

Abstract
Oxidative stress contributes to the pathogenesis of Duchenne muscular dystrophy (DMD). Although they have been a model for DMD, mdx mice exhibit slowly developing cardiomyopathy. We hypothesized that disease process was delayed owing to the development of an adaptive mechanism against oxidative stress, involving glutathione synthesis. At 15 to 20 weeks of age, mdx mice displayed a 33% increase in blood glutathione levels compared with age-matched C57BL/6 mice. In contrast, cardiac glutathione content was similar in mdx and C57BL/6 mice as a result of the balanced increased expression of glutamate cysteine ligase catalytic and regulatory subunits ensuring glutathione synthesis in the mdx mouse heart, as well as increased glutathione peroxidase-1 using glutathione. Oral administration from 10 weeks of age of the glutamate cysteine ligase inhibitor, l-buthionine(S,R)-sulfoximine (BSO, 5 mmol/L), led to a 33% and 50% drop in blood and cardiac glutathione, respectively, in 15- to 20-week-old mdx mice. Moreover, 20-week-old BSO-treated mdx mice displayed left ventricular hypertrophy associated with diastolic dysfunction, discontinuities in β-dystroglycan expression, micronecrosis and microangiopathic injuries. Examination of the glutathione status in four DMD patients showed that three displayed systemic glutathione deficiency as well. In conclusion, low glutathione resource hastens the onset of cardiomyopathy linked to a defect in dystrophin in mdx mice. This is relevant to the glutathione deficiency that DMD patients may suffer.
Duchenne muscular dystrophy (DMD) is a lethal X-linked recessive disorder caused by a defect in dystrophin, a subsarcolemmal structural protein of the cardiac and skeletal muscles that contributes to the integrity of the cellular membrane.1 Dystrophin-deficient mdx mice are the most commonly used experimental model of DMD.2,3,4,5,6,7 In DMD patients, as well as in mdx mice, dystrophin defect alters the sarcolemmal integrity of cardiac and skeletal myocytes,8,9,10 with increased susceptibility to oxidative stress11,12,13 leading to necrotic cell death and inflammation.14,15 Further evidence in support of oxidative stress as a cause of the dystrophic pathophysiology comes from in vivo studies demonstrating reduced signs of muscle damages in mdx mice given antioxidants.16,17
The tripeptide glutathione (l-gamma glutamyl-cysteinyl-glycine) is a major antioxidant, which plays a central role in cell redox regulation and controls multiple cellular processes. In particular, glutathione, through the inhibition of the neutral sphingomyelinase-dependent apoptotic pathway,18,19 determines cardiac myocyte function and survival.20,21,22 Alterations in glutathione homeostasis and metabolism are associated with inflammation23 and characterize several human inflammatory chronic diseases.24,25 Thus, fibroblasts from DMD patients show a low capacity for glutathione synthesis.26 In contrast, a noticeable feature of dystrophic muscles in mdx mice is the up-regulation of glutathione peroxidase and glutathione reductase, which respectively use and recycle reduced glutathione.27 However, this protective mechanism does not entirely compensate the chronic oxidative challenge since glutathione levels in 6- to 8-week-old mdx mice tibialis anterior muscles are decreased by 20% compared with control mice.27 Accordingly, green tea polyphenols, which beyond their inherent antioxidant properties, promote glutathione synthesis, and reduce muscle damage and necrosis in mdx mice.16,17 This does not rule out that the intrinsic increased turnover of glutathione in mdx mice may delay the progression of the dystrophic disease, although it cannot fully protect against it.
Cardiac manifestations of DMD are recognized later than those of skeletal muscles, but are present in 90% of DMD patients by the age of 18 years, progressing to fatal heart failure in 20% of patients.28 Similar to DMD patients, mdx mice experience a progressive development of cardiac defects. However, the disease develops with less severity than in patients and in old mice only; young adult mice show few echocardiographic signs of cardiomyopathy before 10 months of age.7,29 Glutathione plays a central role in protecting cardiac vasculature,30 and cardiac glutathione deficiency is a common feature of cardiomyopathies in animal models,20,21 as in patients.21,22,31
We hypothesized that the progression of cardiomyopathy in mdx mice was slow owing to a boost in glutathione synthesis and resources, set as an adaptative mechanism against oxidative stress. Mice administered oral l-buthionine sulfoximine (BSO), a specific inhibitor of glutamate cysteine ligase (GLCL), the rate-limiting enzyme of glutathione synthesis, are a suitable, well-characterized model for glutathione deficiency.32 From 10 weeks of age, mdx mice experience a continuing cycle of skeletal muscle fiber degeneration and regeneration.33,34 We chose this age to start a 10-week treatment with BSO added to the drinking water at a low dose (5 mmol/L) devoid of toxicity.32 At variance from 20-week-old untreated mdx mice receiving water, 20-week-old BSO-treated mdx mice displayed cardiac left ventricular (LV) structural abnormalities and hypertrophy together with altered diastolic function, reminiscent of the early stage of the cardiomyopathy in DMD patients before the manifestations of clinical symptoms.35,36 To get a first insight into the glutathione status of DMD patients, we measured blood glutathione in four DMD patients with mean age of 28 ± 2 years and on wheelchair dependency. We found that three of them demonstrated systemic glutathione deficiency.
Materials and Methods
Patients
This study, performed prospectively for a period of 7 months (September 2009 to March 2010) at the AP-HP, Groupe Hospitalier Cochin, Service de Cardiologie, Paris, France and at the Hôpital Raymond Poincaré, Service de Réanimation et Unité de Ventilation à domicile, Garches, France conformed to the Declaration of Helsinki and to our institutional ethics committee guidelines. All of the participants gave informed consent. We enrolled four consecutive patients with DMD (four males with mean age of 28 ± 2 years), referred to our institutions for cardiac evaluation. Patients underwent biological tests including the measurement of whole blood glutathione and an echocardiography (ATL HDI 5000 system, ATL Ultrasound, Bothell, WA). Examinations were conformed to the recommendations of the American Society of Echocardiography. Left ventricular ejection fraction (LVEF) was determined according to the Simpson’s method. Blood glutathione of DMD patients was compared with that of four matched healthy volunteers.
Animals and Experimental Design
Experimental procedures were performed in accordance with the European legislation on animal experimentation (L358-86/609/EEC). C57BL/6 (B6) and mdx4Cv (mdx) mice were used. The mdx4Cv mouse mutant, in which a C-to-T nucleotide transition generates a stop codon in exon 53 of the dystrophin gene, has almost no background of revertant fibers in skeletal muscle.4,6,37 B6 and mdx mice were randomly assigned to two groups: 1) an untreated group receiving water; 2) a group receiving BSO (Sigma) via drinking water at the concentration of 5 mmol/L (average dose of 185 mg/kg/day) from 10 to 20 weeks of age. As previously stated by Watanabe et al,32 this dose of BSO did not produce either deaths or pathological signs. Blood was withdrawn from the queue at regular intervals from 3 weeks to 20 weeks of age. At 20 weeks of age, all mice were subjected to echocardiography then euthanized by intraperitoneal injection of overdose anesthetics (xyzaline, 20 mg/kg, and ketamine, 50 mg/kg). Blood was withdrawn and hearts were excised and frozen in liquid nitrogen cooled isopentane. All samples were stored at −80°C until use.
Echocardiography
Transthoracic two-dimensional echocardiography measurements were performed in isoflurane-anesthetized mice using an echocardiograph (Vivid 7, General Electric) equipped with a linear 13 MHz transducer. Body temperature was maintained with a heating pad. Numeric images were stored and reviewed at a slower frame rate by a single observator blinded of the animal treatment and genotype status. Left ventricular end-diastolic diameter (LVEDD), interventricular septal wall thickness in diastole (IVS) and posterior wall thickness in diastole (LVPW) were measured from long axis view by two-dimensional-guided M-mode. LV mass (LVM) was calculated using the formula: LVM = 1.05 [(IVS + LVEDD + LVPW)3 − (LVID)3].38 Left ventricular ejection fraction (LVEF) was measured using two-dimensional parasternal long axis cine-loop.39 Peak mitral early diastolic velocity (E), its deceleration time (DcT) and the peak velocity of the atrial contraction (A) were measured using the trans-mitral pulsed wave Doppler. All measurements were averaged on three consecutive cardiac cycles.
Biochemical Analysis
Total glutathione was measured in patients and mice whole blood collected in EDTA tubes, and in mouse heart homogenates according to a modification of Tietze method40 as previously described.21,22,41 Results are given as means ± SEM of three separate determinations made in duplicates.
Histological and Immunohistochemical Analysis
Cardiomyocyte structure was studied in air-dried frozen heart sections from B6 mice, untreated or BSO-treated mdx mice. After blocking nonspecific staining with AffiniPure Fab and Fc fragments (Jackson ImmunoResearch Europe Ltd. Suffolk, United Kingdom),17 sections were incubated with either a mouse monoclonal antibody to β-dystroglycan (diluted 1:50; Abcam, San Francisco, CA) and the secondary goat anti-mouse Alexa-fluor 555-conjugated antibody (diluted 1:1000; Invitrogen, Carlsbad, CA) or a rat monoclonal antibody to mouse CD68 (diluted 1:250; Serotec, Kidlington, United Kingdom) and the biotinylated sheep secondary anti-rat IgG antibody (diluted 1:500; Serotec). Slides were mounted with Prolong Gold antifade reagent with DAPI (Invitrogen) or Immu-Mount (Thermo Electron Corporation, Courtaboeuf, France) for β-dystroglycan or CD68 staining, respectively.
Electronic Microscopy
Whole hearts were dissected and the apex of the LV was removed, and immediately placed in fix (4% paraformaldehyde, 3% glutaraldehyde in 0.1 mol/L sodium phosphate buffer, pH 7.4) for 1 hour. The samples were transferred to 3% glutaraldehyde in phosphate buffer (0.1 mol/L sodium phosphate, pH 7.4) overnight, then washed for 15 minutes in phosphate buffer, and postfixed for 1.5 hours in phosphate buffer added with in 1% osmium tetraoxide. Following osmium fixation, the samples were rinsed 3 to 5 minutes in phosphate buffer, dehydrated for 10 minutes in successive ethanol washes (50%, 70%, 80%, 95%, 100%, 100%), and rinsed for 15 minutes in propylene oxide. Finally, the samples were embedded in Lowecryl resin, overnight. Sections of 70 to 80 nm were cut on a Reichert UltracutE, stained with 2% uranyl acetate plus Reynold’s lead citrate and viewed using a Phillips CM120 transmission electron microscope.
RNA isolation and Real-Time PCR analysis Total RNA was extracted from frozen hearts using TRIzol reagent (Qiagen, Hilden, Germany) and reverse transcription (RT)-PCR was performed as previously described.42 Concentration of total RNA was dosed using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA) and RNA quality was assessed by electrophoresis. cDNA was synthesized using SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). Real-time PCR was performed on a Light Cycler (Roche Diagnostics, Meylan, France), using the Quantitest SYBR green kit (Qiagen) with sense and anti-sense oligonucleotides primers for the different genes (Table 1). The PCR-amplified products were controlled by sequencing. Relative levels of mRNA expression were calculated according to the ΔΔCT method.43 Individual mRNA expression values were normalized by comparison with 18S mRNA level.
Table 1.
Primers Used for Quantitative RT-PCR
| Gene | Primers | Fragment size (bp) | NCBI reference |
|---|---|---|---|
| GLCLC | 5′−ATCCTCCAGTTCCTGCACAT−3′ | 390 | NM_010295.1 |
| 5′−TGTGAATCCAGGGCCTA−3′ | |||
| GLCLM | 5′−GCCACCAGATTTGACTGCCTTT−3′ | 119 | NM_008129.2 |
| 5′−CAGGGATGCTTTCTTGAAGAGCTT−3′ | |||
| GPX1 | 5′−GACTACACCGAGATGAACGA−3′ | 110 | NM_008160.5 |
| 5′−CTTCATTCTTGCCATTCTCCT−3′ | |||
| COL1A1 | 5′−ACCTGTGTGTTCCCTACTCA−3′ | 322 | NM_007742.2 |
| 5′−GACTGTTGCCTTCGCCTCTG−3′ | |||
| COL1A2 | 5′−TTAAGACTCAGCCACCCAGA−3′ | 267 | NM_007743.2 |
| 5′−GAGCTGAGTTGCCATTTCCT−3′ | |||
| COL3A1 | 5′−AATGGTGGTTTTCAGTTCAGC−3′ | 322 | NM_009930.1 |
| 5′−TGGGGTTTCAGAGAGTTTGGC−3′ | |||
| COL6A1 | 5′−AATTGCCCTGGTCATTACGG−3′ | 169 | NM_009933.3 |
| 5′−CGATAAGCCTTGGCAGGAAA−3′ | |||
| 18S | 5′−GTAACCCGTTGAACCCCATT−3′ | 151 | XO1117 |
| 5′−CCATCCAATCGGTAGTAGCG−3′ |
Abbreviations: GLCLC, glutamate-l-cysteine ligase catalytic subunit; GLCLM, glutamate-l-cysteine ligase modulatory subunit; GPX1, Glutathione peroxidase 1; COL1A1, procollagen type I, alpha 1; COL1A2, procollagen type I, alpha 2; COL3A1, procollagen type III, alpha 1; COL6A1, procollagen type VI, alpha 1.
Statistical Analysis
Data are expressed as means ± SEM. Data were analyzed by Mann-Whitney test or Kruskal-Wallis test combined with Dunn posttest, as appropriate (GraphPad Prism software, La Jolla, CA). Echocardiographic measurements were analyzed by a two-way repeated measure analysis of variance followed by a multiple comparison test (Holm-Sidak method). Statistical significance was assumed at P ≤ 0.05.
Results
Blood Level and Cardiac Content of Glutathione in mdx Mice
B6 mice displayed steady blood glutathione levels from 5 to 20 weeks of age, with an average value of 0.70 ± 0.03 mmol/L. Until the age of 10 weeks, mdx mice had similar blood glutathione levels as B6 mice (Figure 1). However, after 15 weeks of age, blood glutathione levels in mdx mice increased by 33% to 0.93 ± 0.03 mmol/L (Figure 1). At variance from blood glutathione, cardiac glutathione content in 20-week-old mdx mice was not significantly increased as compared with B6 mice (Figure 2), despite significant elevation in cardiac mRNA expression of both GLCL catalytic (GLCLC) and modulatory subunits (GLCLM) that ensure glutathione synthesis (Figure 3, A and B). In fact, increased expression of GLCL subunits in the heart of 20-week-old mdx mice coped with that of glutathione peroxidase-1 (GPx1), which utilizes glutathione to reduce hydroperoxides (Figure 3C).
Figure 1.

Evolution as a function of age of blood glutathione levels in B6 and mdx mice treated or not with BSO. B6 mice received water; mdx mice received either water or 5 mmol/L BSO added to the drinking water from 10 to 20 weeks of age. Blood glutathione levels were measured at 5, 10, 15, and 20 weeks of age. N = 5 in each group. *P < 0.05 vs. 5-week-old mdx mice.
Figure 2.

Cardiac glutathione levels in 20-week-old B6 and mdx mice treated or not with BSO. Glutathione was measured in cardiac samples of 20-week-old B6 and mdx mice having received either water or BSO. N = 4 to 10 in each group. *P < 0.05 vs mdx mice receiving water.
Figure 3.

Cardiac expression of glutathione recycling enzymes in 20-week-old B6 and mdx mice treated or not with BSO. Quantitative real-time PCR analysis of GLCLC (A), GLCLM (B), and GPX1 (C) gene expression, encoding glutathione ligase catalytic subunit (GLCLC), glutathione ligase modulatory subunit (GLCLM) and glutathione peroxidase (GPX1), respectively, in the hearts of 20-week-old B6 and 20- mdx mice having received water or BSO. Individual mRNA expression values were normalized relative to 18S mRNA. Results are expressed as fold change relative to mean cardiac mRNA expression in B6 mice. N = 4 to 6 in each group. *P < 0.05 vs B6 mice; **P < 0.05 vs untreated mdx mice.
Effects of BSO Treatment on Blood and Cardiac Glutathione Content in mdx Mice
BSO was given at a low non-toxic dose (5 mmol/L), ie, a dose that did not produce either deaths or pathological signs.32 BSO treatment, started at the age of 10 weeks, blunted the rise in blood glutathione level in 15- and 20-week-old mdx mice, maintaining it at 0.74 ± 0.04 mmol/L (Figure 1). Cardiac glutathione content in BSO-treated mdx mice was decreased by 50% as compared with untreated mdx mice given water, reaching a level not significantly different from that in B6 mice, whether receiving water or BSO (Figure 2). The decrease in cardiac glutathione content in BSO-treated mdx mice was related to a reduced expression of glutathione enzyme mRNAs as compared with untreated mdx mice, similar to that in B6 mice (Figure 3).
BSO-Treated mdx Mice Display LV Hypertrophy and Diastolic Dysfunction
As compared with age-matched B6 mice hearts, 20-week-old mdx mice hearts featured a smaller body weight-indexed LV mass (iLVM) related to thinner posterior and intraventricular septal wall thickness (LVPW and IVS, respectively) (Figure 4A). In other ways, mdx mice displayed normal LV systolic function, as assessed by the LV ejection fraction (LVEF), and normal LV diastolic function, as assessed by both the ratio between early diastolic filling wave (E) and filling wave illustrating atrial contraction (A), and the E-wave deceleration time (DcT) (Figure 4A). Ten-week BSO-treatment induced LV hypertrophy in mdx mice, as demonstrated by the 21% increase in iLVM (Figure 4A). LV hypertrophy in BSO-treated mdx mice had no impact on LV systolic function, but was associated with impaired diastolic function as characterized by a concomitant E/A decrease and DcT prolongation (Figure 4A). Noteworthy, LV hypertrophy in mdx mice was correlated with cardiac glutathione deficiency (r = 0.66, P = 0.002; Figure 4B).
Figure 4.

Echocardiography of 20-week-old B6 and mdx mice treated or not with BSO. A: Twenty-week-old B6 and mdx mice have received either water or BSO. BW: body weight; HR: heart rate; IVS: intra ventricular septum thickness; LVPW: posterior wall thickness; LVEDD: LV end diastolic diameter; iLVEDD: LVEDD indexed to BW; LVM: LV mass; iLVM: LVM indexed to BW; LVEF: LV ejection fraction. E/A: ratio between early diastolic filling wave (E) and filling wave due to atrial contraction (A); DcT: E-wave deceleration time. #P < 0.05 mdx vs matched B6 group; †P < 0.05 BSO mdx versus H2O mdx group. B: LVM in mdx mice, having received water or BSO, was inversely linearly correlated with cardiac glutathione content (r = 0.66, P = 0.002).
BSO-Treated mdx Mice Display LV Structure Alterations
Immunohistochemical analysis on LV sections of 20-week-old mdx mice given water showed a regular expression of β-dystroglycan along cardiac myocyte membranes, similar to that in LV sections of age-matched B6 mice (Figure 5A). LV of mdx mice receiving water did not present either any histologically discernable abnormality (as assessed by H&E staining, not shown), or fibrosis (assessed by red-sirius staining, not shown), despite increased gene expression of 1a2, 3a1, and 6a1 collagens as compared with B6 mice (Figure 5B). In contrast, LV of BSO-treated mdx mice displayed destabilized β-dystroglycan expression (Figure 5A) with reduced collagen gene expression as compared with mdx mice receiving water (Figure 5B). Foci of micronecrosis infiltrated by CD68+ macrophages (Figure 6, A and B) were also identified in LV sections of BSO-treated mdx mice.
Figure 5.

Sarcolemmal β-dystroglycan immunostaining and cardiac collagen gene expression in 20-week-old B6 mice and mdx mice treated or not with BSO. A: β-dystroglycan expression around cardiac myocyte membranes is regular in B6 mice and mdx mice receiving water, but is destabilized in BSO-treated mdx mice. B: Quantitative real-time PCR analysis of COL1A2, COL3A1, and COL6A1 genes coding for 1a2, 3a1, and 6a1 collagen, respectively, in the hearts of 20-week-old B6 mice and mdx mice having received either water or BSO. Individual mRNA expression values were normalized relative to 18S mRNA. Results are expressed as fold change relative to mean cardiac mRNA expression in B6 mice. N = 4 to 6 in each group. *P < 0.05 vs B6 mice.
Figure 6.

Presence of inflammatory micronecrotic foci in the hearts of 20-week-old BSO-treated mdx mice. CD68 immunostaining showing A: a necrotic cardiomyocyte encircled and phagocytized by CD68+ macrophages and B: typical foci of antiinflammatory infiltrates (original magnification × 400 for both).
Electron Microscopy Reveals Focal Ultrastructural Lesions in LV of BSO-Treated mdx Mice
Electron microscopy analysis of LV of 20-week old mdx mice receiving water showed tightly packed contractile proteins, and did not show any ultrastructural abnormalities (Figure 7B). In contrast, focal ultrastructural lesions were discernible in LV of BSO-treated mdx mice, with degenerative changes including intracellular amorphous accumulations separating contractile bundles, pronounced irregularities of sarcomere elements such as the Z-line (Z streaming), extreme dilation of smooth endoplasmic reticulum profiles and giant mitochondria (Figure 7A). These lesions were spatially associated with microangiopathic lesions characterized by edema in endothelial cells leading to partial obstruction of the lumen, and accompanied by edema in interstitium and neighboring cardiomyocytes (Figure 8A–D).
Figure 7.
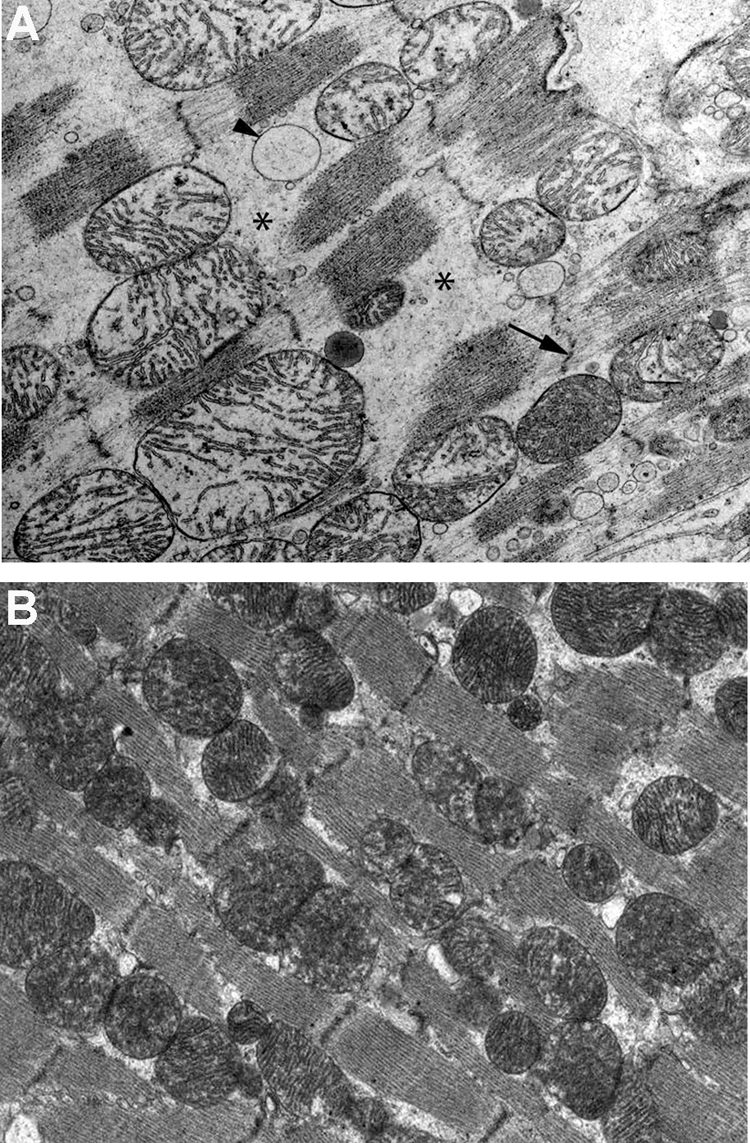
Ultrastructural degenerative alterations in the hearts of 20-week-old BSO-treated mdx mice. A: Regions of 20-week-old BSO-treated mdx mice hearts show cardiomyocytes with cytoplasmic degenerative alterations: intracellular amorphous accumulations (asterisks) separating contractile bundles, pronounced sarcomere irregularities (Z streaming) (arrow), dilatation of smooth endoplasmic reticulum profiles (arrowhead) and giant mitochondria (original magnification ×11,700). B: In contrast, hearts of 20-week-old mdx mice receiving water display normal sarcomere organization illustrated by tightly packed contractile proteins (original magnification ×11,500).
Figure 8.

Vessel associated lesions in the hearts of 20-week-old BSO-treated mdx mice. Hearts of 20-week-old BSO-treated mdx mice display a gamut of vessel associated lesions. A: Massive edema (asterisks) and extravasated erythrocytes (arrow) of interstitium between two cardiomyocytes (arrowheads) (original magnification × 5600); (B and C). Edematous endothelial cells (arrows) accompanied by edema in interstitium and neighboring cardiomyocytes (asterisks), incipient in B (original magnification × 7100) and pronounced in C (original magnification × 5600). D: A lysing capillary endothelial cell characterized by a swollen cytoplasm, (arrowhead), a “ground glass” nucleus (arrow) and a reduced lumen (asterisk), and degenerated mitochondria spilling over from surrounding cardiomycytes (double arrows), suggesting a focus of micronecrosis (original magnification ×7,100).
Glutathione Status in DMD Patients
Next, we intended to get a first insight into the glutathione status of DMD patients. Four wheelchair-dependent DMD patients were enrolled, with a mean age of 28 ± 2 years. They received an anti-angiotensin therapy, consisting in an angiotensin-converting enzyme inhibitor or an angiotensin receptor blocker, and showed a mean LVEF of 45 ± 5%. Compared with healthy volunteers, three out of the four DMD patients displayed 25% to 40% deficiency in systemic glutathione (Figure 9).
Figure 9.
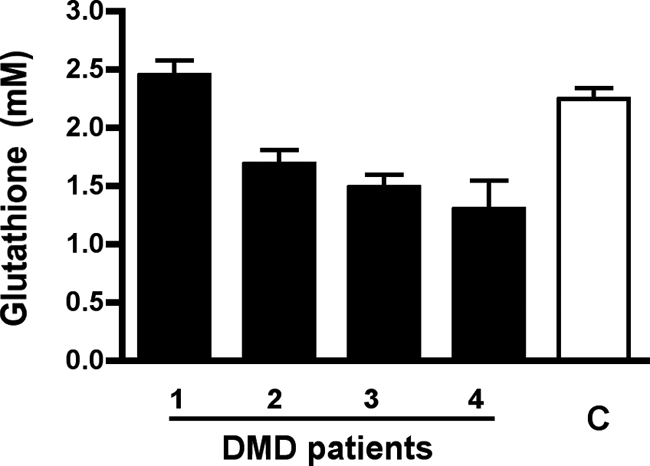
DMD patients may demonstrate systemic glutathione deficiency. Total whole blood glutathione was measured in four DMD patients on wheelchair dependency (mean age of 28 ± 2 years) and in four matched healthy volunteers. Three DMD patients displayed 25% to 40% systemic glutathione deficiency as compared with healthy volunteers.
Discussion
Our current findings are twofold. Firstly, the slow progression of cardiomyopathy in mdx mice is related to a boost in glutathione synthesis and resource. Conversely, administration of the GLCL inhibitor BSO to mdx mice from the age of 10 weeks leads to a decrease in glutathione resource in 20-week-old mdx mice, along with LV hypertrophy, diastolic dysfunction, and structural alterations. Secondly, we point out the occurrence of glutathione deficiency in DMD patients.
Several studies have provided evidence that oxidative stress is implicated in the pathogenesis of the dystrophic disease in DMD patients13,44,45,46 and mdx mice,12,13 and that of cardiomyopathy in mdx mice.47 Oxidative stress in the dystrophic muscles of mdx mice is counteracted by compensatory mechanisms involving up-regulation of glutathione cycle enzymes.27,45,48 We now show a similar adaptation of glutathione cycle enzymes expression in the heart of mdx mice, together with increased systemic glutathione reserve from the age of 15 weeks, when mice undergo a continuing cycle of skeletal muscle fiber degeneration and regeneration.34,49 Such adaptive changes of glutathione metabolism in mdx mice are likely to lessen the severity and/or delay the progression of dystrophic disease and cardiomyopathy. In fact, Dudley et al15 have shown that, as compared with age-matched B6 mice, mdx mice at the age of 14 days before the onset of fiber necrosis demonstrate normal glutathione content in tibialis anterior muscles whereas muscle necrosis in 6- to 8-week-old mdx mice is related to a 20% deficiency in glutathione content, occurring despite increased GPx activity.15 We also found that glutathione content in gastrocnemius of 20-week-old mdx mice was depleted by 33% as compared with B6 mice (0.45 ± 0.06 vs. 0.68 ± 0.07 nmol/mg tissue, respectively, P < 0.05, data not shown). This is in keeping with the significant muscular weakness experienced by mdx mice from 1 month of age throughout their lifespan.50,51 At the difference from the muscular degenerative process, cardiac involvement in mdx mice is usually detected only above 29 weeks of age as an intermediate LV hypertrophic stage, before the manifestations of contractile dysfunction and dilated cardiomyopathy by the age of 42 weeks.29 We now give evidence that the preservation of cardiac structure and function in 20-week-old mdx mice is related to an increased expression of cardiac GLCL and GPx1, likely to compensate for increased glutathione demand ensuing from exacerbated oxidative challenge. This proposal is supported by the finding that BSO-treated mdx mice, which are unable to boost glutathione supply, display an earlier onset of cardiac symptoms than untreated mdx mice. Of note, at the dose of BSO used, BSO-treated mdx mice have similar glutathione blood level and cardiac content as B6 mice. Thus BSO treatment essentially impinges on the adaptive increment of glutathione synthesis, as further attested by the comparable level of glutathione recycling enzymes gene expression in BSO-treated mdx mice and B6 mice.
The corollary of this statement would be that glutathione supplementation prevents the development of cardiomyopathy. As a matter of fact, using a mdx mouse strain that has overt cardiac fibrosis, inflammation and abnormalities in cardiomyocyte Ca2+ handling as early as the age of 9 weeks, Williams and Allen47 reported reduced cardiac abnormalities in the 9 week-old mice having received oral N-acetylcysteine, a glutathione precursor, from weaning. However, before 1 year-old most of the mdx mice from the different strains, including ours, display little cardiac pathology.29,52 The small heart syndrome that we identified in our 20-week-old mdx mice has been previously reported in 10-week-old mdx mice,53 and typifies patients with chronic fatigue.54,55 Interestingly, we found that N-acetylcysteine treatment (1.4 mg/10 g body weight/day), started from weaning, boosted blood glutathione in young mdx mice (but not in older mdx mice with high blood glutathione) and prevented the small heart syndrome in 20-week-old mdx mice (not shown).
From 6 months of age, mdx mice show LV hypertrophy.29,56 This is representative of DMD development since most DMD boys display a hypertrophic stage before clinical symptoms.35,57 An important finding of this study is that glutathione deficiency in BSO-treated mice is associated with LV hypertrophy in 20-week-old mdx mice together with diastolic dysfunction, structural and ultrastructural abnormalities. The presence of collapsed vessels in BSO-treated mdx mice heart is compatible with compromised vascular supply and cardiomyocyte injury due to ischemia. As a matter of fact, a main ultrastructural alteration in the heart of BSO-treated mdx mice consists of mitochondrial abnormalities in cardiomyocytes that are considered as stigmatic of oxidative damage.58,59 The blunting of cardiac collagen genes up-regulation in mdx mice treated with BSO, together with the appearance of necrosis and inflammation foci, are reminiscent of the increased inflammation associated with fibrosis reduction in the diaphragm of mdx mice treated with neutralizing antibody against transforming growth factor-beta1.60 This may be indicative of a role of collagens in stabilizing the extracellular matrix and cellular membranes in mdx mice heart, before the development of fibrosis and subsequent disruption of the cardiac structure.
In conclusion, first, our study shows that BSO-treated mdx mice provide a possible new experimental model for DMD cardiomyopathy, with defined endpoints (left ventricular hypertrophy, diastolic dysfunction, structural abnormalities) that could be used for translational studies to test various therapeutic agents. Second, our results bring to light an adaptive capacity of mdx mice to increase tissue glutathione resource, which faces oxidative stress associated with skeletal muscle degeneration/regeneration cycles and is likely to delay dystrophic and cardiac disease progression. Finally, our preliminary insight into the glutathione status in DMD patients points out the possible occurrence of systemic glutathione deficiency in these patients. This is relevant with the previously reported altered glutathione metabolism in DMD fibroblasts.26 One may also note that the degree of glutathione deficiency in DMD patients is akin to that previously reported in patients with cardiac diseases of non-genetic etiology, whose functional status and structural cardiac abnormalities were related to glutathione deficiency.22 Obviously, more investigation is needed to prove that glutathione deficiency may determine the progression of the cardiac disease in DMD patients. However, these first results encourage further exploration of blood glutathione status in DMD patients at different stages of the disease process. In future, this might open new therapeutic perspectives with molecules targeting glutathione pathway, N-acetylcysteine in particular, which is being launched for the treatment of other inflammatory chronic diseases including idiopathic pulmonary fibrosis.61,62
Acknowledgments
We thank Pr. Bruno Eymard and Dr. David Orlikowski who have examined the patients, Bénédicte Chazaud and Fabrice Chretien for providing us with mdx4Cv mice, and Sophie Lotersztajn for helpful discussions and permanent support.
Footnotes
Address reprint requests to Francoise Pecker, Ph.D., INSERM U955, Institut Mondor de Recherche Biomédicale, Hôpital Henri Mondor, 94010 Créteil France. E-mail: francoise.pecker@inserm.fr.
Supported by the Institut National de la Santé et de la Recherche Médicale, the Assistance Publique des Hôpitaux de Paris (as a Contrat d’Interface to F.P.), the Université Paris-EST and the Association Française contre les Myopathies (as an aide aux études to L.K.).
None of the authors disclosed any relevant financial relationships.
References
- Hoffman EP, Fischbeck KH, Brown RH, Johnson M, Medori R, Loike JD, Harris JB, Waterston R, Brooke M, Specht L, Kupsky W, Chamberlain J, Caskey Y, Shapiro F, Kunkel LM. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne’s or Becker’s muscular dystrophy. N Engl J Med. 1988;318:1363–1368. doi: 10.1056/NEJM198805263182104. [DOI] [PubMed] [Google Scholar]
- Chapman VM, Miller DR, Armstrong D, Caskey CT. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc Natl Acad Sci USA. 1989;86:1292–1296. doi: 10.1073/pnas.86.4.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allamand V, Campbell KP. Animal models for muscular dystrophy: valuable tools for the development of therapies. Hum Mol Genet. 2000;9:2459–2467. doi: 10.1093/hmg/9.16.2459. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Stornaiuolo A, Mavilio F. Failure to correct murine muscular dystrophy. Nature. 2001;411:1014–1015. doi: 10.1038/35082631. [DOI] [PubMed] [Google Scholar]
- Connolly AM, Keeling RM, Mehta S, Pestronk A, Sanes JR. Three mouse models of muscular dystrophy: the natural history of strength and fatigue in dystrophin-, dystrophin/utrophin-, and laminin alpha2-deficient mice. Neuromuscul Disord. 2001;11:703–712. doi: 10.1016/s0960-8966(01)00232-2. [DOI] [PubMed] [Google Scholar]
- Chretien F, Dreyfus PA, Christov C, Caramelle P, Lagrange JL, Chazaud B, Gherardi RK. In vivo fusion of circulating fluorescent cells with dystrophin-deficient myofibers results in extensive sarcoplasmic fluorescence expression but limited dystrophin sarcolemmal expression. Am J Pathol. 2005;166:1741–1748. doi: 10.1016/S0002-9440(10)62484-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D. Challenges and opportunities in dystrophin-deficient cardiomyopathy gene therapy. Hum Mol Genet. 2006;15 Spec No 2:R253–R261. doi: 10.1093/hmg/ddl180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danialou G, Comtois AS, Dudley R, Karpati G, Vincent G, Des Rosiers C, Petrof BJ. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 2001;15:1655–1657. doi: 10.1096/fj.01-0030fje. [DOI] [PubMed] [Google Scholar]
- Townsend D, Yasuda S, Metzger J. Cardiomyopathy of Duchenne muscular dystrophy: pathogenesis and prospect of membrane sealants as a new therapeutic approach. Expert Rev Cardiovasc Ther. 2007;5:99–109. doi: 10.1586/14779072.5.1.99. [DOI] [PubMed] [Google Scholar]
- Rando TA, Disatnik MH, Yu Y, Franco A. Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul Disord. 1998;8:14–21. doi: 10.1016/s0960-8966(97)00124-7. [DOI] [PubMed] [Google Scholar]
- Messina S, Altavilla D, Aguennouz M, Seminara P, Minutoli L, Monici MC, Bitto A, Mazzeo A, Marini H, Squadrito F, Vita G. Lipid peroxidation inhibition blunts nuclear factor-kappaB activation, reduces skeletal muscle degeneration, and enhances muscle function in mdx mice. Am J Pathol. 2006;168:918–926. doi: 10.2353/ajpath.2006.050673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG, Wehling-Henricks M. The role of free radicals in the pathophysiology of muscular dystrophy. J Appl Physiol. 2007;102:1677–1686. doi: 10.1152/japplphysiol.01145.2006. [DOI] [PubMed] [Google Scholar]
- Rando TA. Role of nitric oxide in the pathogenesis of muscular dystrophies: a “two hit” hypothesis of the cause of muscle necrosis. Microsc Res Tech. 2001;55:223–235. doi: 10.1002/jemt.1172. [DOI] [PubMed] [Google Scholar]
- Dudley RW, Danialou G, Govindaraju K, Lands L, Eidelman DE, Petrof BJ. Sarcolemmal damage in dystrophin deficiency is modulated by synergistic interactions between mechanical and oxidative/nitrosative stresses. Am J Pathol. 2006;168:1276–1287. doi: 10.2353/ajpath.2006.050683. quiz 1404–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetler TM, Renard M, Offord EA, Schneider H, Ruegg UT. Green tea extract decreases muscle necrosis in mdx mice and protects against reactive oxygen species. Am J Clin Nutr. 2002;75:749–753. doi: 10.1093/ajcn/75.4.749. [DOI] [PubMed] [Google Scholar]
- Whitehead NP, Pham C, Gervasio OL, Allen DG. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J Physiol. 2008;586:2003–2014. doi: 10.1113/jphysiol.2007.148338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Andrieu-Abadie N, Levade T, Zhang P, Obeid LM, Hannun YA. Glutathione regulation of neutral sphingomyelinase in tumor necrosis factor-alpha-induced cell death. J Biol Chem. 1998;273:11313–11320. doi: 10.1074/jbc.273.18.11313. [DOI] [PubMed] [Google Scholar]
- Pavoine C, Pecker F. Sphingomyelinases: their regulation and roles in cardiovascular pathophysiology. Cardiovasc Res. 2009;82:175–183. doi: 10.1093/cvr/cvp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourraindeloup M, Adamy C, Candiani G, Cailleret M, Bourin MC, Badoual T, Su JB, Adubeiro S, Roudot-Thoraval F, Dubois-Rande JL, Hittinger L, Pecker F. N-acetylcysteine treatment normalizes serum tumor necrosis factor-alpha level and hinders the progression of cardiac injury in hypertensive rats. Circulation. 2004;110:2003–2009. doi: 10.1161/01.CIR.0000143630.14515.7C. [DOI] [PubMed] [Google Scholar]
- Adamy C, Mulder P, Khouzami L, Andrieu-Abadie N, Defer N, Candiani G, Pavoine C, Caramelle P, Souktani R, Le Corvoisier P, Perier M, Kirsch M, Damy T, Berdeaux A, Levade T, Thuillez C, Hittinger L, Pecker F. Neutral sphingomyelinase inhibition participates to the benefits of N-acetylcysteine treatment in post-myocardial infarction failing heart rats. J Mol Cell Cardiol. 2007;43:344–353. doi: 10.1016/j.yjmcc.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Damy T, Kirsch M, Khouzami L, Caramelle P, Le Corvoisier P, Roudot-Thoraval F, Dubois-Rande JL, Hittinger L, Pavoine C, Pecker F. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PLoS ONE. 2009;4:e4871. doi: 10.1371/journal.pone.0004871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad JJ, Harb HL. L-gamma-Glutamyl-L-cysteinyl-glycine (glutathione; GSH) and GSH-related enzymes in the regulation of pro- and anti-inflammatory cytokines: a signaling transcriptional scenario for redox (y) immunologic sensor (s) ? Mol Immunol. 2005;42:987–1014. doi: 10.1016/j.molimm.2004.09.029. [DOI] [PubMed] [Google Scholar]
- Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother. 2003;57:145–155. doi: 10.1016/s0753-3322(03)00043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Schoneveld OJ, Pappa A, Panayiotidis MI. The central role of glutathione in the pathophysiology of human diseases. Arch Physiol Biochem. 2007;113:234–258. doi: 10.1080/13813450701661198. [DOI] [PubMed] [Google Scholar]
- Degl'Innocenti D, Rosati F, Iantomasi T, Vincenzini MT, Ramponi G. GSH system in relation to redox state in dystrophic skin fibroblasts. Biochimie. 1999;81:1025–1029. doi: 10.1016/s0300-9084(99)00334-x. [DOI] [PubMed] [Google Scholar]
- Dudley RW, Khairallah M, Mohammed S, Lands L, Des Rosiers C, Petrof BJ. Dynamic responses of the glutathione system to acute oxidative stress in dystrophic mouse (mdx) muscles. Am J Physiol Regul Integr Comp Physiol. 2006;291:R704–R710. doi: 10.1152/ajpregu.00031.2006. [DOI] [PubMed] [Google Scholar]
- Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology. 2003;99:1–19. doi: 10.1159/000068446. [DOI] [PubMed] [Google Scholar]
- Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord. 2004;14:491–496. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Forgione MA, Cap A, Liao R, Moldovan NI, Eberhardt RT, Lim CC, Jones J, Goldschmidt-Clermont PJ, Loscalzo J. Heterozygous cellular glutathione peroxidase deficiency in the mouse: abnormalities in vascular and cardiac function and structure. Circulation. 2002;106:1154–1158. doi: 10.1161/01.cir.0000026820.87824.6a. [DOI] [PubMed] [Google Scholar]
- Yucel D, Aydogdu S, Cehreli S, Saydam G, Canatan H, Senes M, Cigdem Topkaya B, Nebioglu S. Increased oxidative stress in dilated cardiomyopathic heart failure. Clin Chem. 1998;44:148–154. [PubMed] [Google Scholar]
- Watanabe T, Sagisaka H, Arakawa S, Shibaya Y, Watanabe M, Igarashi I, Tanaka K, Totsuka S, Takasaki W, Manabe S. A novel model of continuous depletion of glutathione in mice treated with L-buthionine (S,R)-sulfoximine. J Toxicol Sci. 2003;28:455–469. doi: 10.2131/jts.28.455. [DOI] [PubMed] [Google Scholar]
- Pastoret C, Sebille A. Age-related differences in regeneration of dystrophic (mdx) and normal muscle in the mouse. Muscle Nerve. 1995;18:1147–1154. doi: 10.1002/mus.880181011. [DOI] [PubMed] [Google Scholar]
- McGeachie JK, Grounds MD, Partridge TA, Morgan JE. Age-related changes in replication of myogenic cells in mdx mice: quantitative autoradiographic studies. J Neurol Sci. 1993;119:169–179. doi: 10.1016/0022-510x(93)90130-q. [DOI] [PubMed] [Google Scholar]
- Nigro G, Comi LI, Palladino A, Petretta VR, Politano L. Cardiomyopathies: diagnosis of types and stages. Acta Myol. 2004;23:97–102. [PubMed] [Google Scholar]
- Crilley JG, Boehm EA, Rajagopalan B, Blamire AM, Styles P, Muntoni F, Hilton-Jones D, Clarke K. Magnetic resonance spectroscopy evidence of abnormal cardiac energetics in Xp21 muscular dystrophy. J Am Coll Cardiol. 2000;36:1953–1958. doi: 10.1016/s0735-1097(00)00960-8. [DOI] [PubMed] [Google Scholar]
- Danko I, Chapman V, Wolff JA. The frequency of revertants in mdx mouse genetic models for Duchenne muscular dystrophy. Pediatr Res. 1992;32:128–131. doi: 10.1203/00006450-199207000-00025. [DOI] [PubMed] [Google Scholar]
- Collins KA, Korcarz CE, Shroff SG, Bednarz JE, Fentzke RC, Lin H, Leiden JM, Lang RM. Accuracy of echocardiographic estimates of left ventricular mass in mice. Am J Physiol Heart Circ Physiol. 2001;280:H1954–H1962. doi: 10.1152/ajpheart.2001.280.5.H1954. [DOI] [PubMed] [Google Scholar]
- Scorsin M, Hagege A, Vilquin JT, Fiszman M, Marotte F, Samuel JL, Rappaport L, Schwartz K, Menasche P. Comparison of the effects of fetal cardiomyocyte and skeletal myoblast transplantation on postinfarction left ventricular function. J Thorac Cardiovasc Surg. 2000;119:1169–1175. doi: 10.1067/mtc.2000.104865. [DOI] [PubMed] [Google Scholar]
- Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2006;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- Defer N, Wan J, Souktani R, Escoubet B, Perier M, Caramelle P, Manin S, Deveaux V, Bourin MC, Zimmer A, Lotersztajn S, Pecker F, Pavoine C. The cannabinoid receptor type 2 promotes cardiac myocyte and fibroblast survival and protects against ischemia/reperfusion-induced cardiomyopathy, FASEB J. 2009;23:2120–2130. doi: 10.1096/fj.09-129478. [DOI] [PubMed] [Google Scholar]
- Ponchel F, Toomes C, Bransfield K, Leong FT, Douglas SH, Field SL, Bell SM, Combaret V, Puisieux A, Mighell AJ, Robinson PA, Inglehearn CF, Isaacs JD, Markham AF. Real-time PCR based on SYBR-Green I fluorescence: an alternative to the TaqMan assay for a relative quantification of gene rearrangements, gene amplifications and micro gene deletions. BMC Biotechnol. 2003;3:18. doi: 10.1186/1472-6750-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter MI, Mohamed JB. Plasma antioxidants and lipid peroxidation products in Duchenne muscular dystrophy. Clin Chim Acta. 1986;155:123–131. doi: 10.1016/0009-8981(86)90273-1. [DOI] [PubMed] [Google Scholar]
- Austin L, de Niese M, McGregor A, Arthur H, Gurusinghe A, Gould MK. Potential oxyradical damage and energy status in individual muscle fibres from degenerating muscle diseases. Neuromuscul Disord. 1992;2:27–33. doi: 10.1016/0960-8966(92)90023-y. [DOI] [PubMed] [Google Scholar]
- Rodriguez MC, Tarnopolsky MA. Patients with dystrophinopathy show evidence of increased oxidative stress. Free Radic Biol Med. 2003;34:1217–1220. doi: 10.1016/s0891-5849(03)00141-2. [DOI] [PubMed] [Google Scholar]
- Williams IA, Allen DG. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am J Physiol Heart Circ Physiol. 2007;293:H1969–H1977. doi: 10.1152/ajpheart.00489.2007. [DOI] [PubMed] [Google Scholar]
- Kar NC, Pearson CM. Catalase, superoxide dismutase, glutathione reductase and thiobarbituric acid-reactive products in normal and dystrophic human muscle. Clin Chim Acta. 1979;94:277–280. doi: 10.1016/0009-8981(79)90076-7. [DOI] [PubMed] [Google Scholar]
- Pastoret C, Sebille A. mdx mice show progressive weakness and muscle deterioration with age. J Neurol Sci. 1995;129:97–105. doi: 10.1016/0022-510x(94)00276-t. [DOI] [PubMed] [Google Scholar]
- Lynch GS, Hinkle RT, Chamberlain JS, Brooks SV, Faulkner JA. Force and power output of fast and slow skeletal muscles from mdx mice 6–28 months old. J Physiol. 2001;535:591–600. doi: 10.1111/j.1469-7793.2001.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe DA, Williams BO, Thomas DD, Grange RW. Molecular and cellular contractile dysfunction of dystrophic muscle from young mice. Muscle Nerve. 2006;34:92–100. doi: 10.1002/mus.20562. [DOI] [PubMed] [Google Scholar]
- Wehling-Henricks M, Jordan MC, Roos KP, Deng B, Tidball JG. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum Mol Genet. 2005;14:1921–1933. doi: 10.1093/hmg/ddi197. [DOI] [PubMed] [Google Scholar]
- Townsend D, Blankinship MJ, Allen JM, Gregorevic P, Chamberlain JS, Metzger JM. Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Mol Ther. 2007;15:1086–1092. doi: 10.1038/sj.mt.6300144. [DOI] [PubMed] [Google Scholar]
- De Lorenzo F, Xiao H, Mukherjee M, Harcup J, Suleiman S, Kadziola Z, Kakkar VV. Chronic fatigue syndrome: physical and cardiovascular deconditioning. QJM. 1998;91:475–481. doi: 10.1093/qjmed/91.7.475. [DOI] [PubMed] [Google Scholar]
- Miwa K, Fujita M. Small heart syndrome in patients with chronic fatigue syndrome. Clin Cardiol. 2008;31:328–333. doi: 10.1002/clc.20227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bia BL, Cassidy PJ, Young ME, Rafael JA, Leighton B, Davies KE, Radda GK, Clarke K. Decreased myocardial nNOS, increased iNOS and abnormal ECGs in mouse models of Duchenne muscular dystrophy. J Mol Cell Cardiol. 1999;31:1857–1862. doi: 10.1006/jmcc.1999.1018. [DOI] [PubMed] [Google Scholar]
- Nigro G, Comi LI, Politano L, Bain RJ. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol. 1990;26:271–277. doi: 10.1016/0167-5273(90)90082-g. [DOI] [PubMed] [Google Scholar]
- Chaiswing L, Cole MP, St Clair DK, Ittarat W, Szweda LI, Oberley TD. Oxidative damage precedes nitrative damage in adriamycin-induced cardiac mitochondrial injury. Toxicol Pathol. 2004;32:536–547. doi: 10.1080/01926230490502601. [DOI] [PubMed] [Google Scholar]
- Shioji K, Kishimoto C, Nakamura H, Masutani H, Yuan Z, Oka S, Yodoi J. Overexpression of thioredoxin-1 in transgenic mice attenuates adriamycin-induced cardiotoxicity. Circulation. 2002;106:1403–1409. doi: 10.1161/01.cir.0000027817.55925.b4. [DOI] [PubMed] [Google Scholar]
- Andreetta F, Bernasconi P, Baggi F, Ferro P, Oliva L, Arnoldi E, Cornelio F, Mantegazza R, Confalonieri P. Immunomodulation of TGF-beta 1 in mdx mouse inhibits connective tissue proliferation in diaphragm but increases inflammatory response: implications for antifibrotic therapy. J Neuroimmunol. 2006;175:77–86. doi: 10.1016/j.jneuroim.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Behr J, Demedts M, Buhl R, Costabel U, Dekhuijzen RP, Jansen HM, MacNee W, Thomeer M, Wallaert B, Laurent F, Nicholson AG, Verbeken EK, Verschakelen J, Flower CD, Petruzzelli S, De Vuyst P, van den Bosch JM, Rodriguez-Becerra E, Lankhorst I, Sardina M, Boissard G. Lung function in idiopathic pulmonary fibrosis–extended analyses of the IFIGENIA trial. Respir Res. 2009;10:101. doi: 10.1186/1465-9921-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, Macnee W, Thomeer M, Wallaert B, Laurent F, Nicholson AG, Verbeken EK, Verschakelen J, Flower CD, Capron F, Petruzzelli S, De Vuyst P, van den Bosch JM, Rodriguez-Becerra E, Corvasce G, Lankhorst I, Sardina M, Montanari M. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005;353:2229–2242. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]