

Abstract
Retinal pigment epithelium (RPE)-derived membranous debris named blebs, may accumulate and contribute to sub-RPE deposit formation, which is the earliest sign of age-related macular degeneration (AMD). Oxidative injury to the RPE might play a significant role in AMD. However, the underlying mechanisms are unknown. We previously reported that hydroquinone (HQ), a major pro-oxidant in cigarette smoke, foodstuff, and atmospheric pollutants, induces actin rearrangement and membrane blebbing in RPE cells as well as sub-RPE deposits in mice. Here, we show for the first time that phosphorylated Heat shock protein 27 (Hsp27), a key regulator of actin filaments dynamics, is up-regulated in RPE from patients with AMD. Also, HQ-induced nonlethal oxidative injury led to Hsp27mRNA up-regulation, dimer formation, and Hsp27 phosphorylation in ARPE-19 cells. Furthermore, we found that a cross talk between p38 and extracellular signal-regulated kinase (ERK) mediates HQ-induced Hsp27 phosphorylation and actin aggregate formation, revealing ERK as a novel upstream mediator of Hsp27 phosphorylation. Finally, we demonstrated that Hsp25, p38, and ERK phosphorylation are increased in aging C57BL/6 mice chronically exposed to HQ, whereas Hsp25 expression is decreased. Our data suggest that phosphorylated Hsp27 might be a key mediator in AMD and HQ-induced oxidative injury to the RPE, which may provide helpful insights into the early cellular events associated with actin reorganization and bleb formation involved in sub-RPE deposits formation relevant to the pathogenesis of AMD.
Age-related macular degeneration (AMD) is the main cause of blindness in the elderly population in the Western societies and has a devastating impact on people’s quality of life.1,2,3,4 An estimated 13 million Americans have some degree of AMD and unless better preventive treatments emerge, this number is expected to climb and even reach epidemic proportions with the overall aging demographics. Although the cause of AMD has not yet been fully determined, this complex multifactorial disease clearly results from the interplay of multiple genetic and environmental risk factors2,5 that lead to progressive destruction of retinal pigment epithelial (RPE) cells and photoreceptors and ultimately to vision loss.
Accumulation of deposits between the RPE and its basement membrane (called basal laminar deposits) is part of the numerous biochemical and anatomical changes associated with aging retina. However, the presence of large numbers and extensive areas of additional deposits (named basal linear deposits and drusen) beneath the RPE and within the inner collagenous layer of Bruch’s membrane (BrM) is a defining histopathological landmark associated with dry AMD,6 the most common type of the disease that affects the greatest number of people. Until now, the underlying pathogenic mechanisms of sub-RPE deposits formation and accumulation as well as their relative contribution to the pathogenesis of AMD are still obscure. However, a growing body of evidence suggests that cumulative oxidative injury is implicated in the pathogenesis of the disease7,8,9 and plays a critical role in deposits formation.10 Cigarette smoking is a major source of oxidative stress and has unequivocally been established as the single greatest environmental risk factor in the onset and development of AMD.11,12,13,14 It has been suggested that cigarette smoking might contribute to the pathogenesis of AMD by causing oxidative damage to the RPE.15 Tar within cigarette smoke contains a large number of pro-oxidant compounds among which hydroquinone (HQ) is the most abundant. HQ is an oxidant of special relevance due to its presence not only in cigarette smoke but also in processed foods, plastic containers, and atmospheric pollutants as well as its widespread occurrence in nature.16,17 Detectable levels of HQ have been measured in the plasma of many nonsmoker individuals living a Western life style.16
We embrace the pathogenic paradigm based on the response-to-injury hypothesis, which proposes that sub-RPE deposits originate from RPE-derived cell membrane blebs18,19 induced by chronic nonlethal injury to the RPE in response to oxidative damage. Clinical studies performed on eyes from patients with AMD have revealed that membranous debris from the basolateral RPE surface traversed the RPE membrane and deposited in the inner and outer collagenous layers of BrM.20 We previously found that exposure to cigarette smoke and HQ resulted in RPE membrane blebbing and sub-RPE deposits in mice.21,22 We also demonstrated that nonlethal HQ-induced oxidative injury in cultured RPE cells results in reorganization of actin cytoskeleton and blebs formation22,23,24,25 relevant to the accumulation of deposits. Blebbing of the plasma membrane is an early morphological sign of cell injury, which occurs immediately after exposure to a wide variety of toxic agents. Oxidative stress induces profound rearrangements of the actin cytoskeleton26,27,28 leading to membrane blebbing through the activation of p38 mitogen activated protein kinase (MAPK)/Heat shock protein 27 (Hsp27) pathway.26,29
Hsp27 (Hsp25 in mice) is a phosphorylation-regulated filamentous actin (F-actin) cap binding protein that plays a key role in modulating actin microfilaments dynamics.30 We previously reported that ARPE-19 cells constitutively express high levels of Hsp27, which are regulated by oxidant-mediated injury.31 We also found that Hsp27 is expressed in extruded blebs confirming that Hsp27 plays a role in actin filaments dynamics. In addition, we demonstrated that prolonged exposure of RPE cells to HQ results in actin rearrangements into aggregates and membrane blebbing, with a concomitant activation of the p38 MAPK pathway showed by immunohistochemistry.25 However, whether HQ-induced oxidative injury can activate other MAPK signaling pathways, including extracellular signal-regulated kinase 1/2 (ERK1/2) cascade, has not been investigated. In addition, this study did not address the question of any posttranslational modifications such as dimerization and phosphorylation of Hsp27 known to regulate F-actin polymerization. Taken together, these data suggest that Hsp27 may be an important mediator of RPE response to HQ-induced oxidative damage and may contribute to injury-induced actin rearrangement and blebbing, which may be crucial in the formation of sub-RPE deposits. The aim of this study was to evaluate the expression of phosphorylated Hsp27 in RPE from patients with AMD and further characterize the contribution of phosphorylated Hsp27 in mediating F-actin cytoskeleton reorganization during nonlethal HQ-induced oxidative stress as well as the involvement of p38 and ERK MAPK pathways. A greater understanding of the sequence of early cellular events associated with HQ-induced F-actin dysregulation in RPE cells is of critical importance to further identify its role in the regulation of RPE blebbing and define its contribution to the accumulation of sub-RPE deposits relevant to the pathophysiology of AMD.
Materials and Methods
Compounds and Drugs
Hydroquinone and dispase were purchased from Sigma-Aldrich (St. Louis, MO). Mouse monoclonal anti-endothelial cell (CD146) antibody was purchased from Chemicon International (Temecula, CA). Mouse monoclonal Hsp25/27 and phospho-Hsp25/27 (Ser82) antibodies were purchased from Stressgen (Ann Arbor, MI). Rabbit p38, mouse phospho-p38 (Thr180/182), mouse monoclonal β-actin, and rabbit monoclonal glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were purchased from Cell Signaling (Danvers, MA). Rabbit ERK1/2 antibody and PCR Master Mix were purchased from Promega (Madison, WI). Mouse phospho-ERK1/2 antibody, horseradish-peroxidase-linked donkey anti-rabbit or anti-mouse antibody, the chemiluminescent solution for Western blots, mouse monoclonal anti-cytokeratin 18 antibody, and Hsp27 small-interfering RNA (siRNA) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Detergent compatible (DC) protein quantification kit, agarose, and iQ SybrGreen Supermix were purchased from BioRad (Hercules, CA). Rhodamine-phalloidin and Alexa Fluor 488 goat anti-mouse IgG antibodies were purchased from Molecular Probes (Eugene, OR). Protein lysis buffer M-PER was purchased from Pierce (Rockford, IL). SB203580 was purchased from Upstate (Lake Placid, NY). Okadaic acid, PD98059, ethidium bromide, bovine serum albumin, SDS, Tris, TRIreagent, and all PCR primers were purchased from Sigma-Aldrich (St. Louis, MO). High-Capacity cDNA Archive Kit was purchased from Applied Biosystems (Foster City, CA). PBS, Dulbecco’s modified Eagle’s medium-Ham’s F12, fetal bovine serum (FBS), l-glutamine, penicillin/streptomycin, Tris-buffered saline, Na2HCO3, and 4, 6-diamidino-2-phenylindole-2-HCL were purchased from Invitrogen-GIBCO (Carlsbad, CA).
Human Tissue Samples
Human RPE lysates were prepared from isolated RPE sheets kindly provided by the Lions Eye Bank (Miami, FL) and Dr. George Inana (University of Miami, Miami, FL; n = 4 patients given a diagnosis of dry AMD and n = 4 control donors with no known history of eye disease). In addition, paraffin-embedded eye cross-sections from donors (n = 3 patients given a diagnosis of AMD and n = 3 controls with no known history of eye disease) were obtained from the National Disease Research Interchange (Philadelphia, PA). Table 1 provides available information about the eye donors. The present study adhered to the tenets of the Declaration of Helsinki.
Table 1.
Human Donor Eyes Information
| Samples | Control donors
|
Dry AMD donors
|
||||
|---|---|---|---|---|---|---|
| Age, year | Sex | Smoker | Age, year | Sex | Smoker | |
| Ocular slices | ||||||
| 1 | 82 | F | No | |||
| 2 | 80 | M | No | |||
| 3 | 79 | F | No | |||
| 4 | 87 | F | Yes | |||
| 5 | 76 | F | Yes | |||
| 6 | 81 | M | Quit at 69 (45 pack/year) | |||
| Isolated RPE sheets | ||||||
| 1 | 64 | M | No | |||
| 2 | 85 | F | No | |||
| 3 | 69 | M | ? | |||
| 4 | 74 | M | ? | |||
| 5 | 70 | M | Yes | |||
| 6 | 68 | M | Yes | |||
| 7 | 81 | M | ? | |||
| 8 | 75 | M | Yes | |||
F, female; M, male.
? denotes information on smoking status was unavailable.
Mice
Eighteen-month-old female C57BL/6 mice (Jackson Laboratories, Bar Harbor, MN) were used in this study. All experiments were conducted according to the Association for Research Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the University of Miami Care and Use Committee. Mice were divided into two groups. Group 1 consisted of control mice receiving regular drinking tap water (n = 6) and group 2 consisted of mice receiving HQ orally in drinking water (0.8% HQ) for 7 months (n = 6). Mice were housed in plastic cages with free access to food and water, and were kept on a 12-hour light–dark cycle. At the end of the experimental period, mice were euthanized and eyes were immediately removed for recovery of the RPE sheets.
RPE Isolation from Mice and Human Donors
RPE sheets were isolated as previously described.32 Briefly, the eyes were enucleated, rinsed with 10% gentamicin for sterilization and twice with PBS 1X, and placed in a dish containing PBS 1X. Using a dissecting microscope, eyes were opened by a circumferential incision at the ora serrata. The anterior segment was removed, and the vitreous-retina was separated from the RPE and choroid eyecup with a round-tipped disposable blade (K20-1504; Katena Products, Inc., Denville, NJ) and Tennant forceps (K5-5230; Katena Products, Inc.). The remaining eyecup was incubated with dispase in PBS 1X at 37°C for 30 minutes. Then, using a Barraquer spatula (K3-2310; Katena Products, Inc.), the RPE monolayer was dissected from BrM and choroid. The RPE sheets were transferred into individual tubes (Eppendorf, Fremont, CA) and homogenized on ice with a pestle. Subsequently, total RNA and protein were extracted and stored at −80°C. Aliquots from the isolated RPE sheets were stained with an anti-cytokeratin 18 antibody as a positive control for epithelial cells and with an anti-endothelial (CD146) monoclonal antibody to ensure that there was no contamination with endothelial cells.
Cell Culture and Treatments
ARPE-19 cells, a human retinal pigment epithelial cell line33 obtained from American Type Culture Collection (Manassas, VA), and ARPE-19 cells stably expressing green fluorescent protein (GFP) targeted to the inner leaflet of the plasma membrane (GFP-c′-rRas-ARPE-19) as described previously31 were maintained and grown to confluence in Dulbecco’s modified Eagle’s medium-Ham’s F12 (1:1 v/v) medium supplemented with 10% FBS, 1 mmol/L l-glutamine, 100 μg/ml penicillin/streptomycin, and 0.348% Na2CO3 in a 5% CO2 humidified air incubator at 37°C. For experiments, cells were split and plated at subconfluent density in six-well plates or two-well chamber slides and grown to confluence. At the time of confluence, the medium was replaced for 48 hours with phenol red-free 10% FBS medium and then with phenol red-free 1% FBS medium for another 24 hours. At this time, cells were treated with 100 μmol/L HQ for various periods of time in phenol red-free 0.1% FBS medium. In some experiments, cells were pretreated for 1 hour with either 20 μmol/L of SB203580, a pyridinyl imidazole highly selective inhibitor of the kinase activity of p38α and p38β MAPK isoforms to block Hsp27 phosphorylation or 60 nmol/L of okadaic acid, a specific inhibitor of protein phosphatases (PP) 1 and 2A used to prevent Hsp27 dephosphorylation, or 40 μmol/L of the ERK inhibitor PD98059. After treatment, cells were washed with PBS and harvested for protein (with lysis buffer M-PER) or RNA (TRI reagent) assessment. All experiments (triplicate wells for each condition) were performed at least in triplicate.
RPE Membrane Blebbing
Confluent GFP-ARPE-19 cells were split and plated onto six-well plates at a density of 200,000 cells per well. At the time of confluence, cells were prepared for the experiments as described previously, and then incubated for 1 hour with or without 20 μmol/L of SB203580 or 60 nmol/L of okadaic acid, and then exposed to 100 μmol/L of HQ for 6 hours in assay medium supplemented with 0.1% FBS. After exposure to HQ, cells were examined under a fluorescence microscope (Axiophot; Carl Zeiss Medith, Inc., Oberkochen, Germany) equipped with the appropriate filters. Images were digitally acquired and the figures composed using image-processing software (Photoshop CS2, San Jose, CA).
Western Blot Analysis
Lysates from ARPE-19 cells, mice, and human RPE sheets were harvested, and protein concentration was determined by a DC protein assay (Bio-Rad, Hercules, CA). Thirty micrograms of protein were resolved on a 4% to 20% SDS polyacrylamide gel (Invitrogen) and transferred in 25 mmol/L Tris, 192 mmol/L glycine, 0.1% SDS, and 20% methanol, pH ∼8.4 to a nitrocellulose membrane (Hybond-ECL; GE Health Care; Piscataway, NJ). The membranes were then blocked in Tris-buffered saline–Tween 20 buffer containing 5% nonfat dry milk for 1 hour at room temperature. The blots were incubated overnight at 4°C with one of the following primary antibodies: Hsp25/27, phospho-Hsp25/27, p38, phospho-p38, ERK1/2, phospho-ERK1/2, and GAPDH or β-actin as loading controls. Since the total and phosphorylated forms of Hsp27, p38, and ERK have similar molecular weight, separate membranes were used for loading controls in some cases. Membranes were washed with Tris-buffered saline–Tween 20 and incubated with horseradish-peroxidase-linked donkey anti-rabbit or anti-mouse antibody for 1 hour at room temperature. After final washes, immunofluorescence bands were detected by exposing the blots to a chemiluminescent solution and exposure to autoradiograph film (Amersham Hyperfilm ECL, GE Health Care Limited, Buckinghamshire, UK). The bands were scanned and quantified by densitometry (ImageJ version 1.34s software available at http://rsb.info.nih.gov/ij/index.html, last accessed May 31, 2010; developed by Wayne Rasband, NIH, Bethesda, MD).
Immunohistochemistry for Phosphorylated Hsp27
Paraffin-embedded cross-sections were deparaffinized and rehydrated from xylene through a graded ethanol series to distilled water. Sections were washed with PBS and then blocked in 1% Triton X-100 containing 5% bovine serum albumin for 1 hour. After washes in PBS, sections were incubated overnight at 4°C with mouse monoclonal anti-phospho-Hsp25/27 (dilution 1:200) antibody. Negative controls were included, omitting the primary antibody to p-Hsp25/27. Subsequently, washes in PBS were followed by incubation with Alexa Fluor 488 goat anti-mouse IgG (dilution 1:500) for 2 hours at room temperature. Cell nuclei were stained with 4, 6-diamidino-2-phenylindole-2-HCL (dilution 1:5000). After several washes in PBS, the slides were mounted in PBS:glycerol 1:1 and examined at ×40 magnification by using a dual-channel laser scanning confocal microscope (Leica, Exton, PA) equipped with the appropriate filters. Images were digitally acquired and the figures were composed by using image-processing software (Photoshop CS2). Immunostaining was performed in three different eye sections per donor.
Immunofluorescence Staining for F-Actin Cytoskeleton and Quantification of Actin Aggregates
ARPE-19 cells grown and treated on chamber slides were fixed in 4% formaldehyde in PBS for 10 minutes at room temperature, washed with PBS, permeabilized in 0.1% Triton X-100 in PBS for 20 minutes at room temperature, and washed with PBS. F-actin was visualized with rhodamine-phalloidin. After several washes in PBS, the slides were mounted in PBS:glycerol 1:1 and examined at ×40 magnification by using a dual-channel laser scanning confocal microscope (Leica) equipped with the appropriate filters. Images were digitally acquired and the figures were composed by using image-processing software (Photoshop CS2). Captured images were imported into NIH Image J version 1.34s software to digitally quantify the actin aggregates by using the particle analysis commands of ImageJ. For each experiment, five images were obtained from three independent experiments. Once images were uploaded onto ImageJ, they were all changed to eight-bit binary images. Particles (aggregates) were extracted from grayscale image by thresholding the image into background and foreground states. Threshold range was adjusted manually for all images and pixel in the image whose values lies in this range was converted to black. Pixels with values outside this range were converted to white. Segmentation settings for image analysis were set so that F-actin aggregates were defined as those having pixel intensity values between 5 and 500, allowing the exclusion of nonaggregate particles. Results were expressed as percentage of either HQ alone or scrambled siRNA.
RNA Extraction and Real-Time PCR
Total RNA was extracted by using TRI reagent (Sigma-Aldrich). RNA concentrations were measured spectrophotometrically (Nanodrop 2000, ThermoScientific, Waltham, MA), and the quality of each RNA sample was confirmed by calculating the ratio of optical density at 260:280 nm. Reverse transcription was performed with random primers on 1.25 μg of total RNA in a final reaction volume of 50 μl by using the High-Capacity cDNA Archive Kit. Quantitative real-time PCR was performed on the BioRad iCycler iQ system by using iQ SybrGreen Supermix as specified by the manufacturers’ instructions, and the fluorescence threshold values were calculated by using iCycle iQ system software. Primer sequences for human Hsp27 were published previously:34 forward 5′-AGGATGGCGTGGTGGAGAT-3′ and reverse 5′-GATGTAGCCATGCTCGTCCTG-3′, yielding a 64-bp product. Primer sequences for mouse Hsp25 were published previously:35 forward 5′-GAGAACCGAACGACCGTCC-3′ and reverse 5′-CCCAATCCTTTGACCTAACGC-3′, yielding a 100-bp product. GAPDH was used as the endogenous control gene. GAPDH primer sequences are available in the public RTPrimerDB database (http://medgen.UGent.be/rtprimerdb/, last accessed May 31, 2010).36 The expected size of the product is 87 bp for human GAPDH and 55 bp for mouse GAPDH. The amplification program consisted of 1 cycle at 95°C for 15 minutes, followed by 40 cycles at 95°C for 15 seconds and 65°C for 1 minute. Regular PCR was performed with the primer sets, and products were analyzed on an ethidium bromide-stained 2% agarose gel to confirm the product size and specificity (data not shown). Melting curves were also acquired and analyzed to ensure specificity of the reaction. Each sample was run in duplicate and normalized to the GAPDH transcript content. The fold change in mRNA levels was calculated by using the 2−ΔΔCT method, with corrections for the housekeeping gene GAPDH.37 Separate control experiments using serial dilutions of cDNA demonstrated that the efficiencies of Hsp25, Hsp27, and GAPDH amplification were similar, hence validating the use of comparative Ct method to determine relative expression of Hsp27 (data not shown).
Transfection with Small Interfering RNA Against Hsp27
ARPE-19 cells grown in six-well plates or two-well chamber slides were allowed to reach ∼90% confluence before transfection. Then, cells were transiently transfected with 60 pmol of either Hsp27 siRNA or scrambled control siRNA according to the manufacturer’s protocol. Twenty-four hours after transfection, cells were prepared for the experiment by replacing the medium with phenol red-free 1% FBS medium for 24 hours. Subsequently, cells were treated with 100 μmol/L of HQ for either 5 minutes or 6 hours in phenol red-free medium supplemented with 0.1% FBS. The Hsp27 siRNA and control siRNA are a pool of four siRNAs targeting Hsp27 or a pool of four nontargeting siRNAs, respectively. The efficiency of siRNA in reducing endogenous Hsp27 protein was confirmed by performing Western blot analysis.
Statistical Analysis
Data are expressed as percentage of control. Results are the mean ± SE of three to six experiments performed in triplicate. Student’s t-test and one-way analysis of variance with the Dunnett multiple comparison posthoc test were performed. A Kruskal-Wallis test and post hoc Wilcoxon-Mann-Whitney U-test were used for Western blot densitometric analysis of the time-course experiments. All analyses were performed with GraphPad Prism version 4.00 (GraphPad Software, San Diego, CA). P < 0.05 was considered statistically significant.
Results
Phosphorylated Hsp27 Is Up-Regulated in RPE from Patients with AMD
Expression of phosphorylated Hsp27 has not been studied in human RPE. With the use of Western blot analysis, we investigated the expression of phosphorylated Hsp27 in RPE from patients who were given a diagnosis of dry AMD and control donors. As shown in Figure 1A, we showed for the first time that phosphorylated Hsp27 expression is robustly up-regulated in RPE from patients with AMD compared with controls. As well, clear expression of phosphorylated Hsp27 was evidenced in human RPE monolayer by immunostaining on paraffin-embedded eye cross-sections from control eyes (Figure 1, D and F). We confirmed that phosphorylated Hsp27 expression was increased in the RPE from patients with AMD (Figure 1, E and G). All of the sections were positively stained for phosphorylated Hsp27. Negative controls did not show any staining for phosphorylated Hsp27 (Figure 1, B and C). These findings demonstrate for the first time that RPE monolayer constitutively expresses phosphorylated Hsp27 and that increased Hsp27 phosphorylation may play a critical role in the pathogenesis of dry AMD.
Figure 1.

Increased phosphorylated Hsp27 (p-Hsp27) expression in human RPE from patient donors with dry AMD. A: Representative Western blot for p-Hsp27, total Hsp27, and GAPDH on RPE lysates from three donors with dry AMD and three controls with no known history of eye disease. B–G: Representative immunofluorescent double staining of p-Hsp27 (green) and nuclei (bleu) in retina sections from human donor eyes with no known eye disease (D), and human donor eyes with dry AMD (E). Negative controls were generated by omission of the primary antibody (B and C). Higher magnification showing RPE and BrM in control (F) and patients with AMD (G). Sections were analyzed by using confocal microscopy (original magnification, ×40 and ×400). INL, inner nuclear layer; ONL, outer nuclear layer; PIS, photoreceptor inner segments; POS, photoreceptor outer segments; Ch, choroid.
Exposure of RPE Cells to HQ Induces Hsp27 Dimerization and Up-Regulates Hsp27 mRNA Expression
Hsp27 in unstressed cells exists as large oligomers, whereas on phosphorylation, Hsp27 reorganizes into small dimeric units. Oxidative injury has been previously shown to regulate Hsp27 expression in RPE cells.31 In the present study, we treated the cells with 100 μmol/L of HQ for 6 hours and investigated whether HQ induces Hsp27 dimerization and modulates Hsp27 expression in cultured ARPE-19 cells by Western blot and real-time PCR, respectively. The Hsp27 antibody detected a band with a molecular weight 54 kDa recognizing Hsp27 dimers (Figure 2A). A basal level of dimerized Hsp27 was expressed in control ARPE-19 cells. Moreover, we found that exposure of cells to 100 μmol/L of HQ for 6 hours resulted in ∼8.9-fold (P < 0.001) increase in Hsp27 dimer formation (Figure 2A). Treatment with 100 μmol/L of HQ for 6 hours also increased Hsp27 mRNA levels by ∼38.7% compared with control cells in response to HQ (P < 0.0001; Figure 2B). These results show that HQ-induced nonlethal oxidative injury up-regulates Hsp27 expression in RPE cells.
Figure 2.
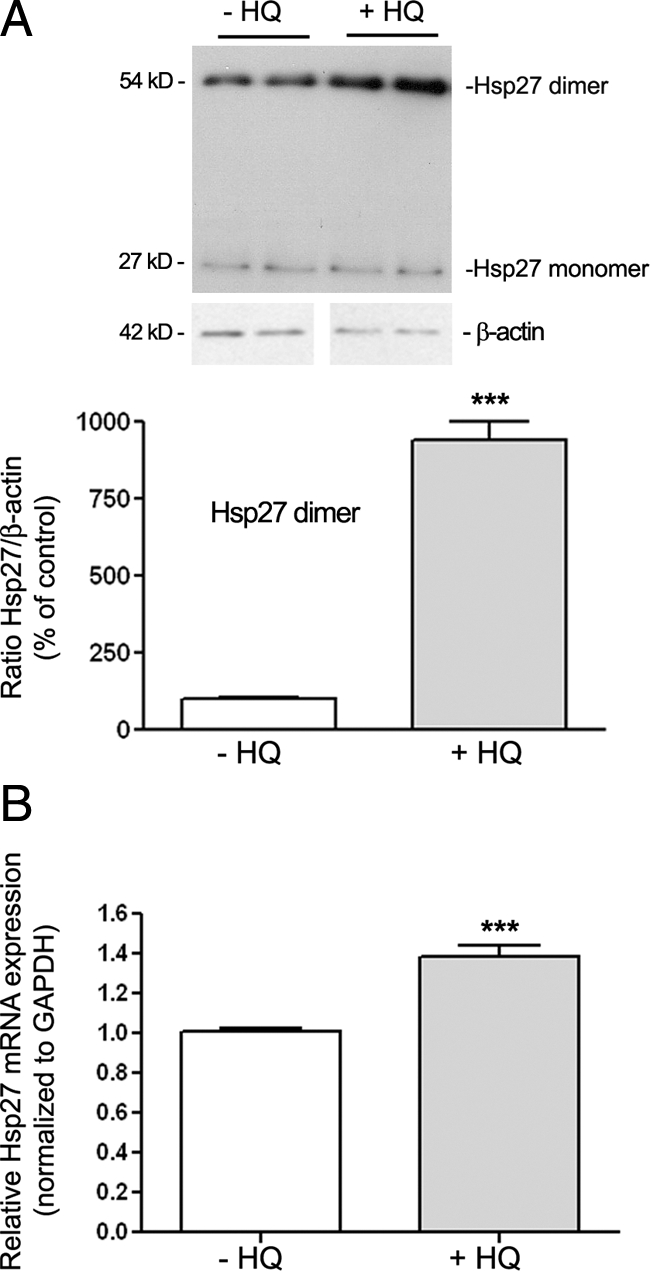
Effect of HQ on Hsp27 in ARPE-19 cells. Confluent serum-starved ARPE-19 cells were treated with or without 100 μmol/L HQ for six hours. A: Hsp27 monomer and dimer expression in ARPE-19 cells before and after treatment with HQ for six hours. Hsp27 expression was assessed by Western blot analysis. Top: Western blot from a representative experiment showing change in Hsp27 dimer protein expression. Number on the left represents protein molecular weight in kilodaltons. Bottom: Average results of three independent experiments run in duplicate. Total Hsp27 protein expression was normalized to β-actin. B: Up-regulation of Hsp27 mRNA expression in ARPE-19 cells exposed to HQ for six hours. Total RNA was extracted and Hsp27 mRNA expression was analyzed by real-time PCR. GAPDH was used as the housekeeping gene. Data represent average results of five independent experiments run in duplicate. Results are expressed as mean ± SE. ***P < 0.001 versus control (−HQ).
HQ Induces p38 MAPK-Mediated Hsp27 Phosphorylation
Next, we sought to elucidate the signaling pathway by which HQ exerts these effects on Hsp27 in ARPE-19 cells. Hsp27 undergoes posttranslational modifications, phosphorylation being the best characterized. The activity of Hsp27 likely depends on the phosphorylation state of serine residues at positions 15, 78, and 82. To investigate whether HQ can regulate this type of modification, we performed a time-course analysis of Hsp27 phosphorylation by Western blot on cell lysates from ARPE-19 cells exposed to 100 μmol/L of HQ for various periods of time by using a specific antibody that recognizes phosphorylated Hsp27 at Serine 82. HQ time-dependently induced a rapid and robust phosphorylation of Hsp27 in ARPE-19 cells that began within 1 minute (147 ± 18.2%, P < 0.05) with a maximum response observed at 5 minutes (321 ± 42.5%, P < 0.001; Figure 3A).
Figure 3.
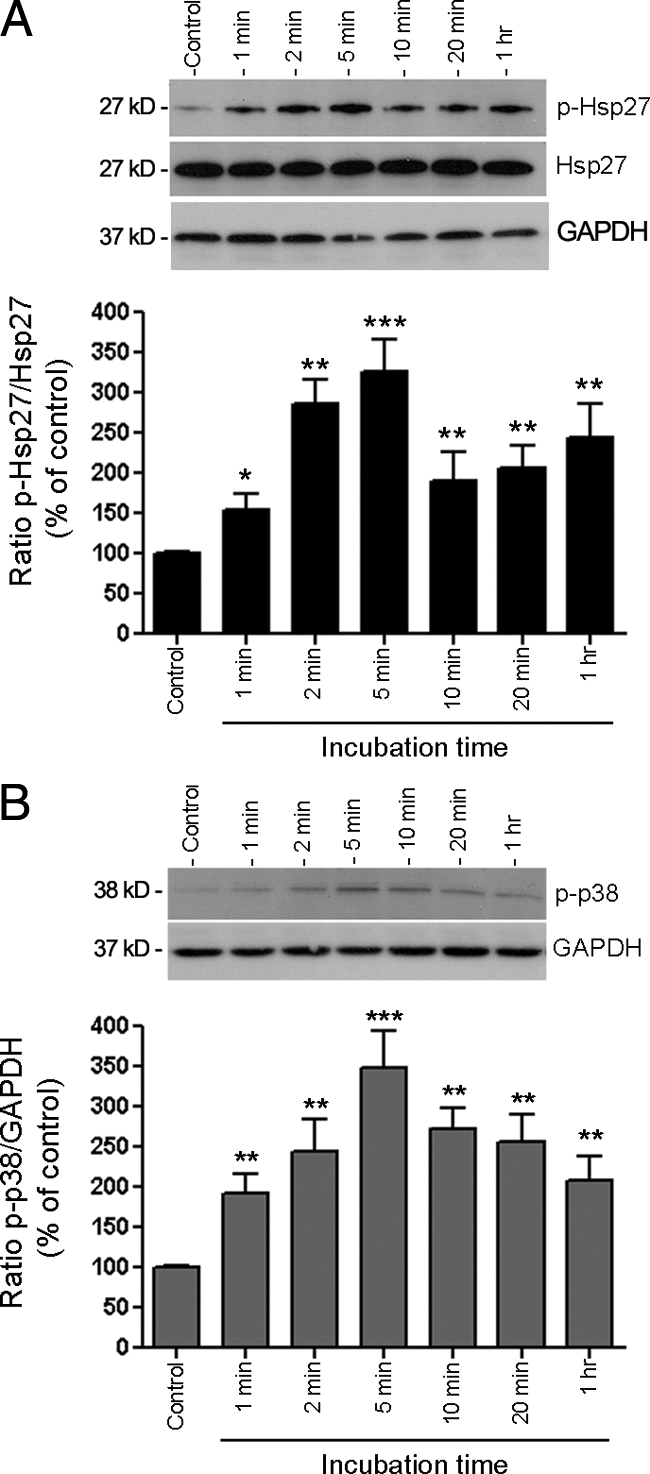
HQ activates Hsp27 and p38 MAPK phosphorylation. A: Time-dependent Hsp27 phosphorylation assessed by Western blot on cell lysates from ARPE-19 cells treated with 100 μmol/L HQ for various periods of time. Top: Western blot from a representative experiment. Number on the left represents protein molecular weight in kilodaltons. Bottom: Average results of three independent experiments run in duplicate. Phospho-Hsp27 (p-Hsp27) protein expression was normalized to total Hsp27. GAPDH was used as loading control for total Hsp27. B: Time-dependent p38 MAPK phosphorylation assessed by Western blot on cell lysates from ARPE-19 exposed to 100 μmol/L HQ for various periods of time. Top: Western blot from a representative experiment. Number on the left represents protein molecular weight in kilodaltons. Bottom: Average results of three independent experiments run in duplicate on ARPE-19 cells. Phospho-p38 (p-p38) MAPK protein expression was normalized to GAPDH. Data are expressed as percentage of control and are means ± SE of three independent experiments run in duplicate. *P < 0.05, **P < 0.01, and ***P < 0.001 versus control.
It is widely recognized that Hsp27 phosphorylation is mediated by the p38 MAPK cascade typically activated on cellular stress. Therefore, we next examined the role of p38 MAPK signaling pathway in HQ-induced Hsp27 phosphorylation in ARPE-19 cells. A time-course study of p38 MAPK activation was first determined by Western blot analysis for phosphorylated p38 MAPK by using a specific antibody that recognizes the dually phosphorylated p38 at threonine 180/tyrosine 182. Exposure to HQ resulted in a rapid increase in p38 MAPK phosphorylation, concomitant to Hsp27 that was maximal at 5 minutes (334.6 ± 44.7%, P < 0.001) and started to decline progressively thereafter (Figure 3B). These data suggest that p38 MAPK mediates HQ-induced Hsp27 phosphorylation in RPE cells exposed to oxidative stress.
Inhibition of Hsp27 Phosphorylation Decreases HQ-Induced Disruption of the F-Actin Cytoskeleton
To confirm that Hsp27 phosphorylation by HQ was dependent on p38 MAPK signaling pathway, ARPE-19 cells were pretreated for 1 hour with 20 μmol/L of SB203580, a pyridinyl imidazole highly selective inhibitor of the kinase activity of p38α and p38β MAPK isoforms, and then exposed to 100 μmol/L of HQ for 5 minutes (based on the kinetics of Hsp27 phosphorylation reported above). Hsp27 phosphorylation was determined by Western blot analysis. Exposure to HQ for 5 minutes resulted in ∼48% (147.6 ± 6.5%, P < 0.001) increase in Hsp27 phosphorylation compared with control cells (Figure 4A). Interestingly, HQ-induced Hsp27 phosphorylation was ∼2.9-fold decreased in response to SB203580 compared with cells treated with HQ alone (P < 0.001; Figure 4A).
Figure 4.

Inhibition of p38 MAPK blocks HQ-induced Hsp27 phosphorylation and focal aggregates formation in ARPE-19 cells. A: Inhibition of HQ-induced Hsp27 phosphorylation by the selective inhibitor of the kinase activity of p38α and p38β MAPK isoforms, SB203580 (SB). ARPE-19 cells were pretreated for one hour with 20 μmol/L of SB, and then exposed to 100 μmol/L HQ for five minutes. Total Hsp27, p-Hsp27, and GAPDH protein expression was assessed by Western blot analysis. Top: Representative Western blot for p-Hsp27. Numbers on the left represent protein molecular mass in kilodaltons. p-Hsp27 protein expression was normalized to total Hsp27 protein. GAPDH was used as loading control for total Hsp27. Bottom: Average densitometry results of three independent experiments run in duplicate. Data are expressed as percentage of control and are means ± SE. **P < 0.01 versus control; ***P < 0.001 versus HQ alone. B: Decreased formation of F-actin aggregates showed by staining for F-actin in ARPE-19 cells pretreated for one hour with or without 20 μmol/L of SB, before they were exposed to 100 μmol/L HQ for six hours. Cells were stained with rhodamine-phalloidin and examined by confocal microscopy by using magnification ×40. White arrowheads show formation of focal aggregates. C: Quantification of F-actin aggregates from three independent experiments run in duplicate. Data are expressed as a percentage of HQ-treated cells and are means ± SE. ***P < 0.001 versus HQ-treated cells.
It has been well documented that phosphorylation of Hsp27 modulates the polymerization of actin and is proposed to play a key role in actin cytoskeleton remodeling during cellular stress. Therefore, to test the biological role of Hsp27 and p38 MAPK in actin cytoskeleton rearrangement in ARPE-19 cells exposed to oxidative injury, cells pretreated with SB203580 before being exposed to HQ for 6 hours were stained for F-actin by using rhodamin-phalloidin and then examined by confocal fluorescence microscopy. As shown in Figure 4B, control cells demonstrated normal actin filaments. Also, nonlethal oxidative injury with HQ preserved most of the cytoskeleton but resulted in formation of focal aggregates compared with the control cells (Figure 4B). Inhibiting p38 MAPK activity and Hsp27 phosphorylation with the specific inhibitor SB203580 almost totally blocked the reorganization of F-actin microfilaments and formation of aggregates in response to HQ as shown by a ∼75% decrease compared with cells treated with HQ alone (P < 0.0001; Figure 4, B and C). Taken together, these results demonstrate that phosphorylated Hsp27 is a key mediator in HQ-induced actin aggregates formation in RPE cells.
Inhibition of Hsp27 Dephosphorylation Increases HQ-Induced Reorganization of F-Actin Filaments
PP2A is responsible for Hsp27 dephosphorylation in vivo.38 To further determine whether phosphorylation of Hsp27 is necessary for its effects on HQ-induced F-actin reorganization, ARPE-19 cells were pretreated for 1 hour with 60 nmol/L of okadaic acid, a potent and specific inhibitor of PP1 and 2A, and then exposed to 100 μmol/L of HQ for 5 minutes. Hsp27 phosphorylation was examined by Western blot analysis. As shown in Figure 5A, inhibition of PP2A activity with okadaic acid resulted in increased phosphorylation of Hsp27 in ARPE-19 cells exposed to HQ for 5 minutes by ∼29% (128.68 ± 16.4%, P < 0.05) compared with cells treated with HQ alone. Also, as shown in Figure 5, B and C, HQ-induced F-actin aggregates formation was robustly increased by ∼75% in the presence of okadaic acid (P < 0.0001). These results confirm that phosphorylated Hsp27 is a key mediator in HQ-induced actin aggregates formation in RPE cells.
Figure 5.

Inhibition of Hsp27 dephosphorylation increases HQ-induced Hsp27 phosphorylation and reorganization of F-actin filaments in ARPE-19 cells. A: Increased HQ-induced Hsp27 phosphorylation by okadaic acid. Confluent serum-starved ARPE-19 cells were pretreated for one hour with 60 nmol/L of okadaic acid, and then exposed to 100 μmol/L HQ for five minutes. Total Hsp27, p-Hsp27, and GAPDH protein expression was evaluated by Western blot analysis. Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. p-Hsp27 protein expression was normalized to total Hsp27 protein. Bottom: Average densitometry results of five independent experiments run in duplicate. Data are expressed as percentage of control and are means ± SE. **P < 0.01 and ***P < 0.001 versus control; *P < 0.05 versus okadaic acid alone; ****P < 0.01 versus HQ alone. B: Increased formation of F-actin aggregates showed by staining for F-actin in ARPE-19 cells pretreated for on hour with or without 60 nmol/L of okadaic acid and then exposed to 100 μmol/L HQ for six hours. Cells were stained with rhodamine-phalloidin and examined by confocal microscopy by using magnification ×40. White arrowheads show formation of focal aggregates. C: Quantification of F-acting aggregates from three independent experiments run in duplicate. Data are expressed as a percentage of HQ-treated cells and are means ± SE. ***P < 0.001 versus HQ-treated cells.
Inhibition of Hsp27 Phosphorylation Decreases HQ-Induced Blebbing Whereas Inhibition of Hsp27 Dephosphorylation Increases Blebs Formation in Response to HQ
Our group previously showed that ARPE-19 cells exhibit a distinct set of physiological responses, including membrane blebbing, when subjected to nonlethal oxidative injury.24 GFP-ARPE-19 cells were pretreated for 1 hour with or without 20 μmol/L of SB203580 or 60 nmol/L of okadaic acid, and then exposed to 100 μmol/L of HQ for 6 hours. Figure 6, A and B, shows the extensive blebbing that occurs in GFP-ARPE-19 cells after exposure to nonlethal concentrations of HQ for 6 hours. Inhibition of p38 MAPK activity and Hsp27 phosphorylation with SB203580 partially blocked bleb formation (Figure 6C). On the other hand, inhibition of PP2 activity with okadaic acid resulted in a large production of cell membrane blebs (Figure 6D). Figure 6, E and F, shows the extensive F-actin aggregates that occur in GFP-ARPE-19 cells after exposure to nonlethal concentrations of HQ for 6 hours. HQ-induced F-actin aggregates formation was partially blocked by SB203580 (Figure 6G) and increased in the presence of okadaic acid (Figure 6H) suggesting a relationship between bleb formation regulation and actin cytoskeleton modifications mediated by phosphorylated Hsp27. These data suggest that regulation of actin cytoskeleton dynamics by phosphorylated Hsp27 plays a key role in the regulation of blebbing.
Figure 6.

Induction of cellular changes in GFP-ARPE-19 cells exposed to HQ. Membrane blebbing (A–D) and formation of F-actin aggregates (E–H). Confluent GFP-ARPE-19 cells were pretreated with or without 20 μmol/L of SB203580 (SB) or 60 nmol/L of okadaic acid, and then exposed to 100 μmol/L HQ for six hours. Cells were directly observed under a fluorescence microscope (top) or stained with rhodamin-phalloidin and then examined under a fluorescent microscope (bottom). A: Control GFP-ARPE-19 cells in which GFP was localized to the membrane. Membrane-derived blebs (thin arrows) after exposure to 100 μmol/L HQ for six hours (B), 20 μmol/L of SB for one hour followed by 100 μmol/L HQ for six hours (C), and 60 nmol/L of okadaic acid for one hour followed by 100 μmol/L HQ for six hours (D). E: Control GFP-ARPE-19 cells showing normal F-actin filaments. F-actin aggregates (thick arrows) after exposure to 100 μmol/L HQ for six hours (F), 20 μmol/L of SB for one hour followed by 100 μmol/L HQ for six hours (G), and 60 nmol/L of okadaic acid for one hour followed by 100 μmol/L HQ for six hours (H). Images are representative of three independent experiments. Original magnification, ×400.
Hsp27 siRNA Decreases HQ-Induced Hsp27 Phosphorylation and F-Actin Microfilaments Disruption
To further evaluate the functional significance of Hsp27 in HQ-induced F-actin rearrangement by another strategy, ARPE-19 cells were transfected with siRNA against Hsp27 to decrease endogenous Hsp27 protein expression. Cells transfected with scrambled siRNA were used as controls. Forty-eight hours later, cells were exposed to 100 μmol/L of HQ for either 5 minutes or 6 hours. The constitutive expression of Hsp27 as evaluated by Western blot was reduced by ∼60% in ARPE 19 cells transfected with Hsp27 siRNA compared with cells transfected with scrambled siRNA (41.0 ± 5.1% versus 100.0 ± 5.7%, P < 0.01; Figure 7A). Next, we tested the effect of Hsp27 silencing on HQ-induced Hsp27 phosphorylation. ARPE-19 cells transfected with Hsp27 siRNA or scrambled siRNA were treated with 100 μmol/L of HQ for 5 minutes, and HQ-induced Hsp27 phosphorylation was examined by Western blot analysis. Treatment with HQ resulted in ∼45% increase in Hsp27 phosphorylation in cells transfected with scrambled siRNA compared with unstimulated cells (P < 0.05). Interestingly, HQ-induced Hsp27 phosphorylation was completely abolished in cells transfected with Hsp27 siRNA (Figure 7B). Finally, rhodamin-phalloidin staining on siRNA transfected cells demonstrated that cells transfected with scrambled siRNA displayed actin aggregates on HQ exposure for 6 hours. Also as shown in Figure 7, C and D, HQ-induced F-actin aggregates formation was robustly decreased by ∼90% when the Hsp27 protein levels were depleted by transfection with Hsp27 siRNA. These data demonstrate that phosphorylated Hsp27 plays an essential role in the regulation of actin cytoskeleton dynamics in response to HQ-induced oxidative injury in RPE cells.
Figure 7.

Effect of Hsp27 gene silencing on HQ-induced Hsp27 phosphorylation and F-actin aggregates formation. ARPE-19 cells were transfected with siRNA against Hsp27 or scrambled siRNA. Forty-eight hours later, cells were exposed to 100 μmol/L HQ for either five minutes or six hours. A: Reduction in endogenous Hsp27 protein expression by siRNA against Hsp27. The efficiency of siRNA against Hsp27 in reducing endogenous Hsp27 protein was confirmed by Western blot analysis. Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Hsp27 protein expression was normalized to GAPDH. Bottom: Average densitometry results of four independent experiments run in duplicate. Data are expressed as percentage of scrambled siRNA and are means ± SE. *P < 0.05 versus scrambled siRNA. B: Effect of Hsp27 siRNA on HQ-induced Hsp27 phosphorylation. Hsp27 phosphorylation was assessed by Western blot analysis and p-Hsp27 protein expression normalized to GAPDH. Top: Western blot from a representative experiment. Number on the left represents protein molecular weight in kilodaltons. Bottom: Average densitometry results of four independent experiments run in duplicate. Data are expressed as percentage of control (−HQ) and are means ± SE. *P < 0.05 versus control (−HQ). C: Inhibition of focal aggregates formation by Hsp27 gene silencing in ARPE-19 cells treated with HQ. Cells were stained with rhodamine-phalloidin and examined by confocal microscopy by using magnification ×40. Arrows show formation of focal aggregates. D: Quantification of F-acting aggregates from three independent experiments run in duplicate. Data are expressed as a percentage of HQ-treated cells transfected with scrambled siRNA and are means ± SE. ***P < 0.001 versus scrambled siRNA.
HQ Activates ERK1/2 MAPK Signaling Pathway
H2O2-induced oxidative stress has been shown to cause ERK1/2 phosphorylation and reorganization of actin cytoskeleton in ARPE-19 cells.39 Although an important contribution of p38 MAPK in HQ-induced Hsp27 phosphorylation and F-actin rearrangement has been evidenced in the present study, we sought to also measure ERK1/2 activation to determine whether HQ-induced phosphorylation of Hsp27 was restricted to the p38 MAPK pathway. Therefore, a time-course of ERK1/2 phosphorylation was analyzed by Western blot in cell lysates from ARPE-19 cells exposed to 100 μmol/L of HQ for various periods of time as described in Material and Methods. Exposure of ARPE 19 cells to HQ induced a rapid and robust ERK1/2 phosphorylation starting at 1 minute (215.32 ± 29.2%, P < 0.01), reaching a peak between 5 (340.05 ± 32.4%, P < 0.001) and 20 minutes (305.6 ± 38.6, P < 0.01), and declining quickly afterward (Figure 8). These results show for the first time that HQ-induced oxidative stress leads to a dual activation of p38 and ERK pathways in RPE cells.
Figure 8.
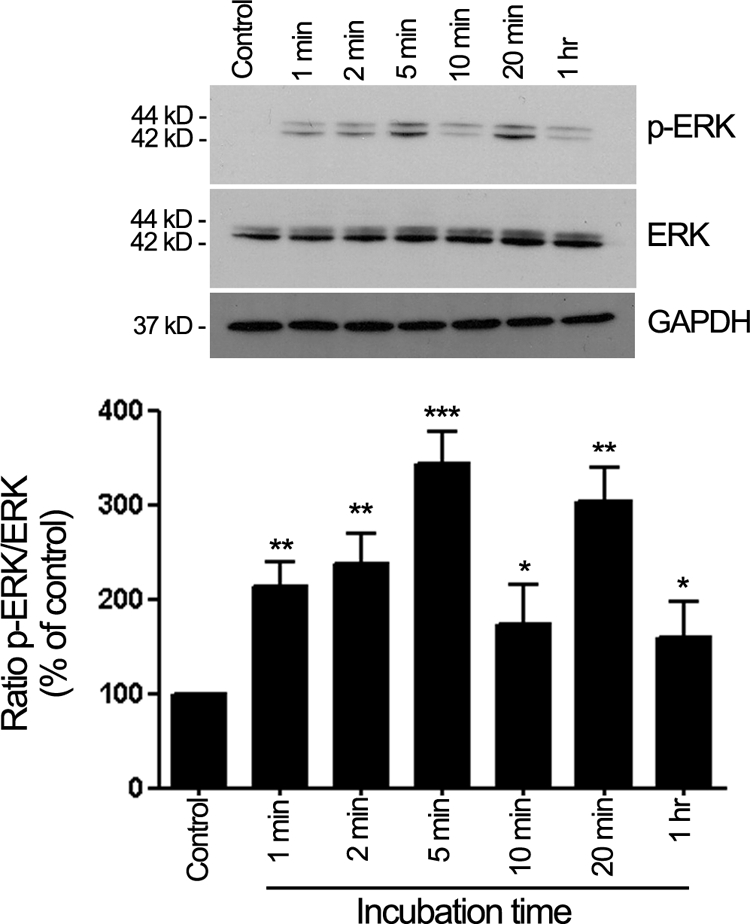
HQ activates ERK1/2 phosphorylation. Time-dependent ERK1/2 phosphorylation assessed by Western blot on cell lysates from ARPE-19 cells treated with 100 μmol/L HQ for various periods of time. Top: Western blot from a representative experiment. Number on the left represents protein molecular weight in kilodaltons. Bottom: Average densitometry results of three independent experiments run in duplicate. Phospho-ERK (p-ERK) protein expression was normalized to total ERK. GAPDH was used as loading control for total ERK. Data are expressed as percentage of control and are means ± SE. *P < 0.05, **P < 0.01, and ***P > 0.001 versus control.
A Cross Talk between p38 and ERK1/2 MAPK Signaling Pathways Mediates HQ Effects
A substantial body of evidence suggests a cross talk between p38 and ERK1/2 MAPK pathways in a variety of cells.40,41,42 Enhancement of ERK1/2 phosphorylation by treatment with pharmacological inhibitor of p38 MAPK kinase SB203580 has been reported in PC12 cells in response to epidermal growth factor.43 Moreover, SB203580 has been shown to activate ERK1/2 transduction pathway in primary cultures of human hepatocytes44 as well as in monocytic cells.45 These observations prompted us to examine the possibility of similar interactions between p38 and ERK1/2 MAPK in ARPE 19 cells treated with HQ by determining whether changes in the activity of either the ERK1/2 or the p38 pathways had a feedback effect on the other parallel pathway. First, we sought to establish whether ERK1/2 activation could be modulated by p38 MAPK. For this purpose, ERK1/2 phosphorylation status was analyzed by Western blot in ARPE-19 cells pretreated with 20 μmol/L of SB203580, then exposed to 100 μmol/L of HQ for 5 minutes. As shown in Figure 9A, HQ-induced phosphorylation of ERK1/2 was dramatically enhanced in the presence of p38 kinase inhibitor SB203580 (323.4 ± 36.4%, P < 0.001) compared with HQ alone (191.6 ± 33.6%, P < 0.01). These data strongly suggest a negative regulation of ERK signaling by p38 MAPK in human RPE cells exposed to HQ-induced oxidative injury.
Figure 9.
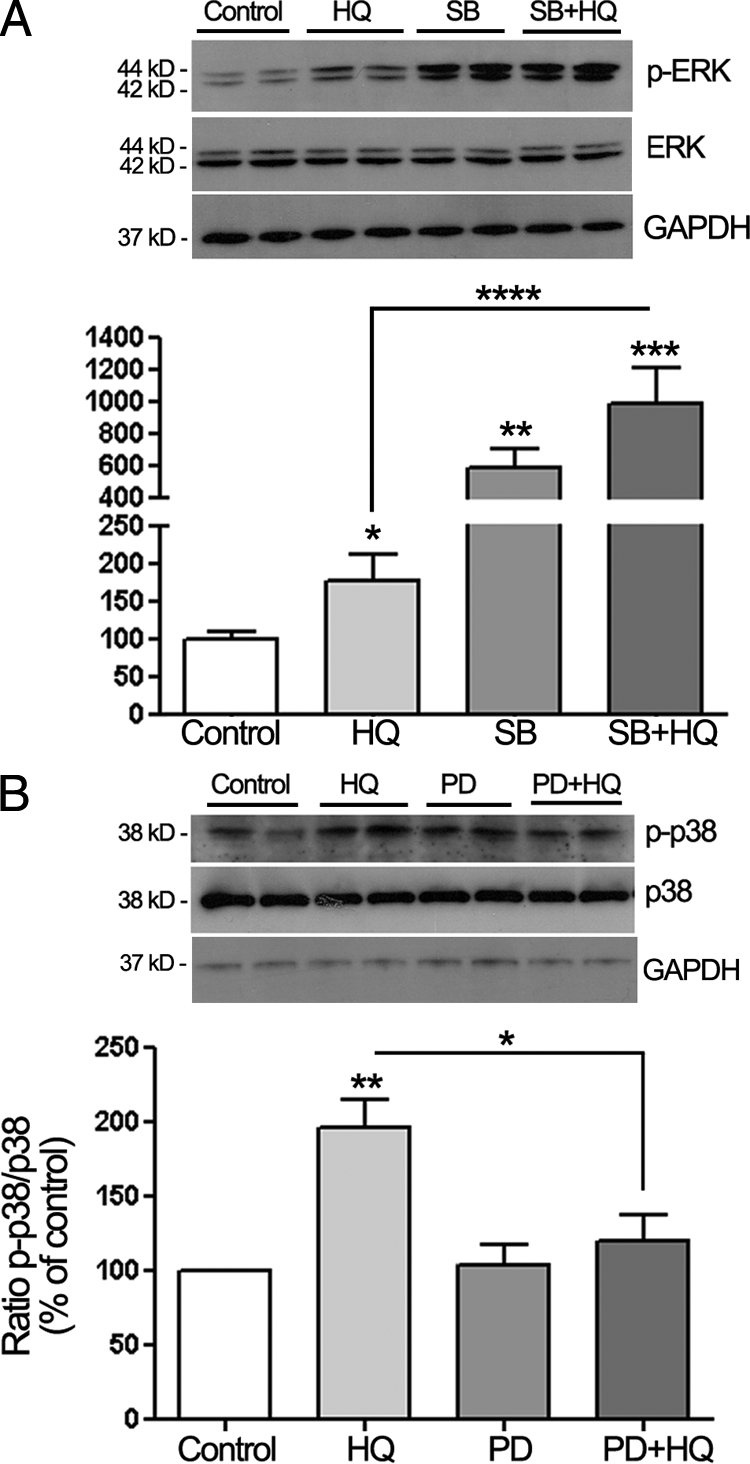
Cross talk between p38 and ERK1/2 MAPK signaling pathways in ARPE-19 cells treated with HQ. A: Inhibition of p38 MAPK pathway potentiates HQ-induced ERK1/2 phosphorylation in ARPE-19 cells. Confluent serum-starved ARPE-19 cells were pretreated for one hour with 20 μmol/L of p38 MAPK inhibitor SB203580 (SB), and then exposed to 100 μmol/L HQ for five minutes. Phospho-ERK (p-ERK) was examined by Western blot and normalized to total ERK. GAPDH was used as loading control for total ERK. B: Inhibition of ERK MAPK pathway with PD98059 (PD) leads to a decrease in HQ-induced p38 phosphorylation in ARPE-19 cells. ARPE-19 cells were pretreated for one hour with 40 μmol/L of ERK inhibitor PD, and then exposed to 100 μmol/L HQ for five minutes. Phospho-p38 (p-p38) was examined by Western blot and normalized to total p38. GAPDH was used as loading control for total p38. Top: Western blot from a representative experiment. Number on the left represents protein molecular weight in kilodaltons. Bottom: Average densitometry results of three independent experiments run in duplicate. Data are expressed as percentage of control and are means ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001 versus control; ****P < 0.01 and *P < 0.05 versus HQ alone.
To further investigate the potential interactions between these two MAPK pathways, we next evaluated the ability of the ERK pathway to modulate p38. ARPE-19 cells were pretreated with 40 μmol/L of ERK inhibitor PD98059 then with HQ for 5 minutes, and p38 phosphorylation was analyzed by Western blot. We observed that pretreatment with PD98059 partially blocked HQ-induced p38 MAPK phosphorylation (Figure 9B). These results suggest a positive regulation of p38 MAPK signaling by ERK in human RPE cells treated with HQ.
Inhibition of ERK Signaling Pathways Blocked HQ-Induced Hsp27 Phosphorylation and F-Actin Reorganization
It is believed that p38 MAPK is the major physiological activator of Hsp27. However, a recent study by Hong et al46 revealed that both p38 and ERK MAPK are upstream regulators of Hsp27 phosphorylation in Glial cell-derived neurotrophic factor (GDNF)-induced neurite outgrowth of dopaminergic neurons. Based on these observations and the cross talk of p38 and ERK MAPK pathways reported above, we next examined whether ERK1/2 could be an upstream regulator of Hsp27 phosphorylation in response to HQ-induced oxidative injury in ARPE-19 cells. To address this question, induction of Hsp27 phosphorylation was analyzed by Western blot in ARPE-19 cells pretreated with 40 μmol/L of the specific ERK inhibitor PD98059, then exposed to 100 μmol/L of HQ for 5 minutes. As shown in Figure 10A, inhibition of ERK signaling pathway with PD98059 totally abolished HQ-induced Hsp27 phosphorylation. These data suggest that Hsp27 might be a downstream common activated protein of p38 and ERK1/2 transduction pathways.
Figure 10.

Inhibition of ERK MAPK pathway blocks HQ-induced Hsp27 phosphorylation and focal aggregates formation. A: Abrogation of HQ-induced Hsp27 phosphorylation by PD98059. Confluent serum-starved ARPE-19 cells were pretreated for one hour with 40 μmol/L of ERK inhibitor PD98059 (PD), and then exposed to 100 μmol/L HQ for five minutes. Total Hsp27, p-Hsp27, and GAPDH protein expression was evaluated by Western blot analysis. Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. p-Hsp27 protein expression was normalized to total Hsp27 protein. Bottom: Average densitometry results of three independent experiments run in duplicate. Data are expressed as percentage of control and are means ± SE. *P < 0.01 versus HQ alone; **P < 0.01 versus control. B: Decreased formation of F-actin aggregates showed by staining for F-actin in ARPE-19 cells pretreated for one hour with or without 40 μmol/L of PD and then exposed to 100 μmol/L HQ for six hours. Cells were stained with rhodamine-phalloidin and examined by confocal microscopy by using magnification ×40. Arrows show formation of focal aggregates. C: Quantification of F-acting aggregates from three independent experiments run in duplicate. Data are expressed as percentage of HQ-treated cells and are means ± SE. ***P < 0.001 versus HQ-treated cells.
Since we observed an induction of ERK1/2 phosphorylation in response to HQ that was increased by SB203580, we were interested in studying the potential role of ERK in HQ-induced actin cytoskeletal alterations in ARPE-19 cells. Therefore, to determine the role of the ERK pathway in mediating the formation of actin aggregates in response to oxidative stress, we examined F-actin immunostaining in cells pretreated with 40 μmol/L of ERK inhibitor PD98059 for 1 hour before being exposed to 100 μmol/L of HQ for 6 hours. PD98059 almost completely abolished F-actin rearrangement induced by HQ (Figure 10, B and C). These data show that F-actin cytoskeleton rearrangement induced by HQ is also modulated by the ERK pathway.
HQ Induces Hsp25, p38, and ERK MAPK Phosphorylation but Decreased Hsp25 Expression in Mice
We used the experimental model for sub-RPE deposits of mice chronically exposed to oral HQ described elsewhere.22,23 As reported earlier, HQ-treated mice had increased blood levels of HQ (10.4 ± 0.7 ng/ml) relative to control mice that showed nondetectable levels.22 Here, we demonstrated for the first time that Hsp25 phosphorylation was increased in RPE from mice exposed to HQ as shown by Western blot (∼1.5-fold, P < 0.0001; Figure 11B). Furthermore, in agreement with our in vitro results, we confirmed that treatment with HQ induced a concomitant robust phosphorylation of p38 (∼4.0-fold increase, P < 0.0001; Figure 12A) and ERK (1.5-fold increase, P < 0.001; Figure 12C) MAPK with no change in total p38 (Figure 12B) and total ERK (Figure 12D). However, we found that HQ administered chronically to mice for 7 months resulted in a dramatic 69% decrease in Hsp25 mRNA (Figure 11A) and 63% decline in Hsp25 protein expression compared with control mice (P < 0.0001; Figure 11C). Together, these data demonstrate that in conjunction with sub-RPE deposits formation reported previously,21 exposure to HQ induces Hsp25 phosphorylation with concomitant activation of both p38 and ERK MAPK pathways in mouse RPE, therefore suggesting a role of phosphorylated Hsp27/25 in mediating deposits formation.
Figure 11.
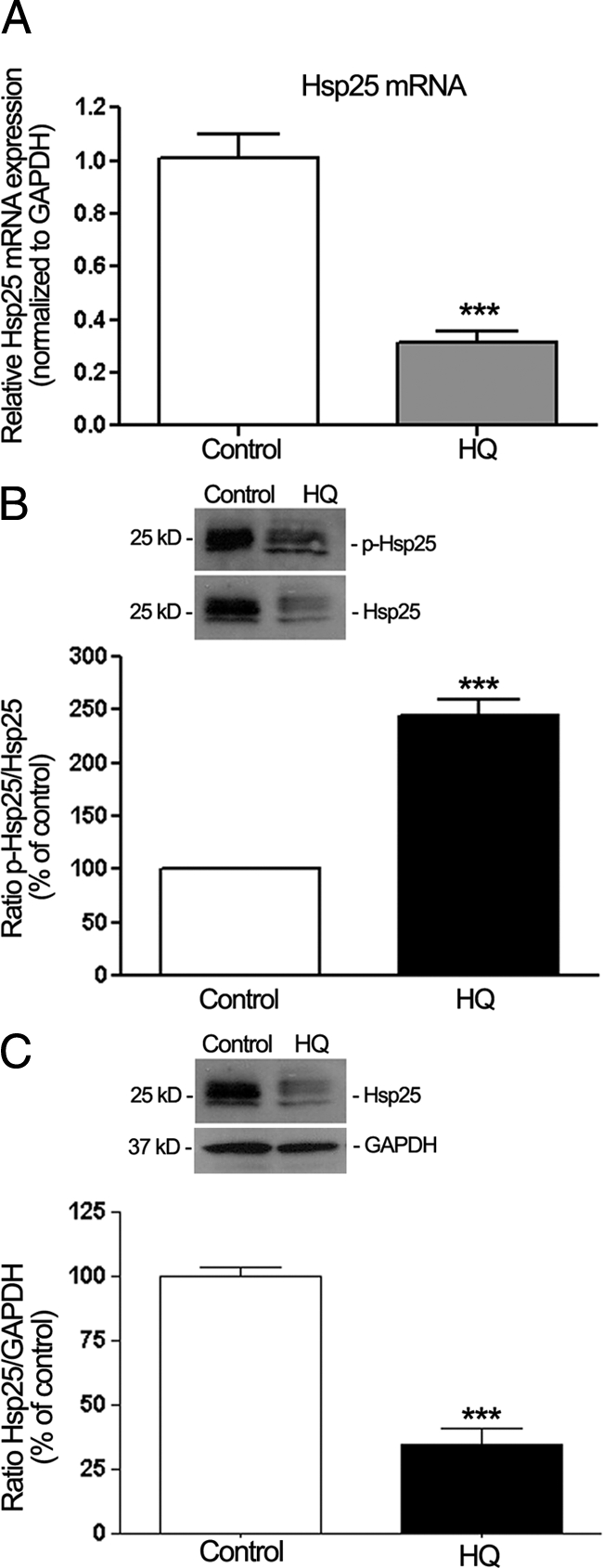
Regulation of Hsp25 and phosphorylated Hsp27 (p-Hsp25) in dissected RPE sheets from mice treated with HQ (0.8%) in drinking water for seven months. Protein and total RNA were extracted from RPE sheets (n = 6 eyes/group). A: Hsp25 mRNA expression analyzed by real-time PCR. GAPDH was used as the housekeeping gene. Data represent average results from six eyes normalized to the housekeeping gene. Results are expressed as mean ± SE. ***P < 0.001 versus control. B: Ratio p-Hsp25/Hsp25 protein expression evaluated by Western blot. Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. p-Hsp25 protein expression was normalized to total Hsp25 protein. Bottom: Data are the average results from six eyes. Data are expressed as percentage of control and shown are mean results ± SE. ***P < 0.001 versus control. C: Ratio Hsp25/GAPDH protein expression evaluated by Western blot. Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Hsp25 protein expression was normalized to GAPDH. Bottom: Data are the average results from six eyes. Data are expressed as percentage of control and shown are mean results ± SE. ***P < 0.001 versus control.
Figure 12.

Regulation of phosphorylated p38 (p-p38), and phosphorylated ERK (p-ERK) in dissected RPE sheets from mice treated with HQ (0.8%) in drinking water for seven months. Protein was extracted from RPE sheets (n = 6 eyes/group). A: p-p38 protein expression determined by Western blot. Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. p-p38 protein expression was normalized to total p38 protein. Bottom: Data are the average results from six eyes. Data are expressed as percentage of control and shown are mean results ± SE. ***P < 0.001 versus control. B: Ratio p38/GAPDH protein expression evaluated by Western blot. Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom: Data are the average results from six eyes. Data are expressed as percentage of control and shown are mean results ± SE. C: pERK protein expression determined by Western blot. Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. p-ERK protein expression was normalized to total ERK protein. Bottom: Data are the average results from six eyes. Data are expressed as percentage of control and shown are mean results ± SE. ***P < 0.001 versus control. D: Ratio ERK/GAPDH protein expression evaluated by Western blot. Top: Western blot from a representative experiment. Numbers on the left represent protein molecular mass in kilodaltons. Bottom: Data are the average results from six eyes. Data are expressed as percentage of control and shown are mean results ± SE.
Discussion
The present study shows for the first time that phosphorylated Hsp27 expression is increased in RPE from patients with AMD. We also demonstrate that HQ-induced Hsp27 phosphorylation and F-actin reorganization required for RPE-derived bleb formation are mediated by a cross talk between p38 and ERK MAPK in ARPE-19 cells. Moreover, we show that mice exposed to HQ in drinking water for 7 months exhibit increased phosphorylated Hsp25, p38, and ERK, and decreased Hsp25 mRNA and protein expression in the RPE.
We previously reported that ARPE-19 cells constitutively express high levels of Hsp27.31 Although Hsp27 expression has been reported in human cornea,47,48 in vivo expression of phosphorylated Hsp27 has not been explored in human RPE. Our results show for the first time that RPE from human eye donors constitutively express phosphorylated Hsp27, and that phosphorylated Hsp27 expression is increased in patients with dry AMD. Hsp27 phosphorylation has been abundantly described in several human diseases.49 Yet, there is a complete lack of information regarding the possible association between phosphorylated Hsp27 and AMD. Increased Hsp27 protein content along with evidence of cellular oxidative stress was reported in human eyes with AMD, but Hsp27 phosphorylation was not investigated.50 Our findings provide novel evidence that phosphorylated Hsp27 may play a major role in the pathogenesis of AMD.
In response to stress, phosphorylated Hsp27 undergoes conformational changes and reorganizes into dimeric units.26,51,52,53,54 Phosphorylated Hsp27 regulates actin filaments dynamics by repressing the ability of Hsp27 to block actin polymerization.55 We previously showed that exposure of ARPE-19 cells to oxidative injury with myeloperoxidase and hydrogen peroxide resulted in a marked increase in Hsp27 mRNA and protein expression.31 The results presented here extend these previous observations by showing that exposure of ARPE-19 cells to a 6-hour HQ nonlethal injury induced the transcriptional activation of Hsp27, accumulation of Hsp27 dimers, and a rapid phosphorylation of Hsp27. Our findings are in agreement with previous studies revealing Hsp27 phosphorylation in different cellular systems in response to oxidative stress.26,29,51,52,56 Several reports have shown that PP2A is involved in Hsp27 dephosphorylation.38,49 Using okadaic acid as an inhibitor of Hsp27 dephosphorylation by PP1 and 2A, we observed an increase in Hsp27 phosphorylation in HQ-stimulated ARPE-19 cells as well as F-actin reorganization and blebs formation. Moreover, we demonstrate that Hsp27 phosphorylation and F-actin aggregates formation are almost completely abolished in cells transfected with siRNA against Hsp27 following treatment with HQ. These data establish a direct correlation between levels of phosphorylated Hsp27 and actin cytoskeleton reorganization in response to HQ-induced oxidative injury in ARPE-19 cells. Collectively, these findings give support to a key role of phosphorylated Hsp27 in the regulation of F-actin filaments dynamics and blebs formation following HQ-induced oxidative stress in RPE cells. Furthermore, in an effort to correlate our in vitro observations to a more physiological environment in vivo, we used the experimental model for sub-RPE deposits.21,22 We previously reported that mice chronically exposed to HQ developed sub-RPE deposits and BrM thickening consistent with changes in early stages of human AMD.57 Here, we found that RPE from these mice showed increased levels of phosphorylated Hsp25. However, in contrast to ARPE-19 cells, our results indicate that Hsp25 mRNA and protein levels were dramatically decreased in mice chronically exposed to HQ. One possible explanation might be that human RPE cells were transiently exposed to HQ for 24 hours, whereas mice were chronically treated for 7 months, which might result in a differential transcriptional regulation of Hsp25/27. Another possibility is that mice might not be able to tolerate chronic activation of the Hsp25 signaling pathway resulting from sustained exposure to HQ. Sustained treatment with HQ might provide a negative feed-back loop that down-regulates Hsp25 expression to accommodate the chronic phosphorylation of Hsp25. Together, these data confirm that phosphorylated Hsp25/27 plays a major role in HQ-induced oxidative injury in the RPE, the key target cell in the pathogenesis of AMD.
Next, we demonstrated that HQ induced a rapid and robust activation of p38 MAPK signaling pathway, which parallels Hsp27 phosphorylation in ARPE-19 cells. Chronic exposure to HQ also resulted in increased p38 phosphorylation in mice. These results together with the observation that SB203580, a specific pharmacological inhibitor of p38 kinase activity,58 efficiently blocked Hsp27 phosphorylation as well as actin cytoskeleton remodeling and blebs formation in response to HQ in RPE cells strongly suggest that p38 activation by HQ modulates F-actin aggregates formation and membrane blebbing through Hsp27 phosphorylation in RPE cells. Our findings are in agreement with prior studies revealing p38 MAPK signaling pathway as an upstream mediator in oxidative stress-induced actin reorganization and Hsp27 phosphorylation.26,29,52
The ERK cascade participates in numerous intracellular signaling pathways in response to environmental stimuli, such as oxidative stress. H2O2-induced oxidative stress causes ERK phosphorylation and reorganization of actin cytoskeleton in ARPE-19 cells.39 In the present study, we found that treatment with HQ led to a robust activation of ERK signaling pathway in ARPE-19 cells as well as in mice. These results together with the observation that PD98059, a specific pharmacological inhibitor of Mitogen-activated ERK Kinase (MEK), completely abolished Hsp27 phosphorylation as well as actin cytoskeleton remodeling in response to HQ strongly suggest that ERK is also a key upstream activator of HQ-induced Hsp27 phosphorylation in RPE cells. Our results not only showed that kinetics of p38 and ERK phosphorylation correlated well with that of Hsp27, but also that Hsp27 phosphorylation and F-actin aggregates formation were decreased after inhibition of either p38 or ERK signaling cascades. These observations suggest that p38 as well as ERK MAPK pathways are required for the optimal activation of Hsp27 leading to F-actin rearrangement and bleb formation in RPE cells in response to HQ. Van Gorp et al59 have reported that H2O2-induced oxidative stress results in p38-mediated membrane blebbing in endothelial cells. However, activation of both p38 and ERK pathways has been described in endothelial cells in response to H2O2-induced oxidative injury.29 But our data are not in agreement with the former study, which revealed that inhibition of the ERK pathway led to an increase in bleb formation after exposure to H2O2. Cell-type specific differences in oxidative stress response may account for such a discrepancy.
Given the concomitant activation of p38 and ERK in response to HQ, we were interested in determining whether interactions might occur between these two signaling pathways. A substantial body of evidence suggests a cross talk between p38 and ERK MAPK cascades in a variety of cells.40,41,42,60 Treatment with p38 inhibitor SB203580 has been shown to enhance ERK phosphorylation in a number of cell lines.43,44,45 Here, we provide evidence that p38 MAPK negatively regulates HQ-induced ERK activation in ARPE-19 cells. HQ-induced ERK phosphorylation was enhanced in response to SB203580. We also showed that p38 MAPK inhibition had the ability to activate the ERK pathway in cells treated with SB203580 alone, suggesting that p38 exerts a tonic inhibition on the ERK pathway under basal conditions. The signaling events responsible for the cross talk between p38 and ERK signaling cascades are unknown. The regulation of MAPK pathways involves a complex interplay between kinases and phosphatases. Therefore, the dynamic balance and the potential interactions between p38 and ERK are important in determining how RPE cells respond to HQ-induced oxidative stress. In this regard, PP2A appears to be the major kinase phosphatase in eukaryotic cells that may play an important role by acting on ERK signaling cascade at multiple levels.61 Interestingly, it has been demonstrated that activation of p38 MAPK can block ERK pathway through activation of protein phosphates 1 and 2A.62 Moreover, PP2A mediates a cross talk between p38 MAPK and ERK in cardiac myocytes, as shown by increased ERK phosphorylation in H2O2-stimulated ventricular myocytes in response to okadaic acid.60 Based on these observations, it can be imagined that p38 MAPK might regulate HQ-induced ERK activation through PP2A in ARPE-19 cells. Future studies will test this hypothesis. Another possibility to be considered is a direct physical interaction between phosphorylated p38 and ERK.63
One-way cross talk between p38 and ERK has been reported in various cells.60,64,65 To further investigate the relationship between p38 and ERK pathways in response to HQ in ARPE-19 cells, we examined the effects of MEK inhibitor PD98059 on HQ-induced phosphorylation of p38. We found that PD98059 decreased HQ-induced p38 MAPK activation indicating that ERK1/2 pathway positively modulates p38 signaling and that ERK is required for HQ-induced activation of p38 MAPK signaling pathway in human RPE cells. These data present evidence of a previously unreported interaction between ERK and p38 during HQ-induced oxidative stress in RPE cells.
In summary, our observations demonstrate that phosphorylated Hsp27 might be a key mediator in human AMD and cigarette smoke-related HQ-induced oxidative injury in RPE. Our study also provides evidence that HQ-induced oxidative stress leads to a dual activation of p38 and ERK pathways, which contribute to HQ-induced Hsp27 phosphorylation, actin rearrangement, and blebs formation in ARPE-19 cells. Furthermore, this study presents ERK as a novel upstream positive regulator of Hsp27 and actin aggregates formation in response to HQ-induced oxidative injury in RPE cells. The cellular mechanisms underlying the regulation of these signaling events have yet to be determined. Given that there is no effective treatment for dry AMD, this study highlights Hsp27 as a potential, disease-related protein as well as biochemical pathways for potential therapeutic strategies.
Acknowledgments
We thank Dr. George Inana, Dr. Sander Dubovy, and co-workers at the Lions Eye Bank, Miami, for donating the human tissues. We also would like to acknowledge Dr. William Feuer for his technical assistance with statistical analysis, and Dr. Gabriel Gaidosh for his expertise and technical assistance with confocal microscopy.
Footnotes
Address reprint requests to Maria-Encarna Marin-Castaño, M.D., Ph.D., Bascom Palmer Eye Institute, University of Miami Miller School of Medicine, 1638 NW 10th Ave, Miami, FL 33136. E-mail: mcastano@med.miami.edu.
Supported by National Eye Institute grants R01 EY015249-01A1, EY015249-01A1S1, and EY014801.
References
- Augood CA, Vingerling JR, de Jong PT, Chakravarthy U, Seland J, Soubrane G, Tomazzoli L, Topouzis F, Bentham G, Rahu M, Vioque J, Young IS, Fletcher AE. Prevalence of age-related maculopathy in older Europeans: the European Eye Study (EUREYE). Arch Ophthalmol. 2006;124:529–535. doi: 10.1001/archopht.124.4.529. [DOI] [PubMed] [Google Scholar]
- Evans JR. Risk factors for age-related macular degeneration. Prog Retin Eye Res. 2001;20:227–253. doi: 10.1016/s1350-9462(00)00023-9. [DOI] [PubMed] [Google Scholar]
- Javitt JC, Zhou Z, Maguire MG, Fine SL, Willke RJ. Incidence of exudative age-related macular degeneration among elderly Americans. Ophthalmology. 2003;110:1534–1539. doi: 10.1016/S0161-6420(03)00495-0. [DOI] [PubMed] [Google Scholar]
- Klein R, Peto T, Bird A, Vannewkirk MR. The epidemiology of age-related macular degeneration. Am J Ophthalmol. 2004;137:486–495. doi: 10.1016/j.ajo.2003.11.069. [DOI] [PubMed] [Google Scholar]
- Katta S, Kaur I, Chakrabarti S. The molecular genetic basis of age-related macular degeneration: an overview. J Genet. 2009;88:425–449. doi: 10.1007/s12041-009-0064-4. [DOI] [PubMed] [Google Scholar]
- Green WR. Histopathology of age-related macular degeneration. Mol Vis. 1999;5:27–36. [PubMed] [Google Scholar]
- Shen JK, Dong A, Hackett SF, Bell WR, Green WR, Campochiaro PA. Oxidative damage in age-related macular degeneration. Histol Histopathol. 2007;22:1301–1308. doi: 10.14670/HH-22.1301. [DOI] [PubMed] [Google Scholar]
- Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- Clemons TE, Milton RC, Klein R, Seddon JM, Ferris FL., 3rd Risk factors for the incidence of advanced age-related macular degeneration in the Age-Related Eye Disease Study (AREDS) AREDS report no. 19. Ophthalmology. 2005;112:533–539. doi: 10.1016/j.ophtha.2004.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen WG, Glynn RJ, Manson JE, Ajani UA, Buring JE. A prospective study of cigarette smoking and risk of age-related macular degeneration in men. JAMA. 1996;276:1147–1151. [PubMed] [Google Scholar]
- Seddon JM, Willett WC, Speizer FE, Hankinson SE. A prospective study of cigarette smoking and age-related macular degeneration in women. JAMA. 1996;276:1141–1146. [PubMed] [Google Scholar]
- Klein R, Klein BE, Moss SE. Relation of smoking to the incidence of age-related maculopathy: the Beaver Dam Eye Study. Am J Epidemiol. 1998;147:103–110. doi: 10.1093/oxfordjournals.aje.a009421. [DOI] [PubMed] [Google Scholar]
- Solberg Y, Rosner M, Belkin M. The association between cigarette smoking and ocular diseases. Surv Ophthalmol. 1998;42:535–547. doi: 10.1016/s0039-6257(98)00002-2. [DOI] [PubMed] [Google Scholar]
- Bertram KM, Baglole CJ, Phipps RP, Libby RT. Molecular regulation of cigarette smoke induced-oxidative stress in human retinal pigment epithelial cells: implications for age-related macular degeneration. Am J Physiol Cell Physiol. 2009;297:C1200–C1210. doi: 10.1152/ajpcell.00126.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisinger PJ, Hill TS, English JC. Human exposure to naturally occurring hydroquinone. J Toxicol Environ Health. 1996;47:31–46. doi: 10.1080/009841096161915. [DOI] [PubMed] [Google Scholar]
- DeCaprio AP. The toxicology of hydroquinone: relevance to occupational and environmental exposure. Crit Rev Toxicol. 1999;29:283–330. doi: 10.1080/10408449991349221. [DOI] [PubMed] [Google Scholar]
- Burns RP, Feeney-Burns L. Clinico-morphologic correlations of drusen of Bruch’s membrane. Trans Am Ophthalmol Soc. 1980;78:206–225. [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Patterson R, Ohnishi Y, Inomata H, Ryan SJ. Formation of drusen in the human eye. Am J Ophthalmol. 1986;101:342–353. doi: 10.1016/0002-9394(86)90830-5. [DOI] [PubMed] [Google Scholar]
- Sarks S, Cherepanoff S, Killingsworth M, Sarks J. Relationship of Basal laminar deposit and membranous debris to the clinical presentation of early age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:968–977. doi: 10.1167/iovs.06-0443. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Suner IJ, Catanuto P, Hernandez EP, Marin-Castano ME, Cousins SW. Cigarette smoke-related oxidants and the development of sub-RPE deposits in an experimental animal model of dry AMD. Invest Ophthalmol Vis Sci. 2006;47:729–737. doi: 10.1167/iovs.05-0719. [DOI] [PubMed] [Google Scholar]
- Marin-Castano ME, Striker GE, Alcazar O, Catanuto P, Espinosa-Heidmann DG, Cousins SW. Repetitive nonlethal oxidant injury to retinal pigment epithelium decreased extracellular matrix turnover in vitro and induced sub-RPE deposits in vivo. Invest Ophthalmol Vis Sci. 2006;47:4098–4112. doi: 10.1167/iovs.05-1230. [DOI] [PubMed] [Google Scholar]
- Alcazar O, Cousins SW, Marin-Castano ME. MMP-14 and TIMP-2 overexpression protects against hydroquinone-induced oxidant injury in RPE: implications for extracellular matrix turnover. Invest Ophthalmol Vis Sci. 2007;48:5662–5670. doi: 10.1167/iovs.07-0392. [DOI] [PubMed] [Google Scholar]
- Marin-Castano ME, Csaky KG, Cousins SW. Nonlethal oxidant injury to human retinal pigment epithelium cells causes cell membrane blebbing but decreased MMP-2 activity. Invest Ophthalmol Vis Sci. 2005;46:3331–3340. doi: 10.1167/iovs.04-1224. [DOI] [PubMed] [Google Scholar]
- Strunnikova N, Zhang C, Teichberg D, Cousins SW, Baffi J, Becker KG, Csaky KG. Survival of retinal pigment epithelium after exposure to prolonged oxidative injury: a detailed gene expression and cellular analysis. Invest Ophthalmol Vis Sci. 2004;45:3767–3777. doi: 10.1167/iovs.04-0311. [DOI] [PubMed] [Google Scholar]
- Huot J, Houle F, Marceau F, Landry J. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ Res. 1997;80:383–392. doi: 10.1161/01.res.80.3.383. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Milzani A, Di Simplicio P, Colombo R. The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic Biol Med. 2001;31:1624–1632. doi: 10.1016/s0891-5849(01)00749-3. [DOI] [PubMed] [Google Scholar]
- Milzani A, DalleDonne I, Colombo R. Prolonged oxidative stress on actin. Arch Biochem Biophys. 1997;339:267–274. doi: 10.1006/abbi.1996.9847. [DOI] [PubMed] [Google Scholar]
- Huot J, Houle F, Rousseau S, Deschesnes RG, Shah GM, Landry J. SAPK2/p38-dependent F-actin reorganization regulates early membrane blebbing during stress-induced apoptosis. J Cell Biol. 1998;143:1361–1373. doi: 10.1083/jcb.143.5.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benndorf R, Hayess K, Ryazantsev S, Wieske M, Behlke J, Lutsch G. Phosphorylation and supramolecular organization of murine small heat shock protein HSP25 abolish its actin polymerization-inhibiting activity. J Biol Chem. 1994;269:20780–20784. [PubMed] [Google Scholar]
- Strunnikova N, Baffi J, Gonzalez A, Silk W, Cousins SW, Csaky KG. Regulated heat shock protein 27 expression in human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2001;42:2130–2138. [PubMed] [Google Scholar]
- Praddaude F, Cousins SW, Pecher C, Marin-Castano ME. Angiotensin II-induced hypertension regulates AT1 receptor subtypes and extracellular matrix turnover in mouse retinal pigment epithelium. Exp Eye Res. 2009;89:109–118. doi: 10.1016/j.exer.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- Kassem H, Sangar V, Cowan R, Clarke N, Margison GP. A potential role of heat shock proteins and nicotinamide N-methyl transferase in predicting response to radiation in bladder cancer. Int J Cancer. 2002;101:454–460. doi: 10.1002/ijc.10631. [DOI] [PubMed] [Google Scholar]
- Cunningham LL, Brandon CS. Heat shock inhibits both aminoglycoside- and cisplatin-induced sensory hair cell death. J Assoc Res Otolaryngol. 2006;7:299–307. doi: 10.1007/s10162-006-0043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Cairns J, Qin S, Philp R, Tan YH, Guy GR. Dephosphorylation of the small heat shock protein Hsp27 in vivo by protein phosphatase 2A. J Biol Chem. 1994;269:9176–9183. [PubMed] [Google Scholar]
- Garg TK, Chang JY. Oxidative stress causes ERK phosphorylation and cell death in cultured retinal pigment epithelium: prevention of cell death by AG126 and 15-deoxy-delta 12, 14-PGJ2. BMC Ophthalmol. 2003;3:5–20. doi: 10.1186/1471-2415-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Yang H, Tachado SD, Capo-Aponte JE, Bildin VN, Koziel H, Reinach PS. Phosphatase-mediated crosstalk control of ERK and p38 MAPK signaling in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2006;47:5267–5275. doi: 10.1167/iovs.06-0642. [DOI] [PubMed] [Google Scholar]
- Chen G, Hitomi M, Han J, Stacey DW. The p38 pathway provides negative feedback for Ras proliferative signaling. J Biol Chem. 2000;275:38973–38980. doi: 10.1074/jbc.M002856200. [DOI] [PubMed] [Google Scholar]
- Kogkopoulou O, Tzakos E, Mavrothalassitis G, Baldari CT, Paliogianni F, Young HA, Thyphronitis G. Conditional up-regulation of IL-2 production by p38 MAPK inactivation is mediated by increased Erk1/2 activity. J Leukoc Biol. 2006;79:1052–1060. doi: 10.1189/jlb.0705418. [DOI] [PubMed] [Google Scholar]
- New L, Li Y, Ge B, Zhong H, Mansbridge J, Liu K, Han J. SB203580 promotes EGF-stimulated early morphological differentiation in PC12 cell through activating ERK pathway. J Cell Biochem. 2001;83:585–596. doi: 10.1002/jcb.1253. [DOI] [PubMed] [Google Scholar]
- Henklova P, Vrzal R, Papouskova B, Bednar P, Jancova P, Anzenbacherova E, Ulrichova J, Maurel P, Pavek P, Dvorak Z. SB203580, a pharmacological inhibitor of p38 MAP kinase transduction pathway activates ERK and JNK MAP kinases in primary cultures of human hepatocytes. Eur J Pharmacol. 2008;593:16–23. doi: 10.1016/j.ejphar.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Numazawa S, Watabe M, Nishimura S, Kurosawa M, Izuno M, Yoshida T. Regulation of ERK-mediated signal transduction by p38 MAP kinase in human monocytic THP-1 cells. J Biochem. 2003;133:599–605. doi: 10.1093/jb/mvg077. [DOI] [PubMed] [Google Scholar]
- Hong Z, Zhang QY, Liu J, Wang ZQ, Zhang Y, Xiao Q, Lu J, Zhou HY, Chen S. Phosphoproteome study reveals Hsp27 as a novel signaling molecule involved in GDNF-induced neurite outgrowth. J Proteome Res. 2009;8:2768–2787. doi: 10.1021/pr801052v. [DOI] [PubMed] [Google Scholar]
- Shi B, Han B, Schwab IR, Isseroff RR. UVB irradiation-induced changes in the 27-kd heat shock protein (HSP27) in human corneal epithelial cells. Cornea. 2006;25:948–955. doi: 10.1097/01.ico.0000224643.43601.5d. [DOI] [PubMed] [Google Scholar]
- Gain P, Thuret G, Chiquet C, Dumollard JM, Mosnier JF, Campos L. In situ immunohistochemical study of Bcl-2 and heat shock proteins in human corneal endothelial cells during corneal storage. Br J Ophthalmol. 2001;85:996–1000. doi: 10.1136/bjo.85.8.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostenko S, Moens U. Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol Life Sci. 2009;66:3289–3307. doi: 10.1007/s00018-009-0086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decanini A, Nordgaard CL, Feng X, Ferrington DA, Olsen TW. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am J Ophthalmol. 2007;143:607–615. doi: 10.1016/j.ajo.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol. 1995;15:505–516. doi: 10.1128/mcb.15.1.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J, Landry J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci. 1997;110 (Pt 3):357–368. doi: 10.1242/jcs.110.3.357. [DOI] [PubMed] [Google Scholar]
- Clerk A, Michael A, Sugden PH. Stimulation of multiple mitogen-activated protein kinase sub-families by oxidative stress and phosphorylation of the small heat shock protein, HSP25/27, in neonatal ventricular myocytes. Biochem J. 1998;333 (Pt 3):581–589. doi: 10.1042/bj3330581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev NB, Bogatcheva NV, Marston SB. Structure and properties of small heat shock proteins (sHsp) and their interaction with cytoskeleton proteins. Biochemistry (Mosc) 2002;67:511–519. doi: 10.1023/a:1015549725819. [DOI] [PubMed] [Google Scholar]
- Schneider GB, Hamano H, Cooper LF. In vivo evaluation of hsp27 as an inhibitor of actin polymerization: hsp27 limits actin stress fiber and focal adhesion formation after heat shock. J Cell Physiol. 1998;177:575–584. doi: 10.1002/(SICI)1097-4652(199812)177:4<575::AID-JCP8>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Huot J, Lambert H, Lavoie JN, Guimond A, Houle F, Landry J. Characterization of 45-kDa/54-kDa HSP27 kinase, a stress-sensitive kinase which may activate the phosphorylation-dependent protective function of mammalian 27-kDa heat-shock protein HSP27. Eur J Biochem. 1995;227:416–427. doi: 10.1111/j.1432-1033.1995.tb20404.x. [DOI] [PubMed] [Google Scholar]
- Curcio CA, Millican CL. Basal linear deposit and large drusen are specific for early age-related maculopathy. Arch Ophthalmol. 1999;117:329–339. doi: 10.1001/archopht.117.3.329. [DOI] [PubMed] [Google Scholar]
- Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- van Gorp RM, Heeneman S, Broers JL, Bronnenberg NM, van Dam-Mieras MC, Heemskerk JW. Glutathione oxidation in calcium- and p38 MAPK-dependent membrane blebbing of endothelial cells. Biochim Biophys Acta. 2002;1591:129–138. doi: 10.1016/s0167-4889(02)00273-2. [DOI] [PubMed] [Google Scholar]
- Liu Q, Hofmann PA. Protein phosphatase 2A-mediated cross-talk between p38 MAPK and ERK in apoptosis of cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;286:H2204–H2212. doi: 10.1152/ajpheart.01050.2003. [DOI] [PubMed] [Google Scholar]
- Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24:186–191. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- Westermarck J, Li SP, Kallunki T, Han J, Kahari VM. p38 mitogen-activated protein kinase-dependent activation of protein phosphatases 1 and 2A inhibits MEK1 and MEK2 activity and collagenase 1 (MMP-1) gene expression. Mol Cell Biol. 2001;21:2373–2383. doi: 10.1128/MCB.21.7.2373-2383.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Shi X, Hampong M, Blanis L, Pelech S. Stress-induced inhibition of ERK1 and ERK2 by direct interaction with p38 MAP kinase. J Biol Chem. 2001;276:6905–6908. doi: 10.1074/jbc.C000917200. [DOI] [PubMed] [Google Scholar]
- Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, Shiojima I, Hiroi Y, Yazaki Y. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest. 1997;100:1813–1821. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RP, Dhawan P, Golden C, Kapoor GS, Mehta KD. One-way cross-talk between p38(MAPK) and p42/44(MAPK). Inhibition of p38(MAPK) induces low density lipoprotein receptor expression through activation of the p42/44(MAPK) cascade. J Biol Chem. 1999;274:19593–19600. doi: 10.1074/jbc.274.28.19593. [DOI] [PubMed] [Google Scholar]