

Abstract
Insig-1 and Insig-2, two closely related proteins, are essential for feedback inhibition of cholesterol biosynthesis. Here, we characterized a line of epidermal-specific, Insig-double knockout (Epi-Insig-DKO) mice lacking both Insigs in epidermis. At birth, Epi-Insig-DKO mice were indistinguishable from control littermates, but thereafter they failed to thrive and died before 6 weeks of age. By 14 days of age, 100% of Epi-Insig-DKO mice exhibited defects in hair growth along with other skin abnormalities, including hyperkeratosis. Hair follicles in Epi-Insig-DKO mice developed normally through postnatal day 7, but they failed to progress to later stages and thus exhibited defects in postnatal hair cycling. Insig deficiency caused a marked buildup of cholesterol precursors in skin associated with a marked increase in 3-hydroxy-3-methylglutaryl coenzyme A reductase protein. Topical treatment of Epi-Insig-DKO mice with simvastatin, an inhibitor of reductase, reduced sterol precursors in skin and corrected the hair and skin defects. We conclude that Insig deficiency in skin causes accumulation of cholesterol precursors, and this impairs normal hair development. These findings have implications for several human genetic diseases in which mutations in cholesterol biosynthetic enzymes lead to accumulation of sterol precursors and multiple cutaneous abnormalities.
INTRODUCTION
Cholesterol has an essential role in mammalian development, and its concentration is tightly regulated by a feedback system that senses the level of cholesterol in cell membranes and modulates the rates of cholesterol synthesis (Brown and Goldstein, 2009). At the core of this feedback regulation are Insig-1 and Insig-2, two closely related endoplasmic reticulum membrane proteins. Under conditions of sterol excess, Insigs limit cholesterol synthesis by enhancing the ubiquitination and degradation of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA reductase), the rate-limiting enzyme of cholesterol biosynthesis. In addition, excess sterols cause Insigs to inhibit the action of Scap, a polytopic membrane protein that transports sterol regulatory element-binding proteins (SREBPs) from the endoplasmic reticulum to the Golgi apparatus (Goldstein et al., 2006).
Sterol regulatory element-binding proteins are membrane bound transcription factors required for transcription of all of the known genes encoding enzymes of the cholesterol biosynthetic pathway (Horton et al., 2002). To enter the nucleus, inactive SREBP precursors must be transported by Scap to the Golgi, where they are processed proteolytically to yield active nuclear fragments (nSREBPs). When sterols accumulate to high levels within cells, Insigs bind and retain the Scap/SREBP complex in the endoplasmic reticulum, preventing the generation of active nSREBPs and thereby reducing transcription of cholesterol biosynthetic genes. By controlling both the stability of HMG-CoA reductase protein and the generation of active nSREBPs, the net result of Insig action is to decrease cholesterol synthesis whenever sterol levels are high (Brown and Goldstein, 2009).
The essential role of Insig-mediated regulation of cholesterol homeostasis in vivo was shown by our previous studies of mice lacking both Insig-1 and Insig-2 (Engelking et al., 2005, 2006). In these Insig double knockout (Insig-DKO) mice, Insig deficiency caused an overproduction of cholesterol and a marked buildup of cholesterol precursors, which led to craniofacial abnormalities. The teratogenic role of sterol intermediates in causing the craniofacial defects was supported by the observation that facial clefting in Insig-DKO mice decreased when pregnant females were treated with lovastatin, an inhibitor of HMG-CoA reductase that lowers the level of cholesterol and its sterol precursors (Engelking et al., 2006). We postulated that the teratogenic effects were caused by the accumulation of sterol precursors, but the mechanism remained unknown. The neonatal lethality of the Insig-DKO mice precluded us from examining whether excess sterol precursors impair developmental processes that occur after birth.
Inasmuch as palatal development requires the interaction between epithelial and mesenchymal cells (Gritli-Linde, 2007), we set out to examine whether the clefting defects in Insig-DKO mice are caused by excess sterol precursors in epithelial and/or mesenchymal cells of the palate. Accordingly, in this study we generated a tissue-specific line of knockout mice lacking both Insig-1 and Insig-2 in the oral epithelium. These mice also lacked Insigs in the epidermis of the skin (termed Epi-Insig-DKO mice). Epi-Insig-DKO mice did not manifest any craniofacial abnormalities, indicating that loss of Insigs in the oral epithelium is not sufficient to produce cleft palate. Instead, Epi-Insig-DKO mice exhibited defects in hair growth and other skin abnormalities, associated with a marked accumulation of sterol precursors in the skin. The causal role of sterol precursors is supported by the observation that the hair and skin defects were completely corrected by topical application of a statin inhibitor of HMG-CoA reductase that lowered the elevated sterol precursors in the skin. These findings have implications for the pathogenesis of skin abnormalities seen in several inborn errors of cholesterol synthesis, such as the CHILD syndrome (congenital hemidysplasia with ichthyosiform nevus and limb defects) and the CDPX2 syndrome (X-linked dominant chondrodysplasia punctata type 2), both of which exhibit a buildup of sterol precursors in skin.
RESULTS
Generation of mice lacking Insig-1 and Insig-2 in the epidermis
Mice carrying floxed Insig-1 and null Insig-2 alleles (Insig-1f/f;Insig-2−/−) (Engelking et al., 2005) were bred to Keratin14-Cre (K14-Cre) transgenic mice in which Cre recombinase is driven by the human Keratin14 promoter that is expressed in the oral epithelium, epidermis, and hair follicles (Dassule et al., 2000; Gritli-Linde et al., 2007). The K14-Cre-mediated deletion of Insig-1 (Figure 1a), in combination with the germ-line deletion of Insig-2, rendered the resulting Insig-1f/f;Insig-2−/−;K14-Cre mice deficient in both Insig-1 and Insig-2 in these tissues. For simplicity, we designated these Insig-1f/f;Insig-2−/−;K14-Cre mice as epidermal-specific Insig-DKO (Epi-Insig-DKO) mice. For all experiments described here, we bred Insig-1f/wt;Insig-2−/−;K14-Cre male and Insig-1f/f;Insig-2−/− female mice to obtain control Insig-1f/f;Insig-2−/− and Epi-Insig-DKO littermates. At 18.5 days post coitum (dpc), Epi-Insig-DKO mice were observed in the expected 1:1:1:1 Mendelian ratio (based on 42 litters). At birth, mutant mice were grossly indistinguishable from wild-type littermates.
Figure 1. Generation of mice lacking Insigs in the epidermis.

(a) Schematic of epidermal-specific deletion of Insig-1. Mice carrying floxed Insig-1 and null Insig-2 alleles (Insig-1f/f;Insig-2−/−) were bred to Keratin14-Cre (K14-Cre) transgenic mice in which Cre recombinase driven by the human Keratin14 promoter was expressed in the oral epithelium, epidermis, and hair follicles. This Cre-mediated deletion of Insig-1, in combination with the germ-line deletion of Insig-2, generated mice that lacked both Insig-1 and Insig-2 in these tissues. Because the epidermis was the major tissue in which Insigs were ablated, we designated these mutant mice as epidermal-specific Insig-double knockout (Epi-Insig-DKO) mice. (b) Total RNA was isolated from back skins and livers of postnatal day 14 (PND 14) littermate control (Insig-1f/f;Insig-2−/−) and Epi-Insig-DKO mice. Relative amount of Insig-1 mRNA was determined by quantitative real-time PCR using the comparative Ct method. Each bar represents the mean ± SEM of data from five mice. The Ct (cycle threshold) numbers are shown inside the bars. (c) Body weights of littermate control and Epi-Insig-DKO mice were measured at various ages. Each bar represents the mean ± SEM of data from the indicated number of mice. Statistical analysis was performed with the two-tailed Student’s t-test.
In Figure 1b, we used real-time PCR to quantify the relative amount of Insig-1 mRNA in the skin and liver from postnatal day 14 (PND 14) control and Epi-Insig-DKO mice. Compared with control mice, the amount of Insig-1 mRNA in the skin of Epi-Insig-DKO mice was reduced by 93%. Liver Insig-1 mRNA levels were comparable between control and Epi-Insig-DKO mice. Therefore, the K14-Cre-mediated recombination led to an efficient and skin-specific ablation of Insig-1 in Epi-Insig-DKO mice, consistent with previous studies in which the same K14-Cre was used to delete other floxed genes in the skin (Wang et al., 2006; Gritli-Linde et al., 2007).
Although Epi-Insig-DKO mice were grossly indistinguishable from control littermates at birth, they failed to thrive, and none survived beyond 6 weeks after birth. The cause of death remains to be determined, but did not appear to result from any detectable gross abnormality in the oral epithelium. Figure 1c shows the body weights of control and Epi-Insig-DKO mice at various ages before death. At 18.5 dpc, Epi-Insig-DKO embryos weighed the same (1.21 g; mean of 99 embryos) as control littermates (1.22 g; mean of 101 embryos). In experiments not shown, both control and mutant 18.5-dpc embryos exhibited an intact epidermal barrier as measured by the X-gal staining assay (Hardman et al., 1998). At PND 4, the mutant mice (1.77 g) were 27% smaller than controls (2.44 g) (Figure 1c). As the mice aged, this difference became more dramatic such that by PND 28 mutant mice (5.93 g) were 63% smaller than controls (15.83 g) (Figure 1c).
Most strikingly, Epi-Insig-DKO mice failed to grow body hair. At PND 14, all of the control mice (44 out of 44) had a full coat of body hair, whereas none of the Epi-Insig-DKO mice (0 out of 48) had any body hair (Figure 2a). Other gross abnormalities in the mutant mice included hyperkeratosis, kinking of the vibrissae (whiskers), and exophthalmos (compare Figure 2c and b). Histological examination revealed abnormalities in the eyes and the meibomian glands. In the former, three out of five Epi-Insig-DKO mice had keratitis; in the latter, five out of five showed gland enlargement. No gross abnormalities were found in the teeth, nails, and internal organs on necropsy.
Figure 2. Gross morphology of control and Epi-Insig-DKO mice.

(a–c) Representative photographs of control and Epi-Insig-DKO littermates at postnatal day (PND) 14. Compared with control mice (a, left), all mutant mice (a, right) were smaller in size, exhibited thickening and flaking of the skin, and failed to grow body hair. Epi-Insig-DKO mice (c) also exhibited exophthalmos and short, kinked vibrissae compared with control mice (b). Scale bars=1.0 cm (a–c).
Histological analyses of skin from control and Epi-Insig-DKO mice
Figure 3 shows histological sections of skin from control and Epi-Insig-DKO mice at PND 14. Whereas the hair follicles of control mice were aligned normally (Figure 3a and inset), those of Epi-Insig-DKO mice were misaligned, unevenly distributed, ectatic, plugged with keratin, and contained distorted hair shafts (Figure 3b and inset). Epi-Insig-DKO mice also showed a loss of hypodermal adipocytes (Figure 3b) and exhibited epidermal hyperplasia and orthokeratotic hyperkeratosis with hypergranulosis (Figure 3d). The hyperplastic epidermis of Epi-Insig-DKO mice was 5- to 7-cell layers thick, whereas the epidermis of control mice was only 2- to 3-cell layers thick. In controls, the dermal papillae were enveloped by follicular epithelial cells to form normal hair bulbs (Figure 3e). In contrast, the dermal papillae of Epi-Insig-DKO mice were condensed and separated from the follicular epithelial cells, associated with degeneration of the hair bulbs (Figure 3f). A slight infiltration of neutrophils was noted in the dermis, but not in the hair follicles.
Figure 3. Histology of skin from control and Epi-Insig-DKO mice.
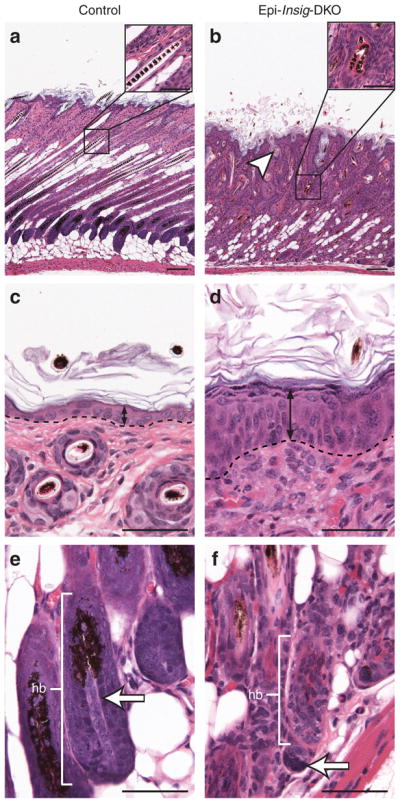
Skin from the upper back of postnatal day (PND) 14 littermate control and Epi-Insig-DKO mice was fixed, sectioned parallel (a, b, e, f) or perpendicular (c, d) to the hair shaft, and stained with hematoxylin and eosin. Shown here are sections representative of five mice in each group. (a, b) Epi-Insig-DKO mice showed a loss of hypodermal adipocytes, misalignment, and ectasia of hair follicles (white arrowhead), and kinking of hair shafts (compare inset of b with the inset of a). (c, d) Epi-Insig-DKO mice exhibited epidermal hyperplasia (black double arrows). The dashed line represents the border between the epidermis and dermis. (e, f) The dermal papillae (white arrows) of Epi-Insig-DKO mice separated from the follicular epithelial cells of the hair bulb (hb), leading to its degeneration. Scale bars=0.1 mm (a, b) and 0.05 mm (c–f, insets of a and b).
Growth of hair follicles is divided into two distinct stages: (1) follicular morphogenesis in which the hair follicle develops and grows from embryonic precursors, and (2) the follicular cycle (or hair cycle) whereby hair follicles grow (anagen), regress (catagen), and rest (telogen), producing new hairs throughout the mammalian lifetime (Alonso and Fuchs, 2006; Schneider et al., 2009). In mice, follicular morphogenesis begins on the upper back at 14.5 dpc. The ectoderm elongates and invaginates, and mesenchymal cells beneath the invaginated ectoderm condense to form the hair placode. The invaginated ectoderm will differentiate into the hair follicle and adjacent epidermis, whereas the mesenchymal cell condensation will become the dermal papilla. After birth, between PNDs 6 and 8, the hair follicles of the back become fully mature, and the cells within the hair bulb continue to proliferate and differentiate, causing the hair shaft to erupt from the skin surface. Around PND 16, when follicular morphogenesis is complete, hair follicles enter the catagen phase of the first follicular cycle and begin to regress. By PND 19, the hair follicles progress to telogen that lasts for 1–2 days. At the end of telogen, the hair follicles enter the first anagen phase of the follicular cycle and remain in this phase until catagen begins again roughly 6 weeks after birth, after which the follicular cycle repeats anew.
To further characterize the hair follicle defects of Epi-Insig-DKO mice, we compared hair follicle development between control and Epi-Insig-DKO mice at PNDs 4, 7, 14, 21, and 28 (Figure 4). At PND 4, hair follicles were histologically similar in control (Figure 4a) and Epi-Insig-DKO (Figure 4f) mice. At PND 7, some hair shafts in Epi-Insig-DKO mice had become kinked (Figure 4g and inset). Near the completion of follicular morphogenesis at PND 14, the hair follicles of control mice were aligned (Figure 4c), whereas the hair follicles of Epi-Insig-DKO had become misaligned and unevenly dispersed (Figure 4h). The hair follicles from these mutant mice also exhibited ectasia, kinked hair shafts, dermal papillae condensation, and hair bulb degeneration. Epi-Insig-DKO mice also showed a loss of hypodermal adipocytes compared with control mice. At PND 21, when hair follicles of control mice were in the telogen phase of the follicular cycle (Figure 4d), those of Epi-Insig-DKO mice were arrested and exhibited progressive ectasia, with keratin accumulation within the follicles (Figure 4i). This arrest of hair follicle cycling persisted in the Epi-Insig-DKO mice at PND 28 with further enlargement of the hair follicles along with minimally detectable scarring (Figure 4j). Because the hair follicles were extremely ectatic at this stage, it was difficult to determine whether follicular drop out had occurred. In control mice at PND 28, hair follicles proceeded into the anagen phase normally (Figure 4e).
Figure 4. Hair follicle morphogenesis and cycling in control and Epi-Insig-DKO mice.

Skin from the upper back of postnatal days (PNDs) 4, 7, 14, 21, and 28 littermate mice was fixed, sectioned parallel to the hair shaft, and stained with hematoxylin and eosin. Shown here are sections representative of five mice in each group. (a, f) At PND 4, hair follicles were histologically identical between control (a) and Epi-Insig-DKO (f) mice. (b, g) At PND 7, some hair shafts in Epi-Insig-DKO mice (g) had become kinked (inset of g). (c, h) By PND 14, the hair follicles of Epi-Insig-DKO mice (h) had become completely misaligned. Furthermore, Epi-Insig-DKO mice also exhibited a loss of hypodermal adipocytes, hyperkeratosis, ectatic hair follicles (white arrowhead), kinking of the hair shafts (compare inset of h with the inset of c), and hair bulb degeneration. (d, e, i, j) At PND 21, when hair follicles of control mice (d) were in the telogen phase of the follicular cycle, those of Epi-Insig-DKO mice (i) were arrested and exhibited progressive ectasia of hair follicles (white arrowheads) with the accumulation of keratin within the follicle. This arrest of hair follicle cycling persisted in the mutant mice (j) at PND 28 with further exacerbation of hair follicle abnormalities; whereas, hair follicles of control mice (e) proceeded into the anagen phase normally. Scale bars=0.1 mm (a–j) and 0.05 mm (insets of b, c, g, h).
Elevation in cholesterol precursors in skin of Epi-Insig-DKO mice and reduction by simvastatin treatment
In our previous studies of germ-line Insig-DKO mice, we showed that Insig deficiency caused an overproduction and marked buildup of sterol precursors of cholesterol that led to craniofacial abnormalities (Engelking et al., 2006). We therefore explored the possibility that the skin abnormalities in Epi-Insig-DKO mice were also due to the accumulation of sterol precursors. As a first step to test this hypothesis, we used oil red O to stain the neutral lipids in skin sections from control and Epi-Insig-DKO mice (Figure 5). At PND 14, sebaceous glands were enlarged and filled with lipids in Epi-Insig-DKO (Figure 5c) compared with those of control (Figure 5a) mice. Quantification of sebaceous gland area revealed that the sebaceous glands of Epi-Insig-DKO mice were 1.7-fold larger in cross-section than those of control mice (2,045 ± 101 vs. 1,236 ± 35 μm2, P<0.001). Lipids also accumulated in the epidermal compartment of the hair bulbs in Epi-Insig-DKO mice (Figure 5d), but not in those of control mice (Figure 5b).
Figure 5. Staining of lipids and immunological visualization of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase in skin from control and Epi-Insig-DKO mice.

(a–d) Skin from the upper back of postnatal day (PND) 14 control and Epi-Insig-DKO mice was fixed, cryosectioned, and stained with oil red O. Shown here are representative sections from four mice per group. (a, c) Sebaceous glands (arrows) were enlarged in mutant (c) compared with control (a) mice. (b, d) Lipids accumulated in the epidermal compartment (arrow) of the hair bulbs in mutant (d) but not in control (b) mice. (e) Immunoblot analysis of HMG-CoA reductase in skin from the upper back of PND 14 control and Epi-Insig-DKO mice. Aliquots of whole-cell lysates (30 μg) were subjected to 8% SDS–PAGE and immunoblot analysis with a rabbit polyclonal antibody against mouse HMG-CoA reductase. Transferrin receptor was used as loading control. Shown are results from four control (lanes 1–4) and four Epi-Insig-DKO (lanes 5–8) mice. (f, g) Immunohistochemical analysis of HMG-CoA reductase in skin from upper back of PND 14 control (f) and Epi-Insig-DKO (g) mice. Shown are sections representative of five mice in each group. Sebaceous glands (arrows) of Epi-Insig-DKO mice (g) showed a massive increase in reductase protein compared with those of control mice (f). Scale bars=0.1 mm (a, c), 0.01 mm (b, d), and 0.02 mm (f, g).
We next carried out immunoblot analysis to determine the protein levels of HMG-CoA reductase in the skin of PND 14 control and Epi-Insig-DKO mice. As shown in Figure 5e, the relative amount of HMG-CoA reductase protein in the skin of Epi-Insig DKO mice was 10-fold higher than that in control mice (mean of four values in each group). These relative values were determined by densitometric quantification of the reductase protein band relative to that of the transferrin protein band (loading control). Figure 5g shows the marked overabundance of reductase protein in the sebaceous glands of the mutant mice, as revealed by immunohistochemistry.
Figure 6a shows an abridged diagram of the cholesterol biosynthetic pathway, depicting the major sterol precursors. Also shown is the enzyme HMG-CoA reductase, which converts HMG-CoA to mevalonate and is the target for the statin class of drugs that inhibit cholesterol synthesis (Goldstein and Brown, 1990). We determined the levels of cholesterol and its precursors in skin from control and Epi-Insig-DKO mice using gas chromatography-mass spectroscopy (GC-MS). In the experiments shown in Figure 6b–g, we treated entire litters of mice daily from PND 2 to PND 13 with topical applications of vehicle or the cholesterol synthesis inhibitor simvastatin. We used a protocol in which acetone is rubbed on the skin to disrupt the permeability barrier before application of the vehicle or simvastatin (Feingold et al., 1990). The drug was applied to a patch of skin 0.5–1 cm2 located between the shoulder blades. At PND 14, after the mice were photographed, skin from the upper back was harvested for sterol measurements, and tail tissues were used for genotyping. The skin of vehicle-treated Epi-Insig-DKO mice had elevated levels of cholesterol and its precursors compared with that of vehicle-treated control mice. As indicated by the black bars in Figure 6, the fold increases ranged from 1.7-fold for desmosterol (Figure 6f) to 7.1-fold for lathosterol (Figure 6e). When treated with simvastatin, the cholesterol content in the skin of control mice was not reduced, whereas the cholesterol content in the skin of Epi-Insig-DKO mice was reduced to a level close to that of control mice (gray bars in Figure 6g). In the skin of control mice, simvastatin treatment significantly reduced the levels of all of the sterol precursors (gray bars in Figure 6b–f). An even greater relative reduction was observed in the skin of the Epi-Insig-DKO mice with the exception of desmosterol, which was not affected by simvastatin treatment (Figure 6f).
Figure 6. Sterol content in the skin of control and Epi-Insig-DKO mice treated with or without simvastatin.

(a) Pathway for cholesterol biosynthesis, showing site of inhibition by simvastatin. (b–g) Littermate control and mutant mice were treated daily with either 1 mg simvastatin or vehicle from postnatal day (PND) 2 to PND 13. At PND 14, skin from the upper back was harvested and subjected to gas chromatography–mass spectroscopy analysis to measure the levels of various sterols. Each bar represents the mean ± SEM of data from five mice. Statistical analysis was performed with the two-tailed Student’s t-test.
Simvastatin-mediated correction of hair defects in Epi-Insig-DKO mice
Figure 7 shows the overall appearances and histological analyses of control and Epi-Insig-DKO mice treated with either vehicle or 1 mg simvastatin. As described earlier, entire litters of mice were treated daily from PND 2 to PND 13 with topical applications of either vehicle or simvastatin. At PND 14, the mice were photographed and scored for hair growth by two independent observers before genotyping. The hair growth and body weights of control mice were not affected by simvastatin treatment (20 out of 20, Figure 7b). All (11 out of 11) of the vehicle-treated Epi-Insig-DKO mice were hairless, the same phenotype exhibited by untreated Epi-Insig-DKO mice (Figure 2). Simvastatin treatment completely corrected the hairless phenotype such that 100% (16 out of 16) of the treated mutant mice had normal hair growth (Figure 7d), no meibomian gland enlargement, and no keratitis. Normalization of hair was observed throughout the body, and not just in the region where the drug was applied. Simvastatin treatment also corrected the low body weight phenotype of Epi-Insig-DKO mice, consistent with a widespread systemic effect. Whereas vehicle-treated Epi-Insig-DKO mice weighed 39% less than vehicle-treated control littermates (4.84 vs. 7.97 g), simvastatin-treated Epi-Insig-DKO and control mice had similar body weights (6.11 vs. 6.90 g). A lower dose of simvastatin (30 μg per day from PND 2 to PND 13) only partially corrected the phenotype; the coat was frizzled and tattered (100% of mutant mice) and the number of hair follicles was markedly reduced and showed abnormalities of the type seen in Figure 3b, d, and f.
Figure 7. Gross morphology and histology of control and Epi-Insig-DKO mice treated with or without simvastatin.
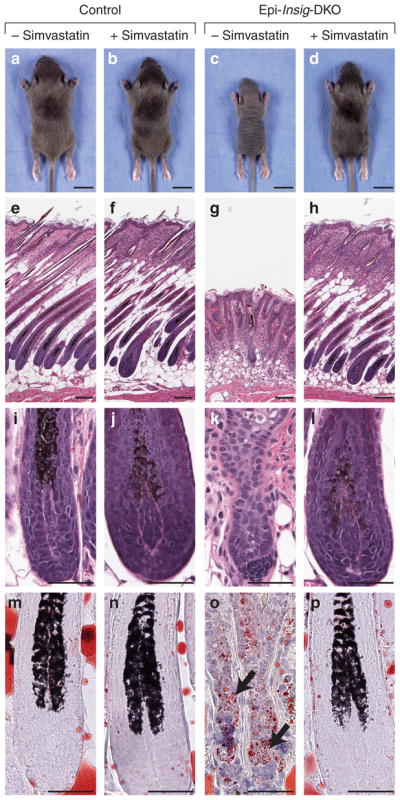
Littermate control and Epi-Insig-DKO mice were treated daily with either 1 mg simvastatin or vehicle from postnatal day (PND) 2 to PND 13. (a–d) At PND 14, animals were photographed and analyzed for hair growth by two independent observers before genotyping. Although none (0 out of 11) of the vehicle-treated Epi-Insig-DKO mice had body hair (c), 100% (16 out of 16) of the simvastatin-treated Epi-Insig-DKO mice exhibited normal hair (d). Hair growth in control mice was not affected by either vehicle (a) or simvastatin (b) treatment. (e–p) Mice were killed at PND 14, and skin from the upper back was fixed and either sectioned for hematoxylin and eosin staining (e–l) or cryosectioned for oil red O staining (m–p). Shown here are sections representative of three mice (m–p) or five mice (e–l) per group. Simvastatin treatment of mutant mice prevented the hair follicle disorganization and degeneration (h vs. g), the hair bulb degeneration and dermal papillae separation (l vs. k), and the accumulation of lipid droplets (black arrows) in the epidermal compartment of hair bulbs (p vs. o). Simvastatin treatment did not have any effect on these histological parameters in control mice (f, j, n vs. e, i, m). Scale bars=1.0 cm (a–d) and 0.1 mm (e–p).
To further evaluate the effect of simvastatin treatment on hair growth, we carried out histological analyses of skin from all four groups of mice described above (Figure 7e–p). Neither vehicle nor simvastatin treatment had any effect on the hair and skin histology of control mice (Figure 7e, f, i, and j). Vehicle-treated Epi-Insig-DKO mice (Figure 7g and k) showed the same gross and histological defects in hair and skin observed in untreated Epi-Insig-DKO mice (Figure 3). All these defects were corrected in simvastatin-treated Epi-Insig-DKO mice (Figure 7h and l). Furthermore, the accumulation of oil red O-stained lipids seen in the epidermal compartment of the hair bulbs of Epi-Insig-DKO mice (Figure 7o) was prevented by simvastatin treatment (Figure 7p). Sebaceous gland hyperplasia seen in Epi-Insig-DKO mice was also ameliorated by simvastatin treatment (data not shown).
Analyses of control and Epi-Insig-DKO mice at different times after stopping simvastatin treatment
In the experiments shown in Figure 7, the mice were treated daily with simvastatin from PND 2 to PND 13 and analyzed at PND 14. To determine whether the mice would regress after cessation of statin treatment, we carried out another study in which control and Epi-Insig-DKO mice were first treated with vehicle or 1 mg simvastatin from PND 2 to PND 13 and then left untreated for the next 5.5 months. The mice were monitored weekly by two independent observers. None (0 out of 4) of the vehicle-treated Epi-Insig-DKO mice survived beyond 6 weeks of age. In contrast, simvastatin-treated Epi-Insig-DKO mice lived to the 6-month end point. Strikingly, though they were only treated with simvastatin from PND 2 to PND 13, the Epi-Insig-DKO mice maintained a full coat of body hair, normal body weight, and normal claws for ~3.5 months (Figure 8a). After 3.5 months, the mutant mice began to lose hair, and body weight decreased. By 6 months of age, the mutant mice showed a near-total loss of body hair (Figure 8b), and their body weight was severely reduced (23.4 ± 0.82 vs. 32.1 ± 1 g in controls, P<0.001). The claws of these mice were deformed and the vibrissae were shortened and kinked. Histological analysis of the skin from Epi-Insig-DKO mice at 6 months of age revealed abnormalities similar to those in Figure 3. Figure 8c–h shows the sterol content of the skin from 6-month old control and Epi-Insig-DKO mice that were treated with simvastatin only from PND 2 to PND 13 and left untreated thereafter. The levels of 24-dihydrolanosterol, zymosterol, lathosterol, and cholesterol were all significantly elevated in the Epi-Insig-DKO mice (Figure 8d–f and h). Lanosterol and desmosterol levels did not differ significantly between control and Epi-Insig-DKO mice (Figure 8c and g).
Figure 8. Morphology and skin sterol content of control and Epi-Insig-DKO mice at 3 and 5.5 months after stopping simvastatin.

(a, b) Five Epi-Insig-DKO mice were treated daily with 1 mg simvastatin from postnatal day (PND) 2 to PND 13. Thereafter, the mice were left untreated for 5.5 months. These photographs show the same Epi-Insig DKO mouse at 3.5 months (a) and 6 months (b) of age. The loss of hair and decrease in body weight that occurred in this mouse between 3.5 and 6 months of age was reproduced in all five mutant mice studied in this experiment. (c–h) The sterol content in the back skin from control and Epi-Insig-DKO mice at 6 months was measured by gas chromatography–mass spectroscopy. Each bar represents the mean ± SEM of data from five mice. Statistical analysis was performed with the two-tailed Student’s t-test.
DISCUSSION
The present studies show that epidermal-specific ablation of the genes encoding Insig-1 and Insig-2, two proteins required for the feedback inhibition of cholesterol synthesis, leads to accumulation of sterol precursors in the skin. The excess sterol precursors impair normal hair development, producing a hairless phenotype along with other skin abnormalities. The causal role of sterol precursors is supported by the observation that the hair and skin defects in the mutant mice were completely corrected by topical application of a statin inhibitor of cholesterol synthesis that lowered the levels of sterol precursors in the skin.
Cholesterol has an essential role in the formation and maintenance of the epidermal permeability barrier (Feingold et al., 1990; Feingold, 2009). To date, three human malformation syndromes caused by inherited enzyme defects in the cholesterol biosynthetic pathway exhibit skin defects (Porter, 2003). These syndromes are CHILD, CDPX2, and desmosterolosis. Inasmuch as the enzymes defective in these syndromes catalyze different steps in the conversion of lanosterol to cholesterol (see Figure 6a), each of these defects leads to a deficiency in cholesterol with a concomitant buildup of various sterol precursors proximal to the defective enzyme. CHILD and CDPX2 patients as well as the corresponding Bpa/Str and Td mice exhibit similar skin defects, including ichthyosis, patchy hyperkeratosis, and alopecia (Liu et al., 1999). Among the two reported cases of human desmosterolosis, one exhibited cutis aplasia (Andersson et al., 2002). The corresponding Dhcr24−/− mice showed wrinkleless taut skin and an impaired epidermal permeability barrier; as a result, the mice died within a few hours after birth (Mirza et al., 2006). The skin defects in these three syndromes are caused, either singly or in combination, by cholesterol deficiency or the accumulation of sterol precursors.
Epi-Insig-DKO mice differ from the aforementioned syndromes in that the accumulation of sterol precursors is not accompanied by cholesterol deficiency. Instead, at PND 14, the skin cholesterol content is elevated 2.2-fold in the Epi-Insig-DKO mice (Figure 6g). Furthermore, the Epi-Insig-DKO mice maintain an intact epidermal barrier at 18.5 dpc and do not die until about 6 weeks after birth, suggesting that skin barrier abnormalities associated with cholesterol deficiency do not underlie the skin and hair defects in Epi-Insig-DKO mice. Together, these results suggest that the defects in the Epi-Insig-DKO mice are caused by a buildup of sterol precursors and not by a deficiency of cholesterol. It is unclear, however, whether these defects are caused by one specific sterol precursor or a combination of sterol precursors.
Hair follicle growth is characterized by two distinct stages: (1) follicular morphogenesis in which the hair follicle develops and grows from embryonic precursors, and (2) the follicular cycle (or hair cycle) whereby hair follicles grow (anagen), regress (catagen), and rest (telogen) to produce new hairs throughout the mammalian lifetime (Alonso and Fuchs, 2006; Schneider et al., 2009). Defects in any one of these steps can lead to anomalies in hair development and growth. The buildup of sterol precursors in the skin of Epi-Insig-DKO mice does not affect the initial stages of follicular morphogenesis (up to PND 7); rather, it appears to interfere with later stages of follicular morphogenesis and the hair cycle (Figure 4).
The mouse mutant, hairless, which is deficient in a transcriptional co-repressor for several genes that regulate keratinocyte differentiation (Benavides et al., 2009), has a hair growth phenotype similar to that of Epi-Insig-DKO mice. However, there are two main phenotypic differences between these two mutants: (1) the defects in hairless appear during the first catagen (after PND 16), whereas those in Epi-Insig-DKO appear during follicular morphogenesis (at PND 7); and (2) hairless mice exhibit dermal cysts, whereas Epi-Insig-DKO mice do not.
The molecular mechanism by which a buildup of sterol precursors interferes with hair follicle growth in the Epi-Insig-DKO mice is unknown. One possible target is the Sonic Hedgehog (Shh) signaling pathway, which has been shown to be important in both follicular morphogenesis and follicular cycling (St-Jacques et al., 1998; Chiang et al., 1999). As shown in Figure 4, Epi-Insig-DKO mice undergo normal follicular morphogenesis up to PND 7, but fail to progress to later stages. These abnormalities are similar but not identical to those in Shh-deficient (Shh−/−) mice that undergo normal follicular morphogenesis up to 15.5 dpc (St-Jacques et al., 1998; Chiang et al., 1999). Whether the accumulation of one or more sterol precursors in Epi-Insig-DKO mice is associated with an inhibition of Shh signaling remains to be determined.
The response of Epi-Insig-DKO mice to topical simvastatin treatment was dramatic. Unfortunately, we were unable to find conditions in which the response was limited exclusively to the region of application, even when we reduced the volume of simvastatin and the surface area to which it was applied. Inasmuch as the drug was applied to skin of suckling pups, the mothers licked the fur of the treated pups, possibly distributing the drug to other regions of the skin. It is likely that the drug was absorbed from the skin, released into the circulation, and taken up by the epidermis where it reduced the synthesis of sterol precursors in the epidermis.
Although further studies of this model are clearly indicated, the current data are sufficient to provide insight into the cutaneous abnormalities that accompany genetic diseases in which sterol precursors accumulate, because of the blocks in the cholesterol biosynthetic pathway. Some of these studies are currently underway in our laboratory.
MATERIALS AND METHODS
Generation of mice lacking Insigs in the epidermis
Mice carrying floxed Insig-1 and null Insig-2 alleles (Insig-1f/f;Insig-2−/−) were generated as described (Engelking et al., 2005). These mice were bred to K14-Cre transgenic mice (stock no. 004782; The Jackson Laboratory, Bar Harbor, ME) to generate Insig-1f/f;Insig-2−/−;K14-Cre mice. For simplicity, we designated these DKO mice lacking Insig-1 and Insig-2 in the epidermis as Epi-Insig-DKO mice. For all experiments described in this paper, we bred Insig-1f/wt;Insig-2−/−;K14-Cre male and Insig-1f/f;Insig-2−/− female mice to obtain control (Insig-1f/f;Insig-2−/−) and Epi-Insig-DKO littermates. Pregnant females were allowed to pup, and the date of birth was denoted as PND 1. Mice were genotyped by PCR using tail genomic DNA with primers described previously (Dassule et al., 2000; Engelking et al., 2005). The genetic background of the studied mice was C57BL6/129SvEv/CBA.
All mice were housed in colony cages with a 12 h light/12 h dark cycle and fed ad libitum Teklad Mouse/Rat Diet 7002 (Harlan Teklad, Indianapolis, IN). All animal experiments were performed with the approval of the Institutional Animal Care and Research Advisory Committee at The University of Texas Southwestern Medical Center at Dallas.
Histology and oil red O staining
For histological analysis, skin from the upper back was collected from mice at PNDs 4, 7, 14, 21, and 28. The skin was fixed in 10% neutral-buffered formalin (catalog no. HT50-1-320; Sigma-Aldrich, St Louis, MO) for 24–72 hours. For hematoxylin and eosin (H&E) staining, skin was embedded in paraffin, sectioned parallel or perpendicular to the hair shaft at 5 μm, and stained with H&E. For oil red O staining, skin was incubated in a 30% aqueous sucrose solution overnight at 4 °C and embedded in Neg-50 mounting medium (catalog no. 6506; Thermo Fisher Scientific, Waltham, MA) and cryosectioned at 10 μm, parallel to the hair shaft. The frozen sections were stained with a 0.18% oil red O solution (catalog no. O0625; Sigma-Aldrich) in 60% (v/v) isopropanol for 10 minutes and counterstained with hematoxylin for 1 second.
Determination of tissue sterol composition
Skin from the upper back was harvested from mice at PND 14, weighed, and placed individually in Folch’s buffer (5 ml) containing 5α-cholestane (4 μg) and epicoprostanol (4 μg) as internal standards. The skins were then homogenized and centrifuged at 260 × g for 5 minutes at room temperature to remove debris. The supernatants were transferred to new glass tubes and then saponified by heating (100 °C) in ethanolic KOH (100 mM) for 2 hours. Lipids were extracted with petroleum ether (final volume of 6 ml), 1/10th of which (0.6 ml) was dried under nitrogen and derivatized with hexamethyldisilazane-trimethylchlorosilane. GC-MS analysis was performed using an Agilent 6890N gas chromatograph coupled to an Agilent 5973 mass selective detector (Agilent Technologies, Santa Clara, CA). The trimethylsilyl-derivatized sterols were separated on a DB-35 (35%-phenyl)-methylpolysiloxane capillary column (30 m × 0.25 mm internal diameter × 0.25 μm film) (catalog no. 122-1932; Agilent Technologies) with carrier gas helium at the rate of 1 ml min−1. The temperature program was 150 °C for 2 minutes, followed by increases of 20 °C per min up to 280 °C, which was then held for 22 minutes. For cholesterol, the injector was operated in a 1:10 split mode. For all other sterols, the injector was operated in splitless mode at 280 °C. The mass spectrometer was operated in the selective ion-monitoring mode. The mass to charge ratios for the extracted ions were 343.3 (desmosterol), 350.4 (7-dehydrocholesterol), 393.4 (lanosterol), 395.0 (24-dihydrolanosterol), 456.4 (zymosterol), 458.4 (lathosterol), and 458.4 (cholesterol). The retention time for lathosterol and cholesterol differed by 2 minutes.
Immunoblot analysis and immunohistochemistry of HMG-CoA reductase
A polyclonal antibody against the C-terminal region (amino acids 426 to 887) of mouse HMG-CoA reductase (GenBank accession no. BC085083) was prepared by immunizing rabbits with purified bacterially expressed (His)6-tagged protein. Immunoblot analysis of mouse reductase was carried out as described (Engelking et al., 2005) except that whole-cell lysates were used. Aliquots of the lysates (30 μg) were subjected to 8% SDS–PAGE and immunoblot analysis with 0.3 μgml−1 anti-reductase (purified IgG fraction). For loading control, identical filters were incubated with 0.25 μgml−1 monoclonal mouse anti-human transferrin receptor (catalog no. 13-6800; Invitrogen Corp, Camarillo, CA).
For immunohistochemistry, skin sections (prepared as described for H&E staining) were heated at 58 °C for 15 minutes, deparaffinized, and quenched in 0.3% hydrogen peroxide for 30 minutes at room temperature. After incubating overnight at 4 °C with 3 μgml−1 anti-reductase antibody, sections were stained with ImmPRESS Reagent and Peroxidase Substrate kits (catalog nos. MP-7401 and SK-4100) from Vector Laboratories (Burlingame, CA).
Simvastatin treatment of neonatal mice
A simvastatin stock solution of 6.4 mgml−1 in propylene glycol/ethanol (PG/E) was prepared in the following manner. Simvastatin (800 mg) (Merck, Whitehouse Station, NJ) was first dissolved in ethanol (16 ml) at 70 °C and converted to its active hydroxyl acid form by the addition of 0.6N NaOH (4 ml). The resulting solution was titrated to pH 7.4, and propylene glycol (catalog no. P-1009; Sigma-Aldrich) was added to give a final PG/E ratio of 7:3 (v/v). Topical treatments of mice with simvastatin were carried out as described (Feingold et al., 1990). Briefly, the back skin of each mouse (0.5–1 cm2) was rubbed for 30 seconds with an acetone-soaked cotton swab to make the skin permeable. Immediately thereafter, 156 μl of PG/E with or without 1 mg simvastatin was applied to the acetone-treated interscapular area. These treatments were carried out daily from PND 2 to PND 13, and the mice were genotyped and photographed at PND 14.
Quantification of sebaceous gland area
Skin from the upper back of PND 14 mice was fixed, cryosectioned, and stained with oil red O as described above. Photographs of all of the sebaceous glands in a section were taken, and their area was quantified using ImageJ (http://rsbweb.nih.gov/ij/index.html). To gain an accurate quantification, we measured the areas of all sebaceous glands (more than 150) from 18 individual skin sections (three mice per group, 6 sections per mouse).
Epidermal barrier assay
The epidermal barrier assay was carried out as described (Hardman et al., 1998). Briefly, embryos were submerged overnight at 30 °C in a solution of 2 mM MgCl2, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, 0.01% (v/v) Triton X, and 1 mgml−1 X-gal in phosphate-buffered saline. The solution was adjusted to pH 4.5 using HCl. In the absence of an intact epidermal barrier, this acidic solution penetrates the epidermal barrier and activates endogenous β-galactosidase to produce a blue precipitate.
Quantitative real-time PCR
Total RNA was prepared from mouse tissues using RNA STAT-60 (Tel-Test, Friendswood, TX). Equal amounts of RNA were pooled from the tissues of five mice and subjected to quantitative real-time PCR as described (Liang et al., 2002). A threshold line of 0.15 was set at the exponential phase of the PCR amplification to determine the number of PCR cycles required (cycle threshold, Ct) for the sample to reach the threshold level. The relative amount of all mRNAs was calculated using the comparative Ct method with cyclophilin as the invariant control. Primers used for real-time PCR have been described previously (Liang et al., 2002; Yabe et al., 2003).
Acknowledgments
We thank the following colleagues for invaluable assistance: Fang Xu and Jonathan Cohen (GC-MS measurements), Jesse Morris (histological sections), and Monica Mendoza and Isis Soto (animal studies). We also thank Dr Paul Bergstresser for critical review of the paper. This research was supported by grants from The Hartwell Foundation, National Institutes of Health (HL-20948), and Perot Family Foundation. Bret M. Evers and Midhat S. Farooqi were supported by Medical Scientist Training Grant GM08014.
Abbreviations
- dpc
days post coitum
- Epi-Insig-DKO mice
epidermal-specific double knockout mice deficient in both Insig-1 and Insig-2
- HMG-CoA reductase
3-hydroxy-3-methylglutaryl coenzyme A reductase
- PND
postnatal day
- Shh
Sonic Hedgehog
- SREBP
sterol regulatory element-binding protein
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- Alonso L, Fuchs E. The hair cycle. J Cell Sci. 2006;119:391–3. doi: 10.1242/jcs.02793. [DOI] [PubMed] [Google Scholar]
- Andersson HC, Kratz L, Kelley R. Desmosterolosis presenting with multiple congenital anomalies and profound developmental delay. Am J Med Genet. 2002;113:315–9. doi: 10.1002/ajmg.b.10873. [DOI] [PubMed] [Google Scholar]
- Benavides F, Oberyszyn TM, VanBuskirk AM, et al. The hairless mouse in skin research. J Dermatol Sci. 2009;53:10–8. doi: 10.1016/j.jdermsci.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Cholesterol feedback: from Schoenheimer’s bottle to Scap’s MELADL. J Lipid Res. 2009;50:S15–27. doi: 10.1194/jlr.R800054-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Swan RZ, Grachtchouk M, et al. Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev Biol. 1999;205:1–9. doi: 10.1006/dbio.1998.9103. [DOI] [PubMed] [Google Scholar]
- Dassule HR, Lewis P, Bei M, et al. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development. 2000;127:4775–85. doi: 10.1242/dev.127.22.4775. [DOI] [PubMed] [Google Scholar]
- Engelking LJ, Evers BM, Richardson JA, et al. Severe facial clefting in Insig-deficient mouse embryos caused by sterol accumulation and reversed by lovastatin. J Clin Invest. 2006;116:2356–65. doi: 10.1172/JCI28988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelking LJ, Liang G, Hammer RE, et al. Schoenheimer effect explained—feedback regulation of cholesterol synthesis in mice mediated by Insig proteins. J Clin Invest. 2005;115:2489–98. doi: 10.1172/JCI25614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold KR. The outer frontier: the importance of lipid metabolism in the skin. J Lipid Res. 2009;50:S417–22. doi: 10.1194/jlr.R800039-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold KR, Mao-Qiang M, Menon GK, et al. Cholesterol synthesis is required for cutaneous barrier function in mice. J Clin Invest. 1990;86:1738–45. doi: 10.1172/JCI114899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A. Molecular control of secondary palate development. Dev Biol. 2007;301:309–26. doi: 10.1016/j.ydbio.2006.07.042. [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A, Hallberg K, Harfe BD, et al. Abnormal hair development and apparent follicular transformation to mammary gland in the absence of hedgehog signaling. Dev Cell. 2007;12:99–112. doi: 10.1016/j.devcel.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman MJ, Sisi P, Banbury DN, et al. Patterned acquisition of skin barrier function during development. Development. 1998;125:1541–52. doi: 10.1242/dev.125.8.1541. [DOI] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–31. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Yang J, Horton JD, et al. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem. 2002;277:9520–8. doi: 10.1074/jbc.M111421200. [DOI] [PubMed] [Google Scholar]
- Liu XY, Dangel AW, Kelley RI, et al. The gene mutated in bare patches and striated mice encodes a novel 3β-hydroxysteroid dehydrogenase. Nat Genet. 1999;22:182–7. doi: 10.1038/9700. [DOI] [PubMed] [Google Scholar]
- Mirza R, Hayasaka S, Takagishi Y, et al. DHCR24 gene knockout mice demonstrate lethal dermopathy with differentiation and maturation defects in the epidermis. J Invest Dermatol. 2006;126:638–47. doi: 10.1038/sj.jid.5700111. [DOI] [PubMed] [Google Scholar]
- Porter FD. Human malformation syndromes due to inborn errors of cholesterol synthesis. Curr Opin Pediatr. 2003;15:607–13. doi: 10.1097/00008480-200312000-00011. [DOI] [PubMed] [Google Scholar]
- Schneider MR, Schmidt-Ullrich R, Paus R. The hair follicle as a dynamic miniorgan. Current Biol. 2009;19:R132–42. doi: 10.1016/j.cub.2008.12.005. [DOI] [PubMed] [Google Scholar]
- St-Jacques B, Dassule HR, Karavanova I, et al. Sonic hedgehog signaling is essential for hair development. Current Biol. 1998;8:1058–68. doi: 10.1016/s0960-9822(98)70443-9. [DOI] [PubMed] [Google Scholar]
- Wang X, Bolotin D, Chu DH, et al. AP-2α: a regulator of EGF receptor signaling and proliferation in skin epidermis. J Cell Biol. 2006;172:409–21. doi: 10.1083/jcb.200510002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe D, Komuro R, Liang G, et al. Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesis. Proc Natl Acad Sci USA. 2003;100:3155–60. doi: 10.1073/pnas.0130116100. [DOI] [PMC free article] [PubMed] [Google Scholar]