2 yielding, at the isosbestic point of 360 nm, a fluorescence quantitatively similar to that measured in Mag-Fura2-AM loaded cells. Rmax was determined by adding known aliquots of MgCl2 until maximal fluorescence was reached. Rmin was determined by adding increments of EDTA until all Mg2+ was complexed [6,22].
ATP measurement
Cellular ATP content was measured by luciferin-luciferase assay [23]. Aliquots of the incubation mixtures were withdrawn at selected time points and sedimented in microfuge tubes. The Mg2+ content of the supernatant was measured by AAS as described previously. The cell pellets were digested with 5% PCA for 5 min on ice. The acid mixture was neutralized by addition of 0.7 ml of 1M KHCO3 and centrifuged at 1200 rpm for 5 min at 4°C. The supernatants were removed and stored at −20°C until used. ATP content in the supernatant was measured by luciferin-luciferase assay (Sigma kit with a detecting sensitivity in the pmol-nmol/ml range) using a LUMAT Berthold LB 9501 luminometer.
Protein measurement
The protein was measured according to the procedure of Bradford [24].
Statistical Analysis
Data are presented as means ± SE. Data were analyzed by 1-way analysis of variance (ANOVA). Multiple means were then compared by Tukey’s multiple comparison test performed with a level of statistical significance designated as p < 0.05.
Results
Mg2+ Efflux upon Chemical Anoxia
The addition of various metabolic inhibitors to suspensions of dispersed hepatocytes resulted in a marked release of cellular Mg2+ into the extracellular milieu (Fig. 1). Mg2+ release was already detectable 10 min after the addition of 2mM NaCN and increased over time in a time-dependent manner. After 20 min from CN− addition 16.2±1.2 nmol Mg2+/mg protein have been released in the extracellular medium. Maximal extrusion was achieved at t = 60 min from the agent administration (36.6±1.1 nmol Mg2+/mg protein). Addition of the mitochondrial uncoupler FCCP (5μg/ml) also resulted in a marked extrusion of Mg2+ from the hepatocytes. In the presence of the uncoupler, however, the amplitude of Mg2+ extrusion was enhanced at all the time points although it reached an end point similar to that measured in the presence of NaCN (Fig. 1). By contrast, Mg2+ extrusion elicited by the addition of the glycolysis inhibitor iodoacetate (2mM) was comparatively smaller (2.62±0.9 nmol/mg protein/10 min) (Fig. 1). Larger doses of iodoacetate (up to 5 mM) did not result in a significant enlargement of Mg2+ extrusion (not shown). The amount of Mg2+ released into the extracellular milieu at t = 20min after the addition of CN− or FCCP (16.7 nmol Mg2+/mg protein) was comparable to the decrease in Mg2+ content in the cell pellets (15.4±0.8 nmol Mg2+/mg protein, n=7), and accounted for approximately 20% of total cellular Mg2+ (Fig. 1). A similar correlation was observed in hepatocytes treated with CN− for 60 min (data not shown)
Figure 1. Mg2+ extrusion from liver cells upon induction of chemical hypoxia.
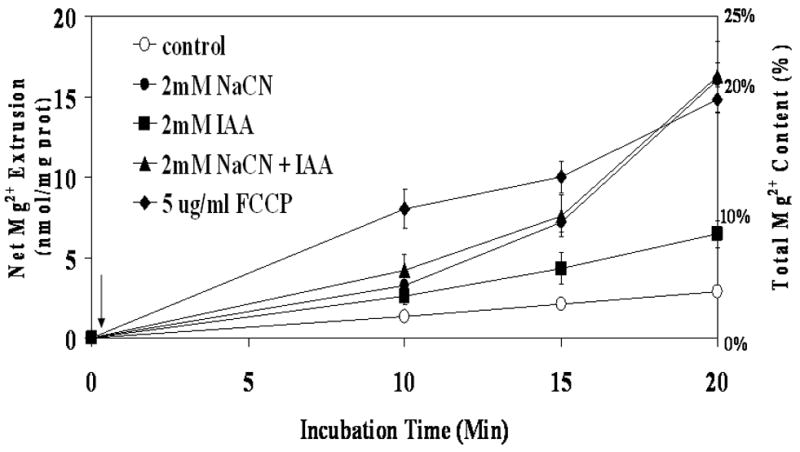
Hepatocytes were isolated and incubated as reported under Materials and Methods. Cells were stimulated by addition of 2mM NaCN (CN−), 2mM Iodoacetic Acid (IAA), NaCN plus IAA, or 5μg/ml FCCP (arrow). The amount of Mg2+ extruded from the cells is reported as the nmol Mg2+/mg protein and as a percent of the total cellular Mg2+ content. Data are means±S.E. of 6 different preparations, each assessed in duplicate. All the data points relative to hepatocytes treated with NaCN, FCCP, IAA and NaCN+IAA are statistical significant vs. corresponding values in Control samples. Labeling is omitted for simplicity.
Addition of 50μg/ml digitonin at t = 60 min after CN− addition resulted in the release of a small residual amount of Mg2+ from the hepatocytes (Fig. 2). In contrast, addition of digitonin to control hepatocytes released more than 30 nmol Mg2+/mg protein, reaching an end-point that was not statistically different from that obtained in CN− treated cell (Fig. 2). Digitonin addition also induced the release of a significant amount of ATP from control hepatocytes (25.3±3.7 nmol ATP/mg protein), but only 1.8±0.6 nmol ATP/mg protein from NaCN treated cells (n=5 for both experimental conditions, p<0.001).
Figure 2. Digitonin-releasable Mg2+ from liver cells treated with NaCN.
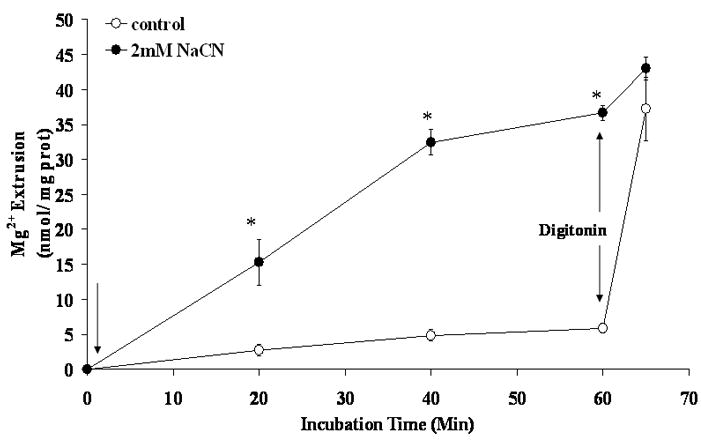
Hepatocytes were isolated and incubated as reported under Materials and Methods. Cells were treated with digitonin (50 μg/ml) following 40 min stimulation with 2 mM NaCN (first arrow). The amount of Mg2+ extruded from the cells is reported as the nmol Mg2+/mg protein. Data are means±S.E. of 6 different preparations, each assessed in duplicate. *Statistical significant vs. corresponding value in NaCN untreated cells.
Under these experimental conditions, cytosolic free [Mg2+]i changed transiently as a result of CN- addition (Fig. 3). A net increase equivalent to 0.4±0.08 mM was observed at time 10 min after CN-addition (Fig. 3A, trace a, and Fig. 3B). This increase in concentration declined towards basal level by time = 20 min from CN-addition (0.1±0.03 mM), remaining to these levels for the remainder of our experimental protocol (Fig. 3A, trace a, and Fig. 3B). When similar experiments were performed on hepatocytes incubated in the absence of extracellular Na+, cytosolic free [Mg2+]i remained elevated for the duration of the experiment (Fig. 3A, trace b, and Fig. 3B) until extracellular Na+ was reintroduced to the system (Fig. 3A).
Figure 3. Changes in cytosolic free [Mg2+]i.
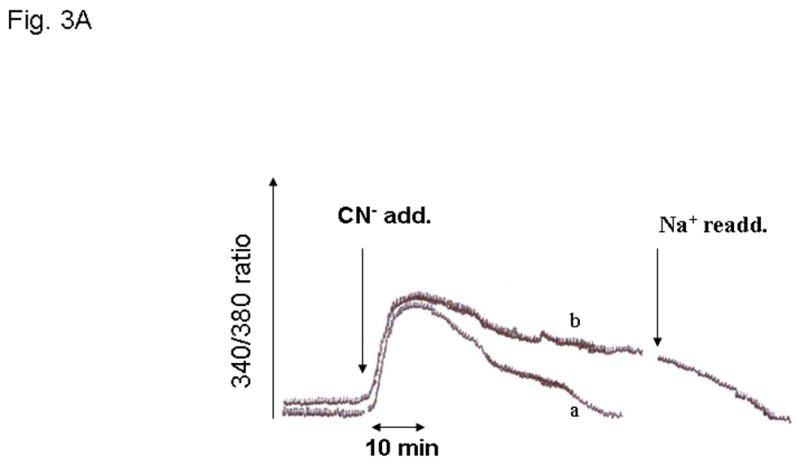
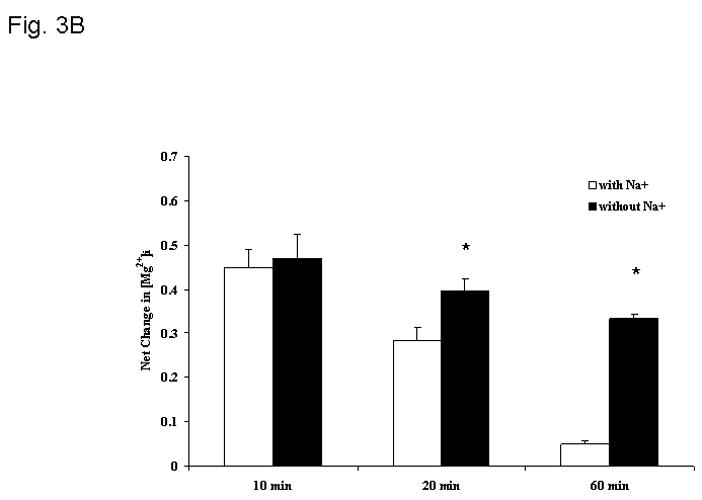
Changes in cytosolic free Mg2+ concentration were determined as described in detail in the Materials and Methods section. Hepatocytes were incubated both in the presence (trace a) and in the absence (trace b) of extracellular Na+. Fig. 3A reports a typical experiment out of 6 different preparations for both experimental conditions, each tested in duplicate. Fig. 3B reports the net change in cytosolic free [Mg2+]i at time 10 min, 20 min and 60 min after CN- addition for hepatocytes incubated in the absence and in the presence of extracellular Na+. Data are means ± S.E. of 6 different preparations, each tested in duplicate for both experimental conditions. *Statistical significant vs. value in the presence of extracellular Na+.
To exclude that Mg2+ extrusion resulted from a nonspecific decrease in permeability, trypan blue exclusion test and determination of LDH release were performed on aliquots of cell suspension. As Table I shows, between 78% to 82% of CN− treated cells (down from the initial 84%–85%) remained trypan blue negative following 20 min of incubation in the absence or in the presence of CN-, irrespective of the absence or the presence of extracellular Na+. No significant release of cytosolic LDH was observed during this period of time (not shown), further excluding the occurrence of cell death, or a non-selective increase in plasma membrane permeability. To minimize possible inconsistencies in our Mg2+ determinations due to variations in the number of dead hepatocytes upon metabolic treatment for the remainder of our study we primarily focused on the 20 min time point.
Table 1.
Cell viability following addition of cyanide or FCCP in the absence or in the presence of extracellular Na+.
| 0 min | 10 min | 20 min | |
|---|---|---|---|
| Experimental Conditions | |||
| Control (with Na+) | 82.9±3.2 | 81.0±4.2 | 78.9±5.7 |
| 2mM NaCN (with Na+) | 82.2±2.8 | 83.4±3.7 | 77.7±6.1 |
| 5μg/ml FCCP (with Na+) | 83.2±3.3 | 81.9±4.5 | 79.5±5.2 |
| Control (without Na+) | 81.8±4.4 | 84.5±2.8 | 80.7±4.7 |
| 2mM NaCN (without Na+) | 85.6±2.3 | 82.3±4.6 | 82.7±2.8 |
Cell viability was assessed by Trypan Blue exclusion test. Values are percent of cell trypan blue negative at the indicated time points. Data are means+S.E. of 5 different experiments, each performed in duplicate.
Dependence of Mg2+ extrusion on extracellular Na+
Previous observation from this and other laboratories indicate that cellular Mg2+ is extruded across the plasma membrane of liver cells through a Na+-dependent and a Na+-independent mechanism under a variety of hormonal stimuli or experimental conditions [4,5,7,25,26]. The possibility that the cyanide-induced Mg2+ extrusion occurred via either mechanism was investigated by selectively modifying the cation composition of the extracellular medium [27]. Removal of extracellular Ca2+ (Fig. 4A) or K+ (not shown) had little or no effect on the amplitude of Mg2+ extrusion induced by CN-. Removal of extracellular Na+, instead, abolished Mg2+ extrusion almost completely (Fig. 4B) as already suggested by the persistent elevation in cytosolic free [Mg2+]i (Fig. 3). The net amount of Mg2+ mobilized into the extracellular medium under the various experimental conditions is reported in Fig. 4C. The selective dependence of Mg2+ extrusion on extracellular Na+ was further supported by the observation that reintroduction of a physiological concentration of extracellular Na+ at a later time point following CN− addition restored the mobilization of Mg2+ into the extracellular compartment (Fig. 5), as indicated by the decrease in intracellular [Mg2+]i (Fig. 3A, trace b). To exclude that the Mg2+ extrusion observed following Na+ reintroduction was due to an osmolarity mismatch, an equiosmolar concentration of sucrose (Fig. 5) or KCl (not shown) was added to the medium instead of the indicated concentration of NaCl. Neither sucrose nor KCl addition was able to elicit Mg2+ extrusion from the hepatocytes.
Figure 4. Na+-dependence of Mg2+ extrusion from liver cells upon induction of chemical hypoxia.
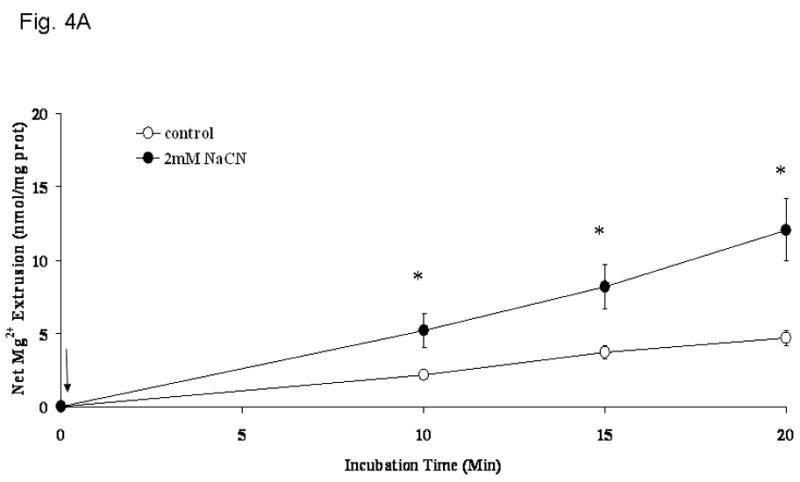
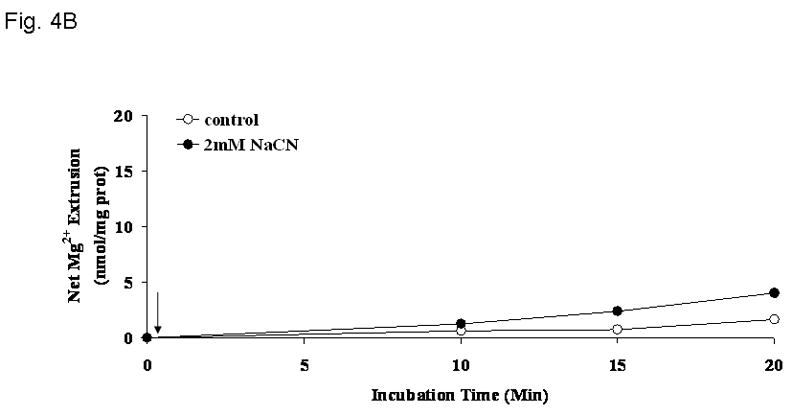
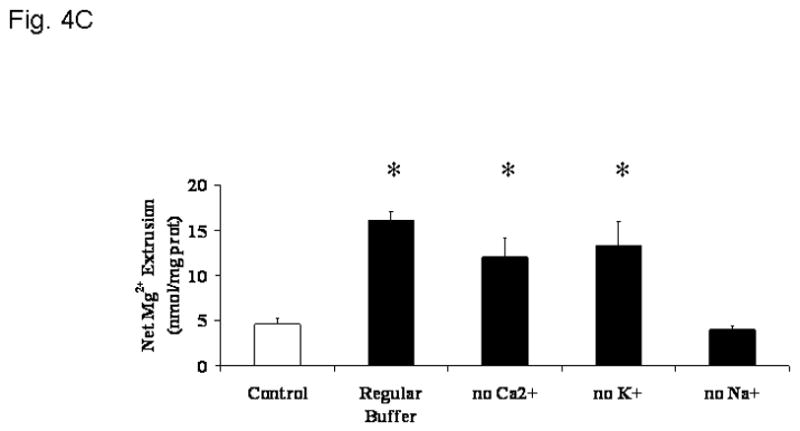
Hepatocytes were isolated and incubated as reported under Materials and Methods. Cells were incubated in a medium devoid of extracellular Ca2+ (Fig. 4A), K+ (not shown), or Na+ (Fig. 4B), and treated with NaCN (arrow = time of addition). Net amount of Mg2+ released in the extracellular medium upon addition of 2mM NaCN is reported as nmol/mg protein in Fig 4C. Data are means±S.E. of 5 different preparations, each assessed in duplicate. *Statistical significant vs. corresponding value in Control cells and cells treated with NaCN in the absence of extracellular Na+.
Figure 5. Effect of Na+-reintroduction on Mg2+ extrusion from liver cells treated with NaCN.
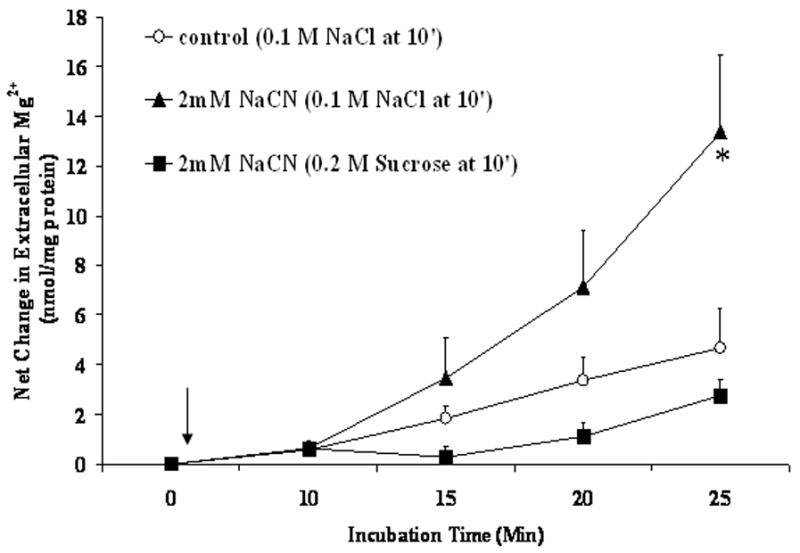
Hepatocytes were isolated and incubated in medium devoid of Na+ as reported under Materials and Methods. Following administration of 2mM NaCN, or 2mM NaCN plus 2mM iodoacetate, 100mM NaCl, 100mM KCl, or an equiosmolar concentration (200mM) of sucrose were added to the incubation system at time = 10 min. Data are means±S.E. of 5 different preparations, each assessed in duplicate for each experimental condition, and are expressed as nmol Mg2+/mg protein. *Statistical significant vs. corresponding value in Control cells.
ATP content
The effectiveness of NaCN in decreasing cellular ATP content was assessed. As Fig. 6 shows, hepatic ATP content decreased to less than 20% of the initial value within 5 min from the administration of NaCN irrespective of the absence or the presence of Na+ in the extracellular milieu. In contrast, ATP level remained relatively stable in untreated cells throughout the duration of the experiment irrespective of the absence or the presence of extracellular Na+.
Figure 6. Change in cellular ATP content upon addition of NaCN.
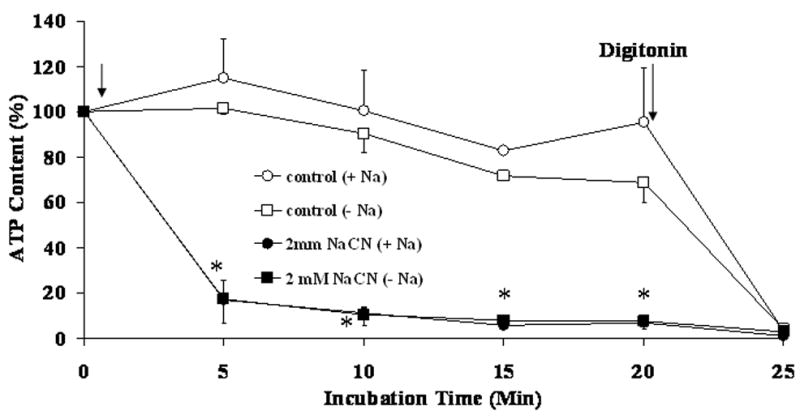
Hepatocytes were isolated and incubated in the absence or in the presence of extracellular Na+ and stimulated by addition of 2mM NaCN (first arrow) as reported under Materials and Methods. Cellular ATP content was assessed by luciferin-luciferase assay as reported previously [23]. Data are means±S.E. of 5 different preparations, each assessed in duplicate. *Statistical significant vs. corresponding value in Control cells.
Pharmacological inhibition of Mg2+ Extrusion
Presently, no specific inhibitor of the putative Na+/Mg2+ exchanger is available. Mg2+ extrusion, however, can be effectively blocked by amiloride, imipramine or quinidine in cells [see ref. 13 as a review] as well as in liver plasma membrane vesicles [11]. As Table 2 shows, the addition of amiloride up to 1mM did not block the extrusion of Mg2+ induced by cyanide (Table 2). By contrast, 100μM imipramine or 1mM quinidine, two agents able to block a variety of Na+ transport mechanisms including Na+ channels, as well as K+ channels, inhibited to a comparable extent (≥75%) the extrusion of Mg2+ induced by cyanide. Experimental evidence indicates that KATP channels are activated by the decrease in cellular ATP content subsequent to chemical hypoxia [17,18]. To test the possibility that quinidine and imipramine prevented Mg2+ extrusion by blocking the KATP channels, glibenclamide [18,28] and pinacidil [29] were used as specific inhibitor and agonist of these channels, respectively. As Table 2 shows, 10μM glibenclamide inhibited the extrusion of Mg2+ induced by cyanide to an extent comparable to that observed in the presence of quinidine and imipramine. By contrast, pinacidil did not elicit an extrusion of Mg2+ per se, nor was effective at potentiating the extrusion elicited by cyanide administration.
Table 2.
Mg2+ extrusion following addition of 2mM NaCN, in the presence of amiloride, quinidine, imipramine and glibencliamide.
| nmol Mg2+/mg protein/20 min | |
|---|---|
| Experimental Conditions | |
| Control | 1.7+0.7 |
| 2mM NaCN | 16.7±1.2a |
| NaCN plus 1mM amiloride | 18.3±3.8a |
| NaCN plus 1 mM quinidine | 4.6±0.5 |
| NaCN plus 100μM imipramine | 4.4±0.2 |
| NaCN plus 100μM glibenclamide | 5.5±0.3 |
| NaCN plus 100 μM Pinacidil | 17.6±1.9a |
Data are means±S.E. of 5 different experiments, each performed in duplicate.
p<0.001 as compared to Control value and to Mg2+ extrusion elicited by NaCN in the presence of quinidine, imipramine or glibenclamide.
pH Regulation of Mg2+ Extrusion
Chemical hypoxia is characterized by a marked decrease (~ 0.35 units) in intracellular pH [30]. An acidic intracellular pH affects the affinity constants of Mg2+ for binding components, mainly phosphonucleotides [30], and results in modest increase in cytosolic free [Mg2+]i [13,31]. On the other hand, acidic extracellular pH has been shown to protect isolated cells and perfused organs from the deleterious effects of anoxia by limiting the entry of Na+ into the cells [20]. Hence, we investigated whether changes in extracellular pH could modulate the amplitude of Mg2+ extrusion. The results reported in Table 3 indicate that either extracellular acidic pH or removal of HCO3− from the incubation medium inhibited by ~80% the amplitude of Mg2+ extrusion. Incubation in an alkaline medium (pH 8.0) tended to enlarge the amplitude of Mg2+ extrusion but the trend did not attain statistical significance.
Table 3.
Dependence of NaCN induced Mg2+ extrusion on extracellular pH.
| nmol Mg2+/mg protein/20 min | |
|---|---|
| Experimental Conditions | |
| pH = 7.2 | 16.7±1.2 |
| pH = 6.5 | 3.4±0.9a |
| pH = 8.0 | 21.3±4.7b |
| without HCO3− | 6.0±1.5a |
Data are means±S.E. of 5 different experiments, each performed in duplicate.
p<0.11 as compared to the pH 7.2 value;
p<0.45 as compared to the pH 7.2 value.
DISCUSSION
Cellular Mg2+ is highly compartmentalized within organelles (namely mitochondria, endoplasmic or sarcoplasmic reticulum, and nucleus [3,32]) and cytoplasm, in which it forms a complex predominantly with ATP [30,33] but also other phosphonucleotides and proteins [33]. The rapid and massive decrease in cytosolic ATP content induced by cyanide or other mitochondria metabolic inhibitors results in a rise in cytosolic free [Mg2+] which is far slower and smaller than it would be expected based upon the rate and amplitude of ATP decrease [30, 34]. Circumstantial evidence has suggested the possibility that the increase in cytosolic free [Mg2+] induced by chemical hypoxia is sufficiently high to displace Ca2+ from its relatively few cytosolic binding sites [30], especially in liver cells. However, the amount of Ca2+ that can be displaced from the hepatocyte’s cytosol is rather limited [30]. Hence, additional mechanisms must be invoked to explain the small rise in cytosolic free Mg2+ observed under these conditions.
In the present study, the possibility that the limited increase in cytosolic free Mg2+ observed upon chemical hypoxia and ATP degradation was due at least in part to a Mg2+ extrusion across the hepatocyte plasma membrane was investigated.
Mg2+ extrusion
Following the addition of cyanide, or FCCP, the cellular level of ATP decreased by 80–85% within 5 min. By contrast, a small but detectable release of Mg2+ from the cell became evident only after a lag phase of 10 min, and increased progressively over time to reach the maximum after ~60 min from the time of the agent addition. At this later time point the addition of digitonin failed to mobilize a significant amount of additional Mg2+ from the cytoplasm, indicating that all cytosolic Mg2+ had already been extruded. Assessment of Mg2+ content within mitochondria and endoplasmic reticulum indicated that these organelles retained the same amount of Mg2+ irrespective of the addition of CN− in the absence or in the presence of external Na+ (data not shown). Hence, these data exclude the possibility of Mg2+ redistribution among cytoplasm and cellular organelles occurred under our experimental conditions, and further emphasize that Mg2+ extrusion is the predominant cause of Mg2+ loss from the cytoplasm. At the present time, we do not have an explanation for the observed lag phase in Mg2+ extrusion. One possibility is that a certain increase in cytosolic free [Mg2+]i has to be attained before the cell ‘senses’ the increase and activates the Mg2+ extrusion mechanisms. Because the nature of this ‘sensor’ is still undetermined, we are not in the position to properly test this hypothesis. An alternative explanation is that the lag phase represents an attempt by the cell to buffer the excess free Mg2+ through cytosolic proteins or ADP and AMP generated from the degradation of ATP. As the levels of ADP and AMP also decline overtime following CN− poisoning, an increased mobilization and extrusion of Mg2+ from the cell would result. The profile of the changes in cytosolic free [Mg2+]i, however, would be consistent with the first of these possibilities, as a larger increase in [Mg2+]i was recorded at a time point after CN- addition at which little Mg2+ extrusion is detected in the extracellular space (compare Fig. 3 and Fig. 1). As for the different extrusion profile observed following CN- or FCCP addition, this could be explained by the marked lipophilicity of FCCP, which results in the rapid redistribution of the uncoupler within the mitochondrial membrane and collapses the proton gradient across the mitochondrial membrane. It has to be noted, however, that the end-point for both CN− and FCCP-induced Mg2+ extrusion is quite similar (Fig. 1), suggesting the draining of the same pool.
The release of Mg2+ from the hepatocyte is a very specific process as attested by the following lines of evidence: 1) Mg2+ extrusion occurs in the absence of LDH release or changes in cell membrane permeability, cell viability, or extracellular osmolarity; 2) Mg2+ is extruded from the cell via the operation of a transport mechanism that is blocked by agents that inhibit Na+ transport (although not specifically), or by the removal of extracellular Na; and 3) reintroduction of Na+ under the latter condition is essential to restore Mg2+ extrusion to an extent comparable to that observed under normal experimental conditions.
Taken together, these results are consistent with the activation of a specific transport mechanism which, based upon its strict Na+ dependence, can be tentatively identified with the Na+/Mg2+ exchanger operating in various cell types including hepatocytes [1,7,8]. Our results are also consistent with numerous experimental observations including work by Flatman et al. [35] and Kubota et al., [25,26] with fluorescent indicators indicating a persistent elevation in cytosolic free [Mg2+]i under conditions in which extracellular Na+ is not available to favor Mg2+ extrusion. The specific inhibition of Mg2+ extrusion by imipramine or quinidine, two agents reported to block the Na+-dependent Mg2+ transport in a variety of cells [11,36], further indicates that the extrusion of Mg2+ induced by the addition of cyanide occurs through a specific transport mechanism. Surprisingly, amiloride, the drug most commonly used to inhibit the Na+-dependent Mg2+ extrusion from liver and other tissues [7] under various experimental conditions, appears to be ineffective at inhibiting the cyanide-induced Mg2+ extrusion even when administered at millimolar concentrations. The reason for this lack of an effect is not clear. At least three explanations are at hand. The first possibility is that amiloride only inhibits Mg2+ transport under conditions in which cellular ATP content is not decreased or is only marginally affected by treatment. Experiments in purified total liver plasma membrane devoid of ATP, however, indicate that this is not the case in that amiloride and imipramine are equally effective at inhibiting Na+-dependent Mg2+ extrusion [11]. A second possibility is that imipramine (or quinidine) and amiloride inhibit different Na+-dependent Mg2+ extrusion mechanisms. This possibility is supported by the experimental evidence that in purified liver plasma membrane vesicles amiloride only inhibits a Na+/Mg2+ exchange mechanism present in the apical domain (bile side) of the hepatocyte, leaving unaffected the one operating in the basolateral domain (blood side) of the cell [11]. In contrast, imipramine inhibits both exchangers to a comparable extent [11]. This observation could explain the results reported here as well as why the co-addition of imipramine and amiloride do not exert an additive inhibitory effect on the Na+-dependent Mg2+ extrusion from intact hepatocytes (Romani, personal observation) or purified liver plasma membranes even in the absence of ATP [37]. A third and not mutually exclusive explanation would be that Na+ is entering the cells through a route insensitive to amiloride. The marked intracellular acidification induced by cyanide treatment [30] activates the Na+/H+ antiporter and the Na+/HCO3− sinporter [38] in the attempt to restore cytosolic pH. While the Na+/H+ antiporter is inhibited by amiloride [39], the Na+/HCO3− sinporter is amiloride-insensitive [40], and can transport Na+ inwardly even in the presence of high concentrations of the inhibitor. As the accumulated Na+ cannot be extruded via the Na+/K+ ATPase for lack of ATP, Mg2+ and possibly K+ [41] would be extruded from the cell for charge compensation.
The observed inhibitory effect of glibenclamide would be consistent with the latter scenario. The plasma membrane of several cells [29] possesses a class of channels regulated by ATP and Mg2+ that extrude K+ when active. These channels, known as KATP channels, become active following the decrease in cellular ATP content induced by metabolic inhibitors [18]. The selective block of these channels by glibenclamide would prevent K+ extrusion and limit, in turn, the entry of Na+ into the cells. The end result will be a decreased in the driving force required for the extrusion of Mg2+ to occur. It has to be noted that the KATP channel agonist, pinacidil is unable to induce a Mg2+ extrusion in the absence of cyanide, and does not enhance the cation extrusion induced by the mitochondrial inhibitor. The most likely explanation for these results is that, in the absence of cyanide, the opening of the KATP channels and the K+ extrusion do not result in a major entry of Na+ because of the compensatory activity of the Na+/K+-ATPase. Under these conditions, cellular ATP content remains a valid Mg2+ buffering system, thus preventing Mg2+ mobilization from the cell. Following cyanide poisoning and the decrease in cellular ATP content, the activation of KATP channels and the mobilization of Mg2+ would be already maximal, de facto rendering the effect of pinacidil undetectable.
Role of pH
One consequence of the decrease in cellular ATP induced by cyanide is the marked cellular acidification [30]. This acidification results from the compensatory activation of the Na+/H+ exchanger to remove the excess cellular Na+ that cannot be extruded via the Na+/K+-ATPase due to the decrease in ATP level [30]. This cellular acidification, however, has a major impact on cytosolic Mg2+ homeostasis in that it changes the affinity constant of ATP, adenine phosphonucleotides and phosphocreatine for Mg2+ [30] at a time in which the content of these complexing moieties is decreasing as a result of cyanide poisoning. The final result is a facilitated release of Mg2+ from its complexes and extrusion from the cell. As indicated by the results reported in Table 3, the difference in transmembrane pH has a major role in regulating cellular Mg2+ extrusion. By decreasing the pH difference across the cell membrane, an extra-cellular acidic pH would reduce the activation of Na+/H+ antiporter and Na+/HCO3− sinporter, and consequently decrease the entry of Na+ that constitute the driving force for Mg2+ extrusion to occur [1]. Similarly, the absence of external HCO3− would prevent the entry of Na+ via the Na+/HCO3−sinporter and hamper the amplitude of Mg2+ extrusion by affecting the driving force required for its transport. This would imply that the not Na+/H+ antiporter but rather the Na+/HCO3− sinporter plays a major role in the Na+-dependent, cyanide-induced Mg2+ extrusion. Support to this hypothesis is provided by the evidence that FCCP, which abolishes the H+ gradient across all biological membranes including the plasma membrane, elicits an extrusion of Mg2+ fairly similar to that induced by cyanide. Thus, it is possible that upon cyanide administration the entry of Na+ occurs mainly via the Na+/HCO3− sinporter, favored by the extrusion of K+ through the KATP channels.
During the preparation of this manuscript, a study by Tashiro et al. [43] reported results in cardiac myocytes undergoing a series of experiments similar to those described here in hepatocytes. At variance of what reported here, Tashiro et al. observed that KCN induced intracellular acidosis and decrease in cellular ATP level below the threshold that elicits rigor cross-bridge formation and cell shortening both contribute to inhibit Mg2+ efflux from the cardiac myocytes [43]. Based on these results, the authors propose an absolute requirement of ATP for the operation of the Na+/Mg2+ exchanger in cardiac myocytes [43], as already proposed by Ebel et al. [44] in red blood cells.
We do not have a clear explanation for the different ATP requirements in cardiac myocytes and hepatocytes. We cannot exclude that spontaneously contracting cells like cardiac myocytes may regulate differently the Na+/Mg2+ exchanger due to its likely involvement in modulating Na+ and Ca2+ homeostasis and consequently cell contractility [45]. On the other hand, it has to be noted that the time of exposure to KCN or Na+ depleted solution reported by Tashiro and Konishi in most experiments (i.e. 70 to 170 min [41]) far exceeded the time utilized in the present study (20 min for the bulk of our reported data).
A last question is whether the Mg2+ extrusion is simply the result of reduced buffering capacity in the cytosol and increased entry of Na+ through the plasma membrane, or it can also operate as an intracellular signal. Hue and collaborators [46] have reported that the decrease in cytosolic ATP content that follows fructose administration to hepatocytes, while modest as compared to that attained following cyanide poisoning, results in an increase in cytosolic free [Mg2+] sufficient to activate glycogen phosphorylase and promote glycegenolysis and glycolysis for energy production purpose. Thus, if we consider cytosolic Mg2+ as an indirect indicator of the metabolic state of the cell, it is possible to envisage a scenario in which the reduction in cellular ATP content induced by cyanide would lead to a rise in cytosolic [Mg2+] which, in turn, may activate anaerobic glycolysis to produce ATP and compensate, or attempt to, for the inability of mitochondria to provide metabolic energy..
Conclusions
Taken together, the data reported here suggest that in liver cells the Na+/Mg2+ exchange mechanism responsible for the extrusion of cellular Mg2+ following hormonal stimulation also plays a key role in extruding Mg2+ under condition in which the cytosolic concentration of the cation rises sensibly as a consequence of a decrease in cellular ATP buffering capacity.
Acknowledgments
Supported by NIAAA- AA11593
Footnotes
The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Flatman PW. Magnesium transport across cell membranes. J Membr Biol. 1984;80:1–14. doi: 10.1007/BF01868686. [DOI] [PubMed] [Google Scholar]
- 2.Grubbs RD, Maguire ME. Magnesium as a regulatory cation: criteria and evaluation. Magnesium. 1987;6:113–127. [PubMed] [Google Scholar]
- 3.Romani A, Scarpa A. Regulation of cell magnesium. Arch Biochem Biophys. 1992;298:1–12. doi: 10.1016/0003-9861(92)90086-c. [DOI] [PubMed] [Google Scholar]
- 4.Romani A. Regulation of magnesium homeostasis and transport in mammalian cells. Arch Biochem Biophys. 2007;458:90–102. doi: 10.1016/j.abb.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Wolf FI, Torsello A, Fasanella S, Cittadini A. Cell physiology of magnesium. Mol Aspects Med. 2003;24:11–26. doi: 10.1016/s0098-2997(02)00088-2. [DOI] [PubMed] [Google Scholar]
- 6.Fatholahi M, Lanoue K, Romani A, Scarpa A. Relationship between total and free cellular Mg2+ during metabolic stimulation of rat cardiac myocytes and perfused hearts. Arch Biochem Biophys. 2000;374:395–401. doi: 10.1006/abbi.1999.1619. [DOI] [PubMed] [Google Scholar]
- 7.Gunther T. Mechanisms and regulation of Mg2+ efflux and Mg2+ influx. Miner Electrolyte Metab. 1993;19:259–265. [PubMed] [Google Scholar]
- 8.Flatman PW. Mechanisms of magnesium transport. Annu Rev Physiol. 1991;53:259–271. doi: 10.1146/annurev.ph.53.030191.001355. [DOI] [PubMed] [Google Scholar]
- 9.Romani A, Scarpa A. Regulation of cellular magnesium. Front Biosci. 2000;5:D720–D734. doi: 10.2741/romani. [DOI] [PubMed] [Google Scholar]
- 10.Gunther T, Vormann J. Na+-independent Mg2+ efflux from Mg2+-loaded human erythrocytes. FEBS Lett. 1989;247:181–184. doi: 10.1016/0014-5793(89)81329-8. [DOI] [PubMed] [Google Scholar]
- 11.Cefaratti C, Romani A, Scarpa A. Differential localization and operation of distinct Mg2+ transporters in apical and basolateral sides of rat liver plasma membrane. J Biol Chem. 2000;275:3772–3780. doi: 10.1074/jbc.275.6.3772. [DOI] [PubMed] [Google Scholar]
- 12.Cefaratti C, Ruse C. Protein kinase A dependent phosphorylation activates Mg2+ efflux in the basolateral region of the liver. Mol Cell Biochem. 2007;297:209–214. doi: 10.1007/s11010-006-9325-1. [DOI] [PubMed] [Google Scholar]
- 13.Harman AW, Nieminen AL, Lemasters JJ, Herman B. Cytosolic free magnesium, ATP and blebbing during chemical hypoxia in cultured rat hepatocytes. Biochem Biophys Res Commun. 1990;170:477–483. doi: 10.1016/0006-291x(90)92116-h. [DOI] [PubMed] [Google Scholar]
- 14.Carroll RG, Thomas JM. Fluorescent monitoring of Jurkatt cell intracellular magnesium during metabolic poisoning. Shock. 1994;1:213–216. doi: 10.1097/00024382-199403000-00009. [DOI] [PubMed] [Google Scholar]
- 15.Grubbs RD, Walter A. Determination of cytosolic Mg2+ activity and buffering in BC3H-1 cells with mag-fura-2. Mol Cell Biochem. 1994;136:11–22. doi: 10.1007/BF00931599. [DOI] [PubMed] [Google Scholar]
- 16.Buck LT, Hochachka PW. Anoxic suppression of Na+-K+-ATPase and constant membrane potential in hepatocytes: support for channel arrest. Am j Physiol. 1993;265:R1020–R1025. doi: 10.1152/ajpregu.1993.265.5.R1020. [DOI] [PubMed] [Google Scholar]
- 17.Leyssens A, Nowicky AV, Patterson L, Crompton M, Duchen MR. The relationship between mitochondrial state, ATP hydrolysis, [Mg2+]i and [Ca2+]i studied in isolated rat cardiomyocytes. J Physiol (London) 1996;496:111–128. doi: 10.1113/jphysiol.1996.sp021669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venkatesh N, Lamp ST, Weiss JN. Sulfonylureas, ATP-sensitive K+ channels, and cellular K+ loss during hypoxia, ischemia, and metabolic inhibition in mammalian ventricle. Circ Res. 1991;69:623–637. doi: 10.1161/01.res.69.3.623. [DOI] [PubMed] [Google Scholar]
- 19.Gores GJ, Nieminen AL, Fleishman KE, Dawson TL, Herman B, Lemasters JJ. Extracellular acidosis delays onset of cell death in ATP-depleted hepatocytes. Am J Physiol. 1988;255:C315–C322. doi: 10.1152/ajpcell.1988.255.3.C315. [DOI] [PubMed] [Google Scholar]
- 20.Carini R, Bellomo G, Benedetti A, Fulceri R, Gamberucci A, Parola M, Dianzani MU, Albano E. Alteration of Na+ homeostasis as a critical step in the development of irreversible hepatocyte injury after adenosine triphosphate depletion. Hepatology. 1995;21:1089–1098. [PubMed] [Google Scholar]
- 21.Romani A, Dowell E, Scarpa A. Cyclic AMP-induced Mg2+ release from rat liver hepatocytes, permeabilized hepatocytes, and isolated mitochondria. J Biol Chem. 1991;266:24376–24384. [PubMed] [Google Scholar]
- 22.Raju B, Murphy E, Levy LA, Hall RD, London RE. A fluorescent indicator for measuring cytosolic free magnesium. Am J Physiol. 1989;256:C540–C548. doi: 10.1152/ajpcell.1989.256.3.C540. [DOI] [PubMed] [Google Scholar]
- 23.Tessman P, Romani A. Acute ethanol administration affects Mg2+ homeostasis in liver cells: Evidence for the activation of a Na+/Mg2+ exchanger. Am J Physiol. 1998;275:G1106–G1116. doi: 10.1152/ajpgi.1998.275.5.G1106. [DOI] [PubMed] [Google Scholar]
- 24.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 25.Kubota T, Tokuno K, Nakagawa J, Kitamura Y, Ogawa H, Suzuli Y, Suzuki K, Oka K. Na+/Mg2+ transporter acts as a Mg2+ buffering mechanism in PC12 cells. Biochim Biophys Res Commun. 2003;303:332–336. doi: 10.1016/s0006-291x(03)00346-2. [DOI] [PubMed] [Google Scholar]
- 26.Kubota T, Shindo Y, Tokuno K, Komatsu H, Ogawa H, Kudo S, Kitamura Y, Suzuki K, Oka K. Mitochondria are intracellular magnesium stores: investigation by simultaneous fluorescent imaging in PC12 cells. Biochim Biophys Acta. 2005;1744:19–28. doi: 10.1016/j.bbamcr.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 27.Fagan TE, Romani A. Activation of Na+- and Ca2+-dependent Mg2+ extrusion by α1- and β-adrenergic agonists in rat liver cells. Am J Physiol. 2000;279:G943–G950. doi: 10.1152/ajpgi.2000.279.5.G943. [DOI] [PubMed] [Google Scholar]
- 28.Virag L, Furukawa T, Hiraoka M. Modulation of the effect of glibenclamide on KATP channels by ATP and ADP. Mol Cell Biochem. 1993;119:209–215. doi: 10.1007/978-1-4615-3078-7_28. [DOI] [PubMed] [Google Scholar]
- 29.Allard B, Lazdunski M, Rougier O. Activation of ATP-dependent K+ channels by metabolic poisoning in adult mouse skeletal muscle: role of intracellular Mg2+ and pH. J Physiol (London) 1995;485:283–296. doi: 10.1113/jphysiol.1995.sp020730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gasbarrini A, Borle AB, Farghali H, Bender C, Francavilla A, Van TD. Effect of anoxia on intracellular ATP, Na+i, Ca2+i, Mg2+i, and cytotoxicity in rat hepatocytes. J Biol Chem. 1992;267:6654–6663. [PubMed] [Google Scholar]
- 31.Silverman HS, Di LF, Hui RC, Miyata H, Sollott SJ, Hanford RG, Lakatta EG, Stern MD. Regulation of intracellular free Mg2+ and contraction in single adult mammalian cardiac myocytes. Am J Physiol. 1994;266:C222–C233. doi: 10.1152/ajpcell.1994.266.1.C222. [DOI] [PubMed] [Google Scholar]
- 32.Gunther T. Functional compartmentation of intracellular magnesium. Magnesium. 1986;5:53–59. [PubMed] [Google Scholar]
- 33.Scarpa A, Brinley FJ. In situ measurements of free cytosolic magnesium ions. Fed Proc. 1981;40:2646–2652. [PubMed] [Google Scholar]
- 34.Li HY, Dai LJ, Krieger C, Quamme GA. Intracellular Mg2+ concentrations following metabolic inhibition in opossum kidney cells. Biochim Biophys Acta. 1993;1181:307–315. doi: 10.1016/0925-4439(93)90037-2. [DOI] [PubMed] [Google Scholar]
- 35.Almulla HA, Bush PG, Steele MG, Ellis D, Flatman PW. Loading rat heart myocytes with Mg2+ using low [Na+] solutions. J Physiol. 2006;575:443–454. doi: 10.1113/jphysiol.2006.109850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feray JC, Garay R. Demonstration of a Na+:Mg2+ exchange in human red cells by its sensitivity to tricyclic antidepressant drugs. Naunyn Schmiedebergs Arch Pharmacol. 1988;338:332–337. doi: 10.1007/BF00173409. [DOI] [PubMed] [Google Scholar]
- 37.Cefaratti C, Romani A, Scarpa A. Characterization of two Mg2+ transporters in sealed plasma membrane vesicles from rat liver [In Process Citation] Am J Physiol. 1998;275:C995–C1008. doi: 10.1152/ajpcell.1998.275.4.C995. [DOI] [PubMed] [Google Scholar]
- 38.Carini R, Bellomo G, Benedetti A, Fulceri R, Gamberucci A, Parola M, Dianzani MU, Albano E. Alteration of Na+ homeostasis as a critical step in the development of irreversible hepatocyte injury after adenosine triphosphate depletion. Hepatology. 1995;21:1089–1098. [PubMed] [Google Scholar]
- 39.Levitsky J, Gurell D, Frishman WH. Sodium ion/hydrogen ion exchange inhibition: a new pharmacologic approach to myocardial ischemia and reperfusion injury. J Clin Pharmacol. 1998;38:887–897. doi: 10.1002/j.1552-4604.1998.tb04383.x. [DOI] [PubMed] [Google Scholar]
- 40.Boron WF, Chen L, Parker M. Modular structure of sodium-coupled bicarbonate transporters. J Exp Biol. 2009;22:1697–1706. doi: 10.1242/jeb.028563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krumschnabel G, Frischmann ME, Schwarzbaum PJ, Wieser W. Loss of K+ homeostasis in trout hepatocytes during chemical anoxia: a screening study for potential causes and mechanisms. Arch Biochem Biophys. 1998;353:199–206. doi: 10.1006/abbi.1998.0646. [DOI] [PubMed] [Google Scholar]
- 42.Vidal G, Durand T, Canioni P, Gallis J-L. Cytosolic pH regulation in perfused rat liver: role of intracellular bicarbonate production. Biochim Biophys Acta. 1998;1425:224–234. doi: 10.1016/s0304-4165(98)00075-0. [DOI] [PubMed] [Google Scholar]
- 43.Tashiro M, Inoue H, Konishi M. Metabolic inhibition strongly inhibits Na+-dependent Mg2+ efflux in rat ventricular myocytes. Biophys J. 2009;96:4941–4950. doi: 10.1016/j.bpj.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ebel H, Kreis R, Günther T. Regulation of Na+/Mg2+ antiport in rat erythrocytes. Biochim Biophys Acta. 2004;1664:150–160. doi: 10.1016/j.bbamem.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 45.Bers DM, Barry WH, Despa S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res. 2003;57:897–912. doi: 10.1016/s0008-6363(02)00656-9. [DOI] [PubMed] [Google Scholar]
- 46.Gaussin V, Gailly P, Gillis JM, Hue L. Fructose-induced increase in intracellular free Mg2+ ion concentration in rat hepatocytes: relation with the enzymes of glycogen metabolism. Biochem J. 1997;326:823–827. doi: 10.1042/bj3260823. [DOI] [PMC free article] [PubMed] [Google Scholar]