

Abstract
Kidney cancer is not a single disease but encompasses a number of different types of cancer that occur in the kidney, each caused by a different gene with a different histology and clinical course that responds differently to therapy. Each of the seven known kidney cancer genes, VHL, MET, FLCN, TSC1, TSC2, FH and SDH, is involved in pathways that respond to metabolic stress and/or nutrient stimulation. The VHL protein is a component of the oxygen and iron sensing pathway that regulates HIF levels in the cell. HGF/MET signaling affects the LKB1/AMPK energy sensing cascade. The FLCN/FNIP1/FNIP2 complex binds AMPK and therefore may interact with the cellular energy and nutrient sensing pathways, AMPK-TSC1/2-mTOR and PI3K-Akt-mTOR. TSC1/TSC2 are downstream of AMPK and negatively regulate mTOR in response to cellular energy deficit. FH and SDH play a central role in the mitochondrial tricarboxylic acid (TCA) cycle whose activities are coupled to energy production through oxidative phosphorylation. Mutations in each of these kidney cancer genes result in dysregulation of metabolic pathways involved in oxygen, iron, energy and/or nutrient sensing suggesting that kidney cancer is a disease of cell metabolism. Targeting the fundamental metabolic abnormalities in kidney cancer provides a unique opportunity for the development of more effective forms of therapy for this disease.
Introduction
Kidney cancer is not a single disease; it comprises a number of different cancers that occur in the kidney, each with a different histology and clinical course, which respond differently to therapy and are caused by mutations in different genes.(1) The study of hereditary kidney cancer syndromes has led to the identification of genes implicated in familial clear cell renal carcinoma, familial chromophobe kidney cancer, familial type 1 and type 2 papillary kidney cancer, familial nonsyndromic renal carcinoma and tuberous sclerosis complex. All cancer is essentially genetic(2) and recently Thompson, et al. have elucidated the central importance of metabolic pathways in cancer.(3–5) Each of the kidney cancer genes identified so far interact with cell metabolism pathways involved in energy, nutrient, iron and/or oxygen sensing (Figure 1).
Figure 1. The genetic basis of kidney cancer: a metabolic disease.

The genes known to cause kidney cancer, VHL, MET, FLCN, FH, SDH, TSC1 and TSC2 share the common feature that each is involved in oxygen, iron, energy and/or nutrient sensing pathways. Kidney cancer is fundamentally a metabolic disease. VHL targets HIF-1α and HIF-2α for ubiquitin-mediated degradation through an oxygen and iron sensing mechanism. The FLCN/FNIP1/FNIP2 complex binds AMPK, the primary energy sensor in the cell, and FLCN is phosphorylated by a rapamycin-sensitive kinase (i.e.,mTORC1). TSC1/TSC2 are phosphorylated by the LKB1/AMPK cascade and help mediate the cell’s response to energy/nutrient sensing. Fumarate hydratase and succinate dehydrogenase are TCA cycle enzymes. When fumarate hydratase or succinate dehydrogenase are deficient, the function of the TCA cycle is impaired and the cell is dependent on glycolysis for energy production. Inactivation of fumarate hydratase or succinate dehydrogenase impairs PHD function and represents a VHL-independent mechanism for dysregulation of HIF degradation. Increased HIF levels lead to increased GLUT1 which enables transport of glucose for ATP production.
Abbreviations: folliculin interacting protein 1 (FNIP1) and folliculin interacting protein 2 (FNIP2), HIF prolyl hydroxylase (PHD). Adapted from Linehan, et al.(1)
The VHL gene pathway is involved in oxygen and energy sensing. The VHL complex targets the hypoxia inducible factors (HIF) for ubiquitin mediated degradation. This is an oxygen and iron sensing mechanism; when the cell is low in oxygen or iron, the VHL complex cannot degrade HIF and HIF over-accumulates. HGF/MET signaling occurs through both the PI3K/Akt/mTOR and the LKB1/AMPK pathways. FLCN, through its interacting partners FNIP1 and FNIP2, binds the bioenergetic sensos, AMP-activated protein kinase (AMPK). In response to energy deficit in the cell, AMPK phosphorylates TSC2 which then complexes with TSC1 and negatively regulates the mTOR pathway. Alterations in any of these genes can affect the energy sensing signaling pathways in the cell.
Hypoxia-inducible factors (HIFs) are oxygen-sensitive basic helix-loop-helix transcription factors, which regulate biological processes that facilitate both oxygen delivery and cellular adaptation to oxygen deprivation. HIF-α, together with the constitutively expressed HIF-β subunit, bind to hypoxia-response elements (HRE) in gene promoters to regulate the expression of genes that are involved in energy metabolism, angiogenesis, erythropoiesis, iron metabolism, cell proliferation, apoptosis and other biological processes. HIF1-a and HIF2-α mediate transcription of a number of downstream genes thought to be important in cancer including transforming growth factor alpha (TGFα), platelet derived growth factor (PDGF), and vascular endothelial growth factor (VEGF). We propose that targeting the metabolic defects in this pathway may provide an opportunity for the development of kidney cancer gene specific therapies that are more durable and more effective than existing treatments. The current targeted therapies for VHL −/− clear cell carcinoma have focused on targeting the genes transcriptionally upregulated by HIF such as vascular endothelial growth factor α (VEGFα), vascular endothelial growth factor receptors (VEGFR), the platelet derived growth factor receptor (PDGFR) or the mTOR/HIF pathway. While most of these agents induce responses in patients with advanced kidney cancer, the responses are usually partial and most patients eventually progress. The fundamental metabolic aspects of these cancer genes may be the Achilles heel that could potentially be exploited to develop more durable and effective forms of therapy. Here we describe the genes that have been identified in clear cell and non-clear-cell kidney cancers, their involvement in cell metabolism pathways and how they might be targeted to provide future therapies.
VHL:von Hippel–Lindau
Positional cloning was used in patients with von Hippel–Lindau to identify the VHL gene locus on the short arm of chromosome 3. (6–12) von Hippel–Lindau is a hereditary cancer syndrome; affected individuals are at risk for the development of renal cysts and clear cell kidney cancer. Germline mutations of the VHL gene (intragenic, deletions or splicing defects) have been found in almost all families with von Hippel–Lindau, (13) and somatic inactivation of VHL by methylation or mutation has been reported in up to 91% of patients with sporadic clear cell renal carcinoma. (14–19)
The VHL protein forms a complex with elongin B, elongin C, and cullin 2(19) that targets HIF for ubiquitin-mediated degradation. (20–22) This process is oxygen-dependent. During normoxia, HIF-α is hydroxylated on a critical proline residue by HIF prolyl hydroxylase (PHD); this process requires molecular oxygen, 2-oxoglutarate, ascorbate and Fe2+ as cofactors, facilitating HIF- α binding to pVHL, and proteasomal degradation by the E3 ubiquitin ligase complex. Under hypoxic conditions, PHD cannot hydroxylate HIF- α, therefore the pVHL/elonginC/elonginB/cullin complex does not bind HIF and HIF- α accumulates. (21;23) (Figure 2) Similarly, when the VHL gene is mutated, mutant pVHL can no longer bind to HIF- α resulting in HIF over accumulation. Although there is considerable overlap in the genes that are transcriptionally regulated by HIF1-α and HIF2-α, in vitro and in vivo studies have indicated that HIF2-α is the critical HIF for tumorigenesis in clear cell kidney cancer. (24–26) Clear cell kidney cancers (with or without VHL mutation) that express both HIF1-α and HIF2-α exhibit enhanced signaling via the mitogen-activated protein kinase (MAPK) and serine/threonine-protein kinase mammalian target of rapamycin (mTOR) pathways, whereas clear cell tumors that express only HIF2-α have elevated c-Myc activity. (27) If HIF2-α is the critical pathway for kidney cancer tumorigenesis, targeting the HIF2-α pathway would likely be more successful than developing agents which solely target the HIF1-α pathway.(Figure 3) Efforts are currently underway to identify agents which downregulate HIF2-α. An increased understanding of VHL signaling has provided the foundation for the development of therapeutic approaches that target this pathway in patients with advanced clear cell kidney cancer. A number of drugs have recently been approved by the FDA that target either the downstream targets of HIF activity (such as the VEGF receptor) or inhibit mTORC1, which provides translational control of HIF1-α.(28) These agents are discussed in detail elsewhere (Karakiewicz Review, June issue). Briefly, bevacizumab is an antibody that targets VEGFA. Sorafenib is a small molecule multikinase inhibitor that targets VEGF and PDGF receptors and the Raf pathway. (29) Sunitinib also inhibits VEGF and PDGF receptors, and is most frequently used as a first-line agent in patients with advanced clear cell kidney cancer. (30) Axitinib and pazopanib are recently developed selective inhibitors of the VEGF receptors. (31;32) Pazopanib has been approved by the US FDA for treatment of metastatic kidney cancer , while axitinib is currently being evaluated in randomized phase III trials. Early clinical experience with the selective VEGF receptor antagonists suggests that these agents may be better tolerated than sunitinib and sorafenib without compromising efficacy; if these observations are confirmed in phase III randomized trials, selective VEGF receptor antoagonists are likely to be increasingly used as first-line agents in patients with advanced clear cell kidney cancer. Temsirolimus and everolimus inhibit mTORC1. Temsirolimus has been shown to prolong survival in previously untreated kidney cancer patients with high-risk features compared to interferon; in patients who have progressed on sunitinib or sorafenib, everolimus is associated with a modest prolongation in progression-free survival compared to placebo and this agent is a reasonable second-line options in patients with advanced kidney cancer who have progressed on front-line therapy targeting the VEGF pathway.
Figure 2. The VHL complex targets HIF: an oxygen/iron sensing pathway.

The VHL E3 ubiquitin ligase complex targets the hypoxia inducible factors for ubiquitin-mediated degradation. This process is mediated by HIF prolyl hydroxylase (PHD) and an essential cofactor, 2-oxoglutarate (2-OG). This is an oxygen/iron sensing system; when the cell is hypoxic or iron levels are low, the VHL complex cannot target and degrade HIF and HIF over-accumulates, driving the transcription of a number of genes important in cancer, such as vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF) and the glucose transporter, GLUT 1.(2–4)
Figure 3. The VHL complex targets HIF-1α and HIF-2α for degradation.

The VHL complex targets HIF-1α and HIF-2α for ubiquitin mediated degradation.(2) When the VHL gene is mutated, as in clear cell kidney cancer, the complex cannot target and degrade HIF. HIF-2α is thought to be a critical pathway for VHL-deficient clear cell kidney tumorigenesis.(5;6)
Most of the therapeutic agents currently approved to treat clear cell kidney cancer target selected downstream components of the HIF pathway including VEGF and the VEGF and PDGF receptors. The responses to these agents observed in patients with advanced clear cell kidney cancer is proof of principle that targeting the VHL/HIF pathway can induce tumor regression in humans. However, these agents target only a small portion of the downstream genes regulated by HIF. An approach to target HIF transcriptional activity or translation of HIF itself, i.e. to affect all of the genes regulated by HIF, could potentially provide a more effective approach to therapy. Topotecan is a topoisomerase 1 inhibitor that represses HIF1-α-dependent transcription. (33;34) This agent is currently under evaluation in a phase 1 clinical trial in patients whose tumors express HIF1-α (Clinicaltrials.org; 888-NCI-1937). A number of groups are currently screening for agents that target transcription of HIF2-α transcriptionally regulated genes. (35) mTOR is part of two distinct signaling complexes. The rapamycin-sensitive mTORC1 complex regulates cell growth and protein synthesis in response to growth factor stimulation by phosphorylating S6 kinase and the eIF-4E binding protein to modulate key regulators of mRNA translation. The rapamycin-insensitive complex mTORC2 regulates actin cytoskeleton organization through phosphorylation of protein kinase Cα and also phosphorylates Akt at S473 to activate the Akt-mTORC1 pathway. The mTORC1 inhibitor temsirolimus and other ‘rapalogs’ suppress tumor growth in VHL-deficient clear cell kidney cancers via inhibition of HIF-1α translation. (36) Most VHL-deficient tumors, however, express both HIF-1α and HIF-2α. (27) Knockout experiments have revealed that HIF1-α translation is dependent on mTORC1 signaling, whereas HIF2-α is downstream of the mTORC2 pathway . (37) New agents that target both the mTORC1 and mTORC2 pathways have the potential to downregulate both HIF1-α and HIF2-α in clear cell kidney cancers and could provide more antitumor activity than temsirolimus and everolimus, which primarily target the mTORC1 pathway. An agent which inhibits both the mTORC1 and mTORC2 pathway, AZD8055, has been recently shown to have potent anti-tumor activity in in vitro and in vivo model systems(38) and it is anticipated that clinical trials evaluating the effectiveness of this agent will be initiated soon.
The development of other strategies that decrease translation of HIF may also have significant potential for patients with advanced kidney cancer. The presence of an iron responsive element (IRE) in the 5’ untranslated region of HIF2-α messenger RNA (mRNA) suggests that components of the iron metabolism pathway might be exploited to this end. (39) Cytoplasmic aconitate hydratase (IRP1) is an iron sensing protein involved in maintaining iron homeostasis. IRP1 binds to the iron responsive elements of HIF2-a mRNA and inhibits its translation. (40;41) Overexpression of IRP1 suppresses growth of lung tumor xenografts, (42) which has prompted the search for small molecules that could decrease HIF2-α translation by enhancing the binding of IRP1 to the 5’ untranslated region of its mRNA.(43) Identification of agents which enhance the binding of IRP1 to the IRE in the 5’ untranslated region of the HIF2-α mRNA or mimic IRP1 activity could provide a novel targeted approach to therapy.
Turcotte et al. (44) have utilized a novel approach to identify agents that target the VHL pathway. They performed a synthetic lethal screen in a set of VHL-negative versus VHL-positive clear cell kidney cancer cell lines and identified STF-62247, a cytotoxic agent that inhibits tumor growth. STF-62247 selectively induced toxicity in VHL −/− cells in a HIF-independent fashion and was found to induce autophagy. This agent was found to have activity in both in vitro and in vivo models, suggesting possible therapeutic potential for this or a related agent in humans with advanced kidney cancer. By targeting autophagy, a central component of the cell’s response to nutrient and energy deprivation, Turcotte and colleagues have developed a new paradigm for cancer therapy. Many chemotherapeutic and targeted tyrosine kinase approaches are limited by their toxicity to non-tumor cells. However, targeting autophagy was found to affect only the VHL −/−cells whereas cells with a wild-type copy of the VHL gene were not affected. (44) This approach, targeting the fundamental metabolic defect in the cell, has the potential to provide a less toxic and more effective form of therapy for patients with this disease.
MET: Hereditary Papillary Renal Cell Carcinoma
The proto-oncogene MET (hepatocyte growth factor receptor) was identified as the gene for hereditary papillary renal carcinoma by genetic linkage analysis in families with this inherited renal cancer syndrome. MET encodes the cell surface receptor for hepatocyte growth factor (HGF), which is involved in mitogenesis, morphogenesis and motogenesis. (45) Activating mutations in the tyrosine kinase domain of MET, have been detected in the germline of affected patients and in a subset of sporadic type 1 papillary kidney cancers. (46;47) The histological patterns of hereditary and sporadic type 1 papillary kidney tumors with MET mutations share a distinct morphological phenotype consisting of papillary or tubulo/papillary architecture with slender short papillae containing delicate fibrovascular cores lined by small cells with low grade basophilic nuclei and scant amphophilic cytoplasm. (48)
One effect of growth factor-dependent activation of the phosphatidylinositol 3-kinase (PI3K) signaling pathway is increased cell surface expression of nutrient transporters increasing uptake of amino acids, glucose and other nutrients.(4) Nutrient-stimulated HGF/MET signaling induces phosphorylation of serine/threonine-protein kinase 11 [STK11; also referred to as LKB1, the upstream kinase of 5’AMP–activated protein kinase (AMPK)] on Ser428 through the RAS-Erk1/2-p90RSK pathway, implicating MET in the LKB1-AMPK-mTOR nutrient and energy sensing pathway.
A clinical trial is currently underway to determine the effect of foretinib, a kinase inhibitor of both MET and VEGF receptors, in patients with papillary kidney cancer (hereditary and sporadic). There is early evidence of efficacy of this agent in patients with germline mutations in the tyrosine kinase domain of MET. (49) It is possible that there would be response to an agent which has activity against MET in tumors that are characterized by a mutation in the tyrosine kinase domain of MET; it is also possible that such an agent would have activity in tumors which have MET amplification. It is not known if an agent such as foretinib would have activity against kidney tumors that are caused by mutation of other genes such as TSC1 or TSC2.
TSC1/TSC2: Tuberous Sclerosis Complex
The tuberous sclerosis genes TSC1 and TSC2 are also involved in the AMPK/mTOR nutrient and energy sensing pathway (Figure 1). Germline mutations in one or other of these genes have been identified in patients with tuberous sclerosis complex, an autosomal dominant disorder associated with the development of cutaneous angiofibromas, pulmonary lymphangiomyomatosis and renal tumors. (50) Angiomyolipoma is the most common renal tumor found in affected individuals; however, other tumor types have been reported, including clear cell renal carcinoma. (51)
TSC1 encodes hamartin and TSC2 encodes tuberin, which form a heterodimer that acts as a GTPase-activating protein toward Rheb, a Ras-family GTPase that activates mTORC1. GTPase activity of the TSC1/2 complex on Rheb results in inhibition of mTOR activity. TSC1 or TSC2 deficient tumors exhibit increased phosphorylation of p70S6 kinase, S6 ribosomal protein and 4E-BP1, downstream effectors of mTORC1 activation, and readouts for initiation of mRNA translation and protein synthesis. (50) Lack of TSC1/2 inhibition would presumably also result in HIF accumulation through increased HIF mRNA translation by activated mTORC1.
Sirolimus has been demonstrated to cause regression of renal angiomyolipomas in patients with tuberous sclerosis complex. Sirolimus forms a complex with FK binding protein 12 and inhibits mTORC1 signaling. (47) Although most of the kidney tumors that regressed on therapy tended to increase in volume after the therapy was stopped, this study provided the foundation for a molecular therapeutic approach for the treatment of renal tumors associated with the TSC1-TSC2 pathway. There are currently a number of trials evaluating the role of sirolimus in patients with TSC. When the role of this agent in TSC patients has been defined, future trials that combine this agent with other agents targeting this pathway will be possible.
Folliculin: Birt-Hogg-Dubé syndrome
Genetic linkage analysis in patients with Birt–Hogg–Dubé syndrome identified folliculin (FLCN), a novel gene on the short arm of chromosome 17, as the causal gene. (52;53) Patients with Birt–Hogg–Dubé syndrome are at risk for the development of benign cutaneous tumors (fibrofolliculomas), (54) pulmonary cysts(54) and chromophobe kidney tumors. (55) (Figure 4) These tumors may be bilateral, multifocal and can metastasize. (56) FLCN gene mutations have been detected in over 90% of families affected by Birt–Hogg–Dubé syndrome. (53–59) Most germline mutations that have been detected in FLCN are predicted to truncate the protein. The results of DNA sequencing to identify somatic mutations in the wild type allele of FLCN in renal tumors from patients with Birt-Hogg-Dubé syndrome revealed loss of heterozygosity of the FLCN locus in 17% of renal tumors and sequence alterations in 53%.(60) These findings support the notion that FLCN is a tumor suppressor gene.
Figure 4. Birt-Hogg-Dubé (BHD) syndrome: kidney cancer, cutaneous fibrofolliculomas and pulmonary cysts.

BHD patients are at risk for the development of benign cutaneous tumors (A, B) that may occur on the face and neck. These cutaneous lesions are benign fibrofolliculomas (C). BHD patients are at risk for the development of renal tumors that can be solitary (D), bilateral, multifocal (E) and can grow to large size and metastasize (F). From Pavlovich, et al.(7)
Folliculin is also involved in the AMPK/mTOR pathway; FLCN forms a complex with folliculin interacting proteins 1 and 2 (FNIP1 and FNIP2), which binds the γ-subunit of AMPK (Figure 5A). (61–63) The functional significance of this interaction and the mechanism by which folliculin may regulate AMPK-mTOR signaling is currently unknown but is under intense investigation by a number of laboratories. One approach to understanding folliculin function is through the study of in vivo animal models in which FLCN is inactivated. Both the mTORC1 and mTORC2 pathways were found to be activated in the kidney tumors of a FLCN knockout mouse. (64) (Figure 5B) These findings suggest that targeting both the mTORC1 and mTORC2 pathways may show promise for patients with Birt-Hogg-Dubé-associated kidney cancers.
Figure 5. The FLCN pathway.

A. FLCN is the gene for the Birt-Hogg-Dubé (BHD) syndrome. Patients affected with BHD are characterized by germline mutation of the FLCN gene. The FLCN/FNIP1/FNIP2 complex binds AMPK and FLCN is phosphorylated by a rapamycin-sensitive kinase (i.e.,mTORC1). B. When FLCN is deficient, AKT, mTORC1 and mTORC2 are activated. From Hasumi, et al.(8)
The FLCN gene has been selectively inactivated in the kidneys of a murine model; affected mice developed enlarged polycystic kidneys and died from renal failure by 3 weeks of age. (65;66) FLCN knockout mice treated with rapamycin had smaller, less cystic kidneys than untreated FLCN knockout mice and a longer median survival time. (65) The results in this in vivo model of BHD would suggest that rapamycin analogues such as sirolimus might be potential therapeutic agents against Birt-Hogg-Dubé-associated renal tumors.
Tricarboxylic Acid Cycle Genes
The tricarboxylic acid (TCA) cycle is part of a metabolic pathway coupled to mitochondrial oxidative phosphorylation that converts nutrients to energy in aerobic cells. The fumarate hydratase (FH) and succinate dehydrogenase (SDH) genes encode mitochondrial TCA cycle enzymes that play an essential role in energy production in the TCA, or Krebs, cycle by catalyzing the conversion of fumarate to malate and succinate to fumarate respectively. Individuals who harbor germline mutations in either of these TCA cycle enzymes have an increased risk for developing renal tumors.
Fumarate Hydratase: Hereditary Leiomyomatosis Renal Cell Carcinoma
Fumarate hydratase (FH) was identified as the gene responsible for hereditary leiomyomatosis renal cell carcinoma (HLRCC). (67) The kidney tumors in affected individuals are most often solitary and unilateral and have a mix of histologic patterns; they can be papillary, tubulopapillary or solid. The hallmark of hereditary leiomyomatosis renal cell carcinoma is the presence of a large nucleus with prominent orangiophilic nucleoli. (68) (Figure 6) Germline mutations of the FH gene have been detected in over 90% of North American individuals with hereditary leiomyomatosis renal cell carcinoma. (69–71) Inhibition of FH leads to increased levels of fumarate that inhibit the activity of HIF prolyl hydroxylase, resulting in HIF-α accumulation and transcriptional upregulation of HIF-target genes, thereby contributing to the highly aggressive nature of HLRCC-associated renal tumors. (72) The inhibition of HIF prolyl hydroxylase by the increased levels of fumarate provides a VHL independent mechanism for dysregulation of HIF degradation in FH- deficient HLRCC-associated kidney cancers. (Figure 7A)
Figure 6. Hereditary leiomyomatosis renal cell cancer.
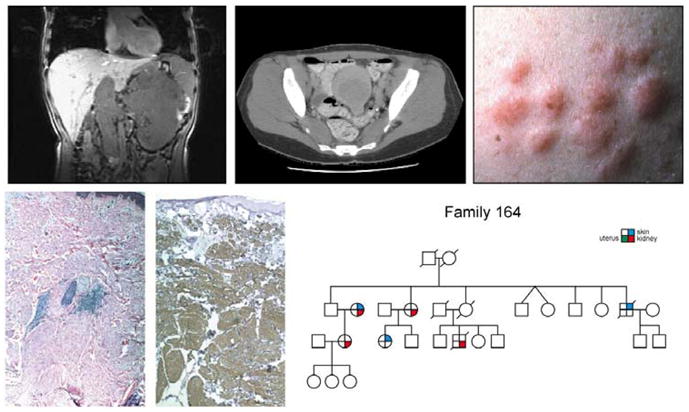
Hereditary leiomyomatosis renal cell cancer (HLRCC) is a hereditary cancer syndrome in which affected individuals are at risk for the development of kidney cancer (upper left panel) and uterine (upper middle panel) and cutaneous (upper right panel) leiomyomas (lower left and center panels). From Linehan et al.(9)
Figure 7. Fumarate Hydratase and Succinate Dehydrogenase Deficient Kidney Cancer: the Warburg Effect.

Fumarate hydratase and succinate dehydrogenase–deficient kidney cancer are examples of the Warburg effect in cancer.(10) Germline fumarate hydratase mutations are found in the germline of patients affected with Hereditary Leiomyomatosis Renal Cell Cancer (HLRCC). HLRCC patients are at risk for the development of an aggressive form of kidney cancer. A) In fumarate hydratase deficient cells, fumarate accumulates and inhibits prolyl hydroxylation by competitively inhibiting HPH (A). HIF accumulates and increases the transcription of vascular endothelial growth factor and the glucose transporter, GLUT-1. Oxidative phosphorylation is impaired by the loss of the TCA cycle enzyme, fumarate hydratase, and the cells depend on glycolysis for energy production.(11;12) B) Succinate dehydrogenase is another TCA cycle enzyme which has also been associated with familial renal carcinoma. Increased levels of succinate have also been shown to inhibit HPH and a similar mechanism for tumorigenesis is likely for SDH-associated kidney cancer.(11)
FH-deficient kidney cancer cell lines are glucose dependent(73;74) and have significantly impaired oxidative phosphorylation. (73;75) Sudarshan et al. (74) have demonstrated that the glucose-mediated generation of cellular reactive oxygen species in an FH-deficient kidney cancer cell line results in stabilization of HIF1-α. The impaired oxidative phosphorylation in FH-deficient kidney cancer results in a nearly total dependence on glycolysis for energy production. Downstream HIF1-α genes such as VEGF and GLUT1 would therefore be critically important to these cancer cells for increasing vasculature and increasing glucose transport. Tumor vasculature and glucose transport could be the Achilles heel for these aggressive cancers that are so dependent on glucose transport for energy production. Whereas targeting the vasculature in patients with advanced VHL-deficient clear cell kidney cancer has resulted in only modest success, targeting the vasculature in a cancer whose metabolism is so completely dependent on glucose transport for energy production could provide a more powerful and effective approach to therapy.
Succinate Dehydrogenase: Familial Renal Cancer
Germline mutations in three of the four succinate dehydrogenase genes (SDHB, SDHC, and SDHD) have been associated with familial paraganglioma and familial pheochromocytoma. (76;77) Individuals with these hereditary conditions are at risk for the development of extra-adrenal paraganglioma or pheochromocytoma and carotid body tumors. Recently early onset renal tumors have been found to develop in individuals with germline SDHB mutations .(77–81) In pre-clinical models increased succinate has been shown to inhibit HIF prolyl hydroxylase and affect HIF stability.(72) Inactivation of succinate dehydrogenase would be expected to severely impair oxidative phosphorylation and lead to glucose dependence of SDH-deficient kidney tumors.(Figure 7B) It is likely that SDH-deficient kidney tumors would be as sensitive to glucose as FH-deficient tumors and an approach to targeting the vasculature and/or glucose transport in these tumors could provide a novel approach to disrupt the fundamental metabolic machinery of these aggressive cancers.
Discussion
Mutations in each of the seven known kidney cancer genes lead to the dysregulation of at least one metabolic pathway that is mediated by oxygen, iron, energy and/or nutrient sensing suggesting that kidney cancer is a disease of dysregulated cellular metabolism. The common endpoint resulting from VHL, FH and SDH mutations is the stabilization of HIF through inactivation of PDH, driving the transcriptional activation of genes that support tumor growth, neovascularization, invasion and metastasis. On the other hand HIF translation is upregulated by activation of the mTOR pathway as a consequence of its dysregulation by TSC1/ TSC2 or FLCN mutations, or oncogenic HGF/MET signaling through RAS-Erk1/2-Mek1/2-p90RSK phosphorylation of LKB1. Targeting HIF and its target genes has provided a first line therapeutic approach to VHL-deficient kidney tumors that may be applied to FH- and SDH-deficient tumors as well.
Progress has been made in this approach to targeted therapy for kidney cancer in recent years. Six novel agents focusing on the VHL pathway have been approved for the treatment of patients with advanced kidney cancer. However, even the most commonly used agent, sunitinib, has a partial response rate of only 31% and a progression free advantage over interferon of 6 months (11 versus 5 months).(82) Most patients with advanced kidney cancer treated with targeted agents eventually progress and there are very few long term complete responses. The need for novel therapeutic approaches for this tumor type is underscored by the eventual failure of the presently available therapeutic treatments. One report describing a novel agent, STF-62247, which targets autophagy selectively in VHL-deficient clear cell kidney cancer is an excellent example of an innovative approach to therapy for VHL-deficient tumors and highlights the role of metabolic defects in this cancer.(44)
It is possible we could spend the next decade performing clinical trials evaluating different combinations of tyrosine kinase inhibitors targeting a limited number of downstream HIF pathway genes with only minimal incremental improvement in outcome. Thinking about kidney cancer as a metabolic disease and targeting the kidney cancer gene metabolic abnormalities provides a potentially different paradigm for the development of therapy for this disease. As we learn more about the metabolic basis of kidney cancer and we start to think about these cancers in a different way, we may find that the importance of the oxidative phosphorylation status of an individual tumor may rival the activation status of downstream HIF targets in guiding rational targeted therapeutic approaches.
The two best examples of a metabolic basis for kidney cancer are fumarate hydratase and succinate dehydrogenase deficient kidney cancers, which represent the “Warburg effect” in cancer.(83) The FH-deficient UOK262 kidney cancer cell line, established from an HLRCC-associated renal tumor, has been shown to have severely impaired oxidative phosphorylation and to be dependent on glycolysis for energy production. This is supported by ultrastructural analysis of UOK262 kidney cancer cells, which revealed abnormal mitochondria with significantly distorted cristae.(73) These FH-deficient tumor cells have significantly increased glucose transport and are extremely dependent on glucose for growth.(73) FH deficient HLRCC tumors in patients express high levels of the glucose transporter, GLUT1, and these tumors characteristically demonstrate high uptake of 18FDG on PET scans.(72;73) Targeting the vasculature and impairing glucose transport in a glucose-addicted tumor could provide a new paradigm for treatment of these lethal malignancies. Interestingly, although the genetic basis of sporadic chromophobe renal carcinoma and oncocytomas is not known, ultrastructural analysis these renal neoplasms reveals abnormal mitochondria with altered cristae also suggestive of compromised mitochondrial function (84). The mitochondrial defects shared by FH-deficient tumors, sporadic chromophobe renal tumors and renal oncocytomas may suggest a common approach to therapy for these tumor types.
Treatment with inhibitors of mTOR activity has been applied to TSC2-deficient tumors and may show promise for FLCN-deficient tumors in the future. Furthermore, there has been renewed interest in agents such as metformin that activate AMPK, which in turn downregulates mTOR activity, as a potential therapeutic treatment for kidney cancer. Metformin use has been associated with a decreased cancer mortality in type II diabetics(85;86) and has been shown to block tumor growth in combination with chemotherapeutic agents in preclinical in vitro and in vivo models of cancer.(87;88) Although metformin is taken by patients with type II diabetics worldwide and is a relatively non-toxic agent, there are risks associated with its long term use.(85) However, if such agents were to be effective components of a targeted therapeutic approach, the benefit would seem to far outweigh the potential risk.
If we think about the factors known to increase the incidence of kidney cancer, it is not surprising that kidney cancer is fundamentally a metabolic disease; numerous studies have shown a significant association between body mass index, obesity and the development of kidney cancer.(89) We have learned much from our study of the metabolic basis for kidney cancer which should enable us to predict the pathways likely to be involved in histologic types of kidney cancer with a lower incidence leading to the design of more effective therapies. Although the genetic basis of more rare types of kidney cancer such as medullary renal carcinoma is not known, it is likely that the origin of medullary renal cell carcinoma is associated with hypoxia. Medullary renal carcinoma is an aggressive form of kidney cancer almost exclusively seen in patients who are affected with sickle cell disease or trait.(90) Sickle cell disease can be associated with a number of nephropathies, including microangiopathy.(91) It would not be surprising to find medullary renal cell carcinoma developing from a hypoxic area of renal parenchyma which may show elevated levels of HIF and may respond to targeted anti-HIF therapies.
Conclusions
Kidney cancer is fundamentally a metabolic disease; each of the inherited kidney cancer syndromes caused by germline mutations in the kidney cancer genes identified to date represent disorders in oxygen, iron, nutrient and/or energy sensing (Figure 1). Identification of the genes for kidney cancer has provided the foundation for development of targeted approaches to therapy for patients with these disorders. While there has been considerable progress in the development of novel therapeutic approaches for patients with kidney cancer, there remains a significant opportunity for targeting the common metabolic defects in these cancer gene pathways.
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institute of Health, National Cancer Institute, Center for Cancer Research and funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The authors acknowledge the outstanding editorial and graphics support by Georgia Shaw.
Biographies
W. Marston Linehan, M.D. is Chief of Urologic Oncology Branch at the National Cancer Institute, Bethesda, Maryland. For the last 28 years he has studied the genetic basis of cancer of the kidney. He and his colleagues have studied the VHL gene (von Hippel Lindau, clear cell renal carcinoma), the gene for Hereditary Papillary Renal Carcinoma (the MET proto-oncogene, type I papillary renal carcinoma), the FLCN gene (Birt Hogg Dubé syndrome, chromophobe renal carcinoma) and the germline fumarate hydratase mutations in the North American families with hereditary leiomyomatosis renal cell carcinoma (HLRCC). They work on the development of therapeutic strategies for the different types of kidney cancer based on understanding the molecular pathway of the specific cancer genes associated with the different types of kidney cancer.
Ramaprasad Srinivasan, M.D., Ph.D is a senior attending clinician in the Urologic Oncology Branch, National Cancer Institute. He obtained his M.B.B.S.degree in 1991 from Bangalore Medical College, India and went on to receive a Ph.D. in Biomedical Sciences from the University of Texas MD Anderson Cancer Center in 1996. He completed his residency training in Internal Medicine at the University of Texas Health Sciences Center, Houston and subsequently completed fellowship training in Medical Oncology and Hematology at the National Institutes of Health. He joined the Urologic Oncology Branch in 2003 and currently leads their targeted therapeutics program. His main research focus is the development of rational targeted therapeutic strategies in urologic malignancies.
Laura S. Schmidt, Ph.D. received her Ph.D. degree in biochemistry from Vanderbilt University and was a Medical Foundation fellow at Tufts University School of Medicine in Boston. She currently holds the position of Principal Scientist in the Urologic Oncology Branch at the National Cancer Institute, National Institutes of Health. For the past 20 years her research interests have focused on the identification of new renal cancer predisposing genes through studies of families with inherited kidney cancer. Currently, her research is centered on understanding the function of the protein encoded by the folliculin gene and how germline mutations in folliculin increase risk for renal neoplasia in Birt-Hogg-Dube' syndrome.
Footnotes
Competing interests
The authors have no competing interests
References
- 1.Linehan WM, Walther MM, Zbar B. The genetic basis of cancer of the kidney. J Urol. 2003 Dec;170(6 Pt 1):2163–72. doi: 10.1097/01.ju.0000096060.92397.ed. [DOI] [PubMed] [Google Scholar]
- 2.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004 Aug;10(8):789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 3.Thompson CB. Attacking cancer at its root. Cell. 2009 Sep 18;138(6):1051–4. doi: 10.1016/j.cell.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009 Mar 1;23(5):537–48. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009 May 22;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seizinger BR, Rouleau GA, Ozelius LJ, Lane AH, Farmer GE, Lamiell JM, et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature. 1988 Mar 17;332(6161):268–9. doi: 10.1038/332268a0. [DOI] [PubMed] [Google Scholar]
- 7.Lerman MI, Latif F, Glenn GM, Daniel LN, Brauch H, Hosoe S, et al. Isolation and regional localization of a large collection (2,000) of single copy DNA fragments on human chromosome 3 for mapping and cloning tumor suppressor genes. Hum Genet. 1991;86:567–77. doi: 10.1007/BF00201543. [DOI] [PubMed] [Google Scholar]
- 8.Glenn GM, Linehan WM, Hosoe S, Latif F, Yao M, Choyke P, et al. Screening for von hippel-lindau disease by DNA-polymorphism analysis. JAMA. 1992;267:1226–31. [PubMed] [Google Scholar]
- 9.Latif F, Modi WS, Duh FM, Schmidt LS, Li H, Geil L, et al. Molecular and genetic characterization and physical mapping of 11 new markers detecting multiallele restriction fragment length polymorphisms on the short arm of human chromosome 3. Hum Genet. 1992;90(1–2):17–22. doi: 10.1007/BF00210739. [DOI] [PubMed] [Google Scholar]
- 10.Maher ER, Bentley E, Payne SJ, Latif F, Richards FM, Chiano M, et al. Presymptomatic diagnosis of von hippel-lindau disease with flanking DNA markers. J Med Genet. 1992;29:902–5. doi: 10.1136/jmg.29.12.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crossey PA, Maher ER, Jones MH, Richards FM, Latif F, Phipps ME, et al. Genetic linkage between von Hippel-Lindau disease and three microsatellite polymorphisms refines the localisation of the VHL locus. Hum Mol Genet. 1993 Mar;2(3):279–82. doi: 10.1093/hmg/2.3.279. [DOI] [PubMed] [Google Scholar]
- 12.Latif F, Tory K, Gnarra JR, Yao M, Duh F-M, Orcutt ML, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993 May 28;260(5112):1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 13.Stolle C, Glenn GM, Zbar B, Humphrey JS, Choyke P, Walther MM, et al. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 1998;12(6):417–23. doi: 10.1002/(SICI)1098-1004(1998)12:6<417::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 14.Gnarra JR, Tory K, Weng Y, Schmidt LS, Wei MH, Li H, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nature Genetics. 1994 May;7(1):85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- 15.Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008 Aug 1;14(15):4726–34. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shuin T, Kondo K, Torigoe S, Kishida T, Kubota Y, Hosaka M, et al. Frequent somatic mutations and loss of heterozygosity of the von Hippel-lindau tumor suppressor gene in primary human renal cell carcinomas. Cancer Res. 1994;54:2852–5. [PubMed] [Google Scholar]
- 17.Duan DR, Pause A, Burgess WH, Aso T, Chen DY, Garrett KP, et al. Inhibition of transcription elongation by the VHL tumor suppressor protein. Science. 1995 Sep 8;269(5229):1402–6. doi: 10.1126/science.7660122. [DOI] [PubMed] [Google Scholar]
- 18.Pause A, Lee S, Worrell RA, Chen DY, Burgess WH, Linehan WM, et al. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci U S A. 1997 Mar 18;94(6):2156–61. doi: 10.1073/pnas.94.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kibel A, Iliopoulos O, DeCaprio JA, Kaelin WG., Jr Binding of the von Hippel-Lindau tumor suppressor protein to elongin B and C. Science. 1995 Sep 8;269(5229):1444–6. doi: 10.1126/science.7660130. [DOI] [PubMed] [Google Scholar]
- 20.Iliopoulos O, Jiang C, Levy AP, Kaelin WG, Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A. 1996;93(20):10595–9. doi: 10.1073/pnas.93.20.10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999 May 20;399(6733):271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 22.Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002 Sep;2(9):673–82. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 23.Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002 Sep;2(9):673–82. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 24.Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG., Jr Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002 Apr;1(3):237–46. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 25.Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr Inhibition of HIF2alpha Is Sufficient to Suppress pVHL-Defective Tumor Growth. PLoS Biology. 2003 Dec;1(3):E83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD. The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002 Apr;1(3):247–55. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]
- 27.Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008 Dec 9;14(6):435–46. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006 Jan;12(1):122–7. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 29.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004 Oct 1;64(19):7099–109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 30.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003 Jan;9(1):327–37. [PubMed] [Google Scholar]
- 31.Rixe O, Bukowski RM, Michaelson MD, Wilding G, Hudes GR, Bolte O, et al. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol. 2007 Oct 22;8:975–84. doi: 10.1016/S1470-2045(07)70285-1. [DOI] [PubMed] [Google Scholar]
- 32.Harris PA, Boloor A, Cheung M, Kumar R, Crosby RM, vis-Ward RG, et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-m ethyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem. 2008 Aug 14;51(15):4632–40. doi: 10.1021/jm800566m. [DOI] [PubMed] [Google Scholar]
- 33.Rapisarda A, Uranchimeg B, Scudiero DA, Selby M, Sausville EA, Shoemaker RH, et al. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002 Aug 1;62(15):4316–24. [PubMed] [Google Scholar]
- 34.Rapisarda A, Uranchimeg B, Sordet O, Pommier Y, Shoemaker RH, Melillo G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. Cancer Res. 2004 Feb 15;64(4):1475–82. doi: 10.1158/0008-5472.can-03-3139. [DOI] [PubMed] [Google Scholar]
- 35.Woldemichael GM, Vasselli JR, Gardella RS, McKee TC, Linehan WM, McMahon JB. Development of a cell-based reporter assay for screening of inhibitors of hypoxia-inducible factor 2-induced gene expression. Journal of Biomolecular Screening. 2006 Sep;11(6):678–87. doi: 10.1177/1087057106289234. [DOI] [PubMed] [Google Scholar]
- 36.Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006 Jan;12(1):122–7. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 37.Toschi A, Lee E, Gadir N, Ohh M, Foster DA. Differential dependence of HIF1alpha and HIF2alpha on mTORC1 and mTORC2. J Biol Chem. 2008 Oct 22; doi: 10.1074/jbc.C800170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010 Jan 1;70(1):288–98. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 39.Sanchez M, Galy B, Muckenthaler MU, Hentze MW. Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. Nat Struct Mol Biol. 2007 May;14(5):420–6. doi: 10.1038/nsmb1222. [DOI] [PubMed] [Google Scholar]
- 40.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006 Aug;2(8):406–14. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 41.Rouault TA, Tong WH. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008 Jul 5; doi: 10.1016/j.tig.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen G, Fillebeen C, Wang J, Pantopoulos K. Overexpression of iron regulatory protein 1 suppresses growth of tumor xenografts. Carcinogenesis. 2007 Apr;28(4):785–91. doi: 10.1093/carcin/bgl210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zimmer M, Ebert BL, Neil C, Brenner K, Papaioannou I, Melas A, et al. Small-molecule inhibitors of HIF-2a translation link its 5'UTR iron-responsive element to oxygen sensing. Mol Cell. 2008 Dec 26;32(6):838–48. doi: 10.1016/j.molcel.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell. 2008 Jul 8;14(1):90–102. doi: 10.1016/j.ccr.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peruzzi B, Bottaro DP. Targeting the c-Met signaling pathway in cancer. Clin Cancer Res. 2006 Jun 15;12(12):3657–60. doi: 10.1158/1078-0432.CCR-06-0818. [DOI] [PubMed] [Google Scholar]
- 46.Schmidt LS, Duh FM, Chen F, Kishida T, Glenn GM, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nature Genetics. 1997 May;16(1):68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt LS, Junker K, Nakaigawa N, Kinjerski T, Weirich G, Miller M, et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene. 1999 Apr 8;18(14):2343–50. doi: 10.1038/sj.onc.1202547. [DOI] [PubMed] [Google Scholar]
- 48.Lubensky IA, Schmidt LS, Zhuang Z, Weirich G, Pack S, Zambrano N, et al. Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol. 1999 Aug;155(2):517–26. doi: 10.1016/S0002-9440(10)65147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Srinivasan R, Linehan WM, Vaishampayan U, Logan T, Shankar ST, Sherman LJ, et al. A phase II study of two dosing regimens of GSK 1363089 (GSK089), a dual MET/VEGFR2 inhibitor, in patients (pts) with papillary renal carcinoma (PRC) Journal of Clinical Oncology. 2009;(Supplement 27):15s. [Google Scholar]
- 50.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006 Sep 28;355(13):1345–56. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 51.Bjornsson J, Short MP, Kwiatkowski DJ, Henske EP. Tuberous sclerosis-associated renal cell carcinoma. Clinical, pathological, and genetic features. Am J Pathol. 1996 Oct;149(4):1–8. [PMC free article] [PubMed] [Google Scholar]
- 52.Schmidt LS, Warren MB, Nickerson ML, Weirich G, Matrosova V, Toro JR, et al. Birt-Hogg-Dube syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. 2001 Oct;69(4):876–82. doi: 10.1086/323744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn GM, Turner ML, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002 Aug;2(2):157–64. doi: 10.1016/s1535-6108(02)00104-6. [DOI] [PubMed] [Google Scholar]
- 54.Birt AR, Hogg GR, Dube WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977 Dec;113(12):1674–7. [PubMed] [Google Scholar]
- 55.Pavlovich CP, Walther MM, Eyler RA, Hewitt SM, Zbar B, Linehan WM, et al. Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol. 2002 Dec;26(12):1542–52. doi: 10.1097/00000478-200212000-00002. [DOI] [PubMed] [Google Scholar]
- 56.Pavlovich CP, Grubb RL, Hurley K, Glenn GM, Toro J, Schmidt LS, et al. Evaluation and Management of Renal Tumors in the Birt-Hogg-Dube Syndrome. J Urol. 2005 May;173(5):1482–6. doi: 10.1097/01.ju.0000154629.45832.30. [DOI] [PubMed] [Google Scholar]
- 57.Toro JR, Pautler SE, Stewart L, Glenn GM, Weinreich M, Toure O, et al. Lung Cysts, Spontaneous Pneumothrorax and Genetic Associations in 89 Families with Birt-Hogg-Dubé Syndrome. Am J Respir Crit Care Med. 2007 Feb 22;175(10):1044–53. doi: 10.1164/rccm.200610-1483OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmidt LS, Nickerson ML, Warren MB, Glenn GM, Toro JR, Merino MJ, et al. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet. 2005 Jun;76(6):1023–33. doi: 10.1086/430842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Toro JR, Wei MH, Glenn GM, Weinreich M, Toure O, Vocke CD, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008 Jun;45(6):321–31. doi: 10.1136/jmg.2007.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vocke CD, Yang Y, Pavlovich CP, Schmidt LS, Nickerson ML, Torres-Cabala CA, et al. High Frequency of Somatic Frameshift BHD Gene Mutations in Birt-Hogg-Dube-Associated Renal Tumors. J Natl Cancer Inst. 2005 Jun 15;97(12):931–5. doi: 10.1093/jnci/dji154. [DOI] [PubMed] [Google Scholar]
- 61.Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, Iwamatsu A, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006 Oct 17;103(42):15552–7. doi: 10.1073/pnas.0603781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hasumi H, Baba M, Hong SB, Hasumi Y, Huang Y, Yao M, et al. Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene. 2008 May 31;415(1–2):60–7. doi: 10.1016/j.gene.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takagi Y, Kobayashi T, Shiono M, Wang L, Piao X, Sun G, et al. Interaction of folliculin (Birt-Hogg-Dube gene product) with a novel Fnip1-like (FnipL/Fnip2) protein. Oncogene. 2008 Jul 28; doi: 10.1038/onc.2008.261. [DOI] [PubMed] [Google Scholar]
- 64.Hasumi Y, Baba M, Ajima R, Hasumi H, Valera VA, Klein ME, et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci U S A. 2009 Nov 3;106(44):18722–7. doi: 10.1073/pnas.0908853106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baba M, Furihata M, Hong SB, Tessarollo L, Haines DC, Southon E, et al. Kidney-targeted Birt-Hogg-Dubé gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst. 2008 Jan 16;100(2):140–54. doi: 10.1093/jnci/djm288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen J, Futami K, Petillo D, Peng J, Wang P, Knol J, et al. Deficiency of FLCN in mouse kidney led to development of polycystic kidneys and renal neoplasia. PLoS ONE. 2008;3(10):e3581. doi: 10.1371/journal.pone.0003581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002 Apr;30(4):406–10. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 68.Merino MJ, Torres-Cabala C, Pinto PA, Linehan WM. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007;31(10):1578–85. doi: 10.1097/PAS.0b013e31804375b8. [DOI] [PubMed] [Google Scholar]
- 69.Stewart L, Glenn GM, Stratton P, Goldstein AM, Merino MJ, Tucker MA, et al. Association of germline mutations in the fumarate hydratase gene and uterine fibroids in women with hereditary leiomyomatosis and renal cell cancer. Arch Dermatol. 2008 Dec;144(12):1584–92. doi: 10.1001/archdermatol.2008.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Toro JR, Nickerson ML, Wei MH, Warren MB, Glenn GM, Turner ML, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003 Jul;73(1):95–106. doi: 10.1086/376435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wei MH, Toure O, Glenn GM, Pithukpakorn M, Neckers L, Stolle C, et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet. 2006 Jan;43(1):18–27. doi: 10.1136/jmg.2005.033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005 Aug;8(2):143–53. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 73.Yang Y, Valera VA, Padilla-Nash HM, Sourbier C, Vocke CD, Vira MA, et al. UOK 262 cell line, fumarate hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: in vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet Cytogenet. 2010 Jan 1;196(1):45–55. doi: 10.1016/j.cancergencyto.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sudarshan S, Sourbier C, Kong HS, Block K, Romero VV, Yang Y, et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and HIF-1{alpha} stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009 May 26;15:4080–90. doi: 10.1128/MCB.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM, et al. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther. 2009 Mar;8(3):626–35. doi: 10.1158/1535-7163.MCT-08-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000 Feb 4;287(5454):848–51. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 77.Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet. 2004 Jan;74(1):153–9. doi: 10.1086/381054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004 Aug 25;292(8):943–51. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- 79.Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, et al. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008 Sep 3;100(17):1260–2. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- 80.Srirangalingam U, Walker L, Khoo B, MacDonald F, Gardner D, Wilkin TJ, et al. Clinical manifestations of familial paraganglioma and phaeochromocytomas in succinate dehydrogenase B (SDH-B) gene mutation carriers. Clin Endocrinol (Oxf) 2008 Oct;69(4):587–96. doi: 10.1111/j.1365-2265.2008.03274.x. [DOI] [PubMed] [Google Scholar]
- 81.Henderson A, Douglas F, Perros P, Morgan C, Maher ER. SDHB-associated renal oncocytoma suggests a broadening of the renal phenotype in hereditary paragangliomatosis. Fam Cancer. 2009 Jan 29;8(3):257–60. doi: 10.1007/s10689-009-9234-z. [DOI] [PubMed] [Google Scholar]
- 82.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, et al. Overall Survival and Updated Results for Sunitinib Compared With Interferon Alfa in Patients With Metastatic Renal Cell Carcinoma. J Clin Oncol. 2009 Jun 1; doi: 10.1200/JCO.2008.20.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Warburg O. On the origin of cancer cells. Science. 1956 Feb 24;123(3191):309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 84.Krishnan B, Truong LD. Renal epithelial neoplasms: the diagnostic implications of electron microscopic study in 55 cases. Hum Pathol. 2002 Jan;33(1):68–79. doi: 10.1053/hupa.2002.30210. [DOI] [PubMed] [Google Scholar]
- 85.Landman GW, Kleefstra N, van Hateren KJ, Groenier KH, Gans RO, Bilo HJ. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care. 2010 Feb;33(2):322–6. doi: 10.2337/dc09-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005 Jun 4;330(7503):1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009 Oct 1;69(19):7507–11. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, et al. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008 Jun 1;412(2):211–21. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- 89.Chow WH, Gridley G, Fraumeni FJ, Jarvholm B. Obesity, Hypertension, and the Risk of Kidney Cancer in Men. N Engl J Med. 2000 Nov 2;343(18):1305–11. doi: 10.1056/NEJM200011023431804. [DOI] [PubMed] [Google Scholar]
- 90.Leitao VA, da SW, Jr, Ferreira U, Denardi F, Billis A, Rodrigues NN., Jr Renal medullary carcinoma. Case report and review of the literature. Urol Int. 2006;77(2):184–6. doi: 10.1159/000093918. [DOI] [PubMed] [Google Scholar]
- 91.Davis CJ, Jr, Mostofi FK, Sesterhenn IA. Renal medullary carcinoma. The seventh sickle cell nephropathy. Am J Surg Pathol. 1995 Jan;19(1):1–11. doi: 10.1097/00000478-199501000-00001. [DOI] [PubMed] [Google Scholar]
- 92.Linehan WM, Bratslavsky G, Pinto PA, Schmidt LA, Neckers L, Bottaro D, et al. Molecular Diagnosis and Therapy of Kidney Cancer. Annual Review of Medicine. 2010;10(61):329–43. doi: 10.1146/annurev.med.042808.171650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nature Reviews Cancer. 2002 Sep;2(9):673–82. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 94.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001 Apr;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]