

Abstract
Five Anopheles marajoara Galvão and Damasceno populations, representing diverse ecological conditions, were sampled throughout Colombia and analyzed using nine hypervariable DNA microsatellite loci. The overall genetic diversity (H = 0.58) was lower than that determined for some Brazilian populations using the same markers. The Caquetá population (Colombia) had the lowest gene diversity (H = 0.48), and it was the only population at Hardy–Weinberg equilibrium. Hardy–Weinberg disequilibrium in the remaining four populations was probably caused by the Wahlund effect. The assignment analyses showed two incompletely isolated gene pools separated by the Eastern Andean cordillera. However, other possible geographical barriers (rivers and other mountains) did not play any role in the moderate genetic heterogeneity found among these populations (FST = 0.069). These results are noteworthy, because this species is a putative malaria vector in Colombia.
Introduction
Colombia has ecological conditions over 85% of its land area suitable for permanent malaria transmission.1,2 Anopheles darlingi is the primary malaria vector in Colombia, but the occurrence of a sympatric cryptic species such as An. marajoara, Galvão and Damasceno, adds additional complexity to the study of the dynamics of malaria transmission in this country. An. marajoara plays a local role in malaria transmission and can be defined as a secondary vector with a local importance in Colombia, although it is certainly not a primary vector like An. darlingi.
The geographic distribution of An. marajoara ranges from Costa Rica (Central America) to Bolivia (South America), including Trinidad (West Indies) and the major part of the Brazilian Amazon basin (Figure 1). An. marajoara has been found infected by Plasmodium falciparum,3 was experimentally infected with P. vivax,4 and showed physiological resistance to organochlorine dichlorodiphenyltrichloroethane (DDT) insecticide.5 Additionally, An. marajoara was shown to be the most abundant and widely distributed anopheline in a lot of urban areas in Colombia, with an apparently high adaptation to human environments.6 As such, it could be playing an important role as secondary malaria vector in some areas in Colombia and Brazil.3,6–9 In fact, in the Amapá state in northeastern Brazil, this was the primary malaria vector.9 Additionally, An. mararajoara is also a possible vector of Wuchereria bancrofti and Mansonella ozzardi.10 There is perhaps a risk of transmitting W. bancrofti but in Guyana only, because the extent of this parasite has been greatly reduced in the neotropical region.
Figure 1.
Map with the overall distribution of An. marajoara in Latin America.
An. marajoara belongs to the An. albitarsis complex, which also includes An. albitarsis, An. deaneorum Rosa-Freitas, An. oryzalimnetes Wilkerson and Motoki, An. janconnae Wilkerson and Sallum, and An. albitarsis F.10 An. marajoara and An. albitarsis F are the only ones recorded for this complex in Colombia.11
Previously, three genetic studies have been carried out with An. marajoara in Brazil. The first used allozymes and restriction fragment-length polymorphism (RFLP) of mtDNA. Three allopatric populations were detected, with a noteworthy heterozygote deficit and unique haplotypes in each of the populations studied.12 The second study, using the whole mtCOI gene,13 found multiple signatures of a demographic expansion event in the eastern Brazilian Amazon. More recently, a third study was done. The population genetic structure of An. marajoara in 15 populations in Brazil and 1 population in Trinidad was described using homologous microsatellite markers (J. Conn and others, unpublished data). A high degree of genetic heterogeneity was found among the populations studied (FST = 0.198–0.401), which suggested some incipient allopatric speciation with partial barriers to gene flow. The observed heterozygosity (H = 0.604) was lower than the expected heterozygosity (H = 0.772), which could be explained by the Wahlund (= subdivision) effect.
In Colombia, only one previous genetic study has been conducted with this species. Analysis of 42 polymorphic random amplified polymorphism DNA (RAPD) markers showed an overall expected heterozygosity of 0.294, moderate genetic heterogeneity among populations (φ = 0.083), and gene-flow estimates ranging from 1.5 to 7.5 individuals per generation.14
The goals of the present study were: (1) to determine the gene diversity levels and the Hardy–Weinberg equilibrium status by means of nine homologous DNA microsatellite markers short tandem repeat of polymorphism (STRP) of five Colombian An. marajoara populations distributed in different geographic and ecological areas of this country, (2) to determine the degree of genetic heterogeneity among these populations and correlate this heterogeneity with geographic barriers, such as the Andean mountains, large aquatic environments, like the Magdalena and the Cauca rivers, the Macarena Mountains, and different ecological habitats, and (3) to determine with different assignment population analyses if the multi-locus genotypic profiles of each one of the mosquitoes analyzed were correctly assigned to its own population. This latter aim is especially important to assess potential interchange of genes of public-health importance, such as those that confer insecticide resistance or refractoriness to infection by human Plasmodia.
Materials and Methods
Samples and molecular analyses.
Five populations distributed across the geographic range of An. marajoara in Colombia were sampled (Figure 2). These populations were from Cáceres, Antioquia Department, 07°34′N, 75°21′W, 100 m above sea level (N = 38 individuals analyzed); El Banco, Magdalena Department, 09°00′N, 73°58′W, 35 m above sea level (N = 36); El Zulia, Norte de Santander Department, 07°56′N, 72°36′W, 200 m above sea level (N = 40); Villavicencio, Meta Department, 04°09′W, 73°38′W, 467 m above sea level (N = 39); and Cartagena del Chairá, Caquetá Department, 01°21′N, 74°50′W, 350 m above sea level (N = 40). All localities are situated on the banks of large rivers. Cáceres is placed at the lower Cauca near to the western area of the San Lucas Mountain and between the Nechí and Cauca rivers with a mean temperature of 28°C. The main human economic activities are the gold extraction and livestock production. El Banco is placed at the confluence of the Magdalena and Cesar rivers. The mean temperature is around 29°C, but in the hottest period, the temperature could be near to 40°C. The clime is tropical with wet and dry periods. The main economic activities are based on agriculture and fishing. El Zulia is at 14 km from the city of Cúcuta. This place has an average annual temperature of 28°C, and its main economic activities are those related to rice and sugar as well as mineria (calizas, carbon, y arena). Villavicencio is the capital of the Meta Department and is near to the Guatiquia River. The mean temperature is 27°C, and the main activities are livestock (one of the most important areas for this activity in Colombia) and petroleum extraction. Cartagena del Chairá is placed at the Caguán River in the Colombian Amazon (neotropical rain forest) with an average annual temperature of 30°C. The main human activity is the agriculture (bananas, maize, and yucca) and fishing. These sites have relative humidity > 80%. The collections were based on accessibility to the localities and confirmation of local security because of the question of the guerrillas in Colombia.
Figure 2.
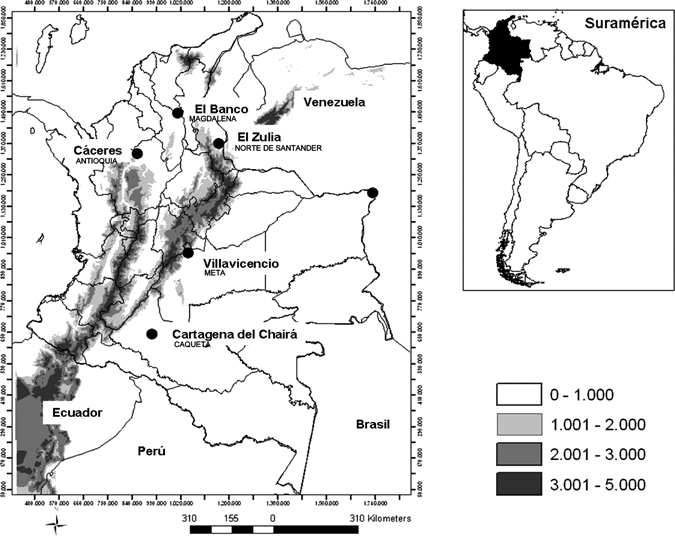
Map with the An. marajoara populations sampled in Colombia. Altitude is in meters.
All specimens (N = 193) were collected between July and September 2004. These were identified as An. marajoara using morphological taxonomic keys15 and were then maintained in absolute ethanol at 4°C. All the specimens analyzed (except three individuals sampled in the Magdalena population that were identified as An. triannulatus) were confirmed as An. marajoara using inter space 2 ribosomal DNA (ITS2) (rDNA)16 and the white gene sequences.17 DNA was extracted by a phenol-chloroform method18 that yielded approximately 2 μg of DNA per specimen. The STRP markers used were those designed by Li and others19 specifically for the species studied here, and they worked perfectly: M9-2, M1-31, M8-2-2, M2, M7-1, M6-8, M5-71, M10-1, and M11-4. One of the primers from each pair was labeled with fluorescein amidite (FAM), 5′hexachloro-flurescein-CE phosphoramidite (HEX), or fluorescein benzoxanthene (NED) (AB Prism, Washington, DC). Touch-down polymerase chain reaction (PCR) was conducted in a 20-μL reaction containing 1 × buffer (10 mM Tris-HCl, pH 8.3, 50 mM KCl), 2.5 mM MgCl2, 200 μL dideoxy nucleoside triphosphates (dNTP), 5 pmol each primer, 0.5 U Taq polymerase (AB), and 5 ng DNA template. PCRs were performed on a GeneAmp PCR System 9700 (AB). With the exception of M5-71, the reaction conditions were 3 min at 95°C followed by two cycles of 94°C for 30 s, 60.7°C for 30 s to annealing temperature, and 72°C for 30 s; 14 cycles using a touchdown protocol (94°C for 15 s, 15 s at 58.6°C, temperature decreased by 1.7°C every other cycle to a touchdown annealing temperature, 72°C for 3 s) were followed by 24 cycles with a standard protocol (94°C for 15 s, 50°C as annealing temperature for 15 s, and 72°C for 30 s) and were closed with 10 min at 72°C to reduce stutter bands. The annealing temperature of M5-71 marker was 66.7°C for the first cycle, 66°C for the second cycle, and 55°C for the third cycle. Products were mixed (Mix 1: M9-2, M1-31, M8-2-2, M2, and M7-1; Mix 2: M6-8, M5-71, M10-1, and M11-4) and run in an ABI3100 (AB). Data were automatically collected and analyzed by Genotyper software 3.7v (AB). The samples that showed ghost bands or rare alleles (alleles that were only found in one of the populations studied) were run a second time to confirm the results obtained.
Population genetics analyses.
The mean number of alleles per locus and the expected heterozygosity (H) 20 were calculated for each of the populations, and differences among these estimates were statistically analyzed with a Student's t test. The heterozygosity data were arcsine transformed before analysis, as proposed by Archie.21 This analysis will permit us to determine if some of the populations analyzed have lost gene diversity by gene drift or founder effect.
The Hardy–Weinberg equilibrium (HWE) was estimated using the Weir and Cokerham22 1984 F (W–C F) statistic. This procedure was used to calculate the excess of homo- and heterozygous genotypes within each population, and the Markov chain method with 1,000 dememorization, 50 batches at 1,000 iterations per batch,23 was used to measure the probabilities. Fisher's method was used to analyze the global HWE estimates (by loci and populations) with GenePop 3.4 version.24 Genotypic linkage disequilibrium was analyzed by marker pairs in each population, and for each marker pair, all of the populations were taken together. Markov Monte Carlo chains with 1,000 dememorization, 50 batches, and 1,000 iterations per batch were applied to determine the significance of the probabilities.
Several strategies were used to calculate genetic heterogeneity globally and among population pairs. First, the gene frequencies of the nine microsatellites were tested with Markov chains with 1,000 dememorizations, 20 batches, and 1,000 iterations per batch. Second, Wright's25 1951 F statistics, with Weir and Cokerham's22 1983 procedure, were applied. The standard deviations of the F statistics and the confidence intervals (95% and 99%) were calculated with jackknifing and bootstrapping (10,000) over loci, respectively. The significance of FST (genetic heterogeneity) was calculated with the G test (10,000 allele randomizations, random mating assumed) and the log-likelihood G test (10,000 genotype randomizations, random mating not assumed) for among populations.26 The significance of FIS and FIT were similarly calculated (10,000 randomizations of alleles within and overall for the populations analyzed). For this second task, the FSTAT 2.9.3.2 version software was used.27
In a complementary way, Nei's gene diversity analysis20 was used to measure the gene heterogeneity between populations as well as the RST statistic.28,29 Rho, as an unbiased estimator of RST, was calculated for individual loci and for all the loci taken simultaneously for all the populations studied and population pairs using RST software.30 From these heterogeneity statistics, theoretical gene-flow estimates were obtained by means of an infinite island model31–33 and an n-dimensional island model.34,35 Another gene-flow estimate was obtained with the private allele method of Slatkin36 and Barton and Slatkin.37
Finally, an assignment analysis was applied to the 193 multi-locus genotype profiles obtained. This assignment analysis was applied by means of the Structure 2.1 software.38 This method, which employs Markov Chain Monte Carlo (MCMC) procedures and the Gibbs sampler, uses multilocus genotypes to infer population structure, and individuals are simultaneously assigned to specific populations. The model considers K populations, where K may be unknown, and the individuals are assigned tentatively to one population or jointly to two or more populations if their genotypes are considered to be admixed. The posterior K probabilities are calculated assuming uniform prior values of K (USEPOPINF = 0). The presence of the most probable number of gene pools within data considered is revealed by the increasing likelihood. After the most likely number of populations is found, the analysis is repeated but with introducing the model with prior geographic population information (USEPOPINF = 1). Different parameter conditions were considered. No admixture model and admixture model were analyzed. In the first case, the individuals exclusively belong to one population, whereas, in the second model, one individual could simultaneously belong to different populations. In each case, another parameter was analyzed: gene frequencies correlated among the populations or independent gene frequencies correlated among the populations. With correlated gene frequencies, two FST values were considered (0.001, the default value of the program, and 0.069, the value empirically obtained for the present study). In the case of correlated gene frequencies, two migration rates (0.01 and 0.05) were considered as well. With each condition, the program was run with 1 million iterations after a burn period of 100,000 iterations. Each analysis was performed two times with convergent results.
Results
All mosquitoes analyzed belonged to An. marajoara based on morphological characters and ITS2 (rDNA) and white gene sequences, with the exception of three individuals sampled in El Banco, Magdalena; these were deleted from the population genetics analyses. Therefore, the homozygote excess was not caused by the mix of different albitarsis complex species in our sample.
The nine STRP markers studied were polymorphic globally and for each of the locations analyzed. Globally, M9-2 and M5-71 yielded the highest number of alleles (10), whereas M8-2-2 had the lowest (6) (Table 1). For populations, the markers with the highest number of alleles were M5-71 (9; Caquetá), M5-71 (9; Meta), M5-71 (7; Norte de Santander), M1-31 and M10-1 (7 each; Magdalena), and M5-71, M6-8, and M7-1 (6 each; Antioquia). In Table 1, the 15 rare alleles are shown (alleles that were only found in one of the populations studied) that were found in this study.
Table 1.
Alleles found in the total sample and for each one of the An. marajoara samples analyzed for the nine STRP used
| Locus | M9-2 | M-2 | M1-31 | M8-2-2 | M10-1 | M11-4 | M5-71 | M6-8 | M7-1 |
|---|---|---|---|---|---|---|---|---|---|
| A | 10 | 8 | 8 | 6 | 8 | 7 | 10 | 7 | 8 |
| B | 93–117 | 150–171 | 198–219 | 115–130 | 142–158 | 192–213 | 172–202 | 107–128 | 142–156 |
| Caquetá (N = 40) | 102, 108, 111 | 153, 162, 165, 168, 171 | 204, 210, 213, 216 | 118, 124, 127, 130 | 144, 148, 150, 156, 158* | 207, 210 | 178, 181, 184, 187, 190, 193, 196, 199, 202 | 113, 122, 125 | 142, 144, 146, 148, 150, 152 |
| Antioquia (N = 38) | 99, 105, 108, 111, 114 | 156, 159, 162, 165, 168 | 204, 207, 210, 213, 216 | 118, 124, 127, 130 | 144, 146, 148, 150, 152 | 207, 210 | 187, 190, 193, 196, 199, 202 | 107, 113, 116, 119, 122, 125 | 144, 148, 150, 152, 154, 156* |
| Magdalena (N = 36) | 99, 105, 108, 111, 114, 117 | 150,* 156, 162, 165, 168 | 198, 204, 207, 210, 213, 216, 219* | 115,* 118, 124, 127, 130 | 142, 144, 146, 148, 150, 152, 156 | 192,* 195,* 204,* 207, 210 | 187, 190, 193, 196, 199, 202 | 113, 116, 122, 125 | 144, 148, 150, 152, 154 |
| Norte de Santander (N = 40) | 102, 105, 108, 111, 114, 117 | 153, 156, 162, 165, 168, 171 | 198, 204, 207, 210, 213, 216 | 118, 124, 127, 130 | 144, 148, 150, 156 | 198, 207, 210 | 184, 187, 190, 193, 196, 199, 202 | 113, 116, 119, 122, 125, 128* | 142, 144, 146, 148, 150, 152 |
| Meta (N = 39) | 93,* 96,* 102, 105, 108, 111 | 153, 156, 159, 162, 165, 168, 171 | 198, 201,* 204, 207, 210, 213, 216 | 118, 121,* 124, 127, 130 | 142, 144, 148, 150, 156 | 198, 207, 210, 213* | 172,* 178, 181, 187, 190, 193, 196, 199, 202 | 107, 113, 116, 119, 122, 125 | 144, 146, 148, 150, 152 |
N = sample size; A = number of alleles for the global sample; B = range of alleles by size (base pairs) in the global sample.
Rare alleles.
The average number of alleles per population was relatively uniform in all populations. The two populations with values slightly lower were Caquetá (nA = 4.56 ± 2.07) and Antioquia (nA = 4.89 ± 1.27); Norte de Santander (nA = 5.33 ± 1.32) and Magdalena (nA = 5.53 ± 1.07) yielded intermediate values, and Meta (nA = 6.11 ± 1.54) had the highest value. Nevertheless, no statistically significant differences were detected between any pair of populations.
Globally, the expected mean heterozygosity (gene diversity) was H = 0.578, which is an intermediate value. Per location, all the populations showed very similar values (Magdalena, H = 0.585; Antioquia, H = 0.557; Norte de Santander, H = 0.555; Meta, H = 0.547) with Caquetá presenting a slightly lower value (H = 0.481), which was significantly lower than that for Magdalena. Therefore, for both statistics (mean allele number and expected heterozygosity), the Caquetá population had the lowest genetic diversity. By marker, M5-71 (H = 0.729), M8-2-2 (H = 0.649), and M10-1 (H = 0.622) yielded the highest levels of gene diversity, whereas M11-4 (H = 0.371) clearly showed the lowest diversity.
HWE.
In Table 2, the HWE results are shown. Globally, only one STRP marker was in HWE (M8-2-2; P = 0.77). The remaining eight markers were clearly in HW disequilibrium by a significant excess of homozygotes (Weir and Cokerham22 F statistic of 0.104–0.320; P = 0.0146–0.0002). Therefore, the total Fisher test done simultaneously for the nine STRP markers and the global sample detected a very high homozygote excess with regard to the HWE (χ2 = infinite, 8 df, P < 0.00000). By population, only Caquetá was in overall HWE (F = 0.006; P = 0.219). In this population, only one STRP was not in HWE but by heterozygote excess (M8-2-2), although when the Bonferroni correction was applied, this significance disappeared. The other four populations were globally in HW disequilibrium. Antioquia presented a homozygote excess (F = 0.065; P = 0.0007) mainly from M9-2, M1-31, and M7-1. With the Bonferroni correction, only the last two markers presented significant homozygote excess. Magdalena showed a homozygote excess (F = 0.131; P = 0.0004) mainly from M2, M1-31, M8-2-2, and M7-1, although only M1-31 was significant after the Bonferroni correction. Norte de Santander yielded a significant homozygote excess (F = 0.093; P = 0.0000) mainly from the contribution of M1-31 and M6-8. Finally, Meta showed a noteworthy homozygote excess (F = 0.096; P = 0.0000) from the considerable contributions of M9-2 and M2. Such as it was shown, each population was in HW disequilibrium by the contribution of different STRP markers.
Table 2.
FIS values for each locus and each Colombian An. mararajoara population according to Weir and Cokerham22
| Locus | Caquetá | Antioquia | Magdalena | Norte de Santander | Meta | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FIS | SD | P | FIS | SD | P | FIS | SD | P | FIS | SD | P | FIS | SD | P | |
| M9-2 | 0.183 | 0.000 | 0.3442 | 0.152 | 0.002 | 0.0146* | 0.174 | 0.010 | 0.1326 | 0.060 | 0.013 | 0.1321 | 0.507 | 0.000 | 0.0000* |
| M2 | 0.059 | 0.013 | 0.1196 | −0.182 | 0.008 | 0.1191 | 0.132 | 0.004 | 0.0400* | 0.080 | 0.013 | 0.2137 | 0.383 | 0.000 | 0.0000* |
| M1-31 | 0.082 | 0.000 | 0.3944 | 0.355 | 0.002 | 0.0077* | 0.071 | 0.002 | 0.0034* | 0.369 | 0.000 | 0.0000* | −0.005 | 0.008 | 0.0517 |
| M8-2-2 | −0.318 | 0.000 | 0.0207* | −0.069 | 0.000 | 1.0000 | 0.250 | 0.004 | 0.0287* | −0.082 | 0.000 | 0.9960 | −0.050 | 0.008 | 0.7776 |
| M10-1 | 0.100 | 0.015 | 0.6317 | 0.137 | 0.0146 | 0.3985 | 0.111 | 0.015 | 0.2093 | −0.037 | 0.000 | 0.9604 | 0.063 | 0.017 | 0.6248 |
| M11-4 | 0.000 | 0.000 | 0.0000* | −0.100 | 0.000 | 0.8240 | 0.093 | 0.011 | 0.2380 | −0.039 | 0.000 | 1.0000 | −0.053 | 0.000 | 1.0000 |
| M5-71 | 0.011 | 0.049 | 0.5652 | 0.071 | 0.019 | 0.3526 | −0.132 | 0.000 | 1.0000 | 0.054 | 0.014 | 0.4971 | 0.007 | 0.012 | 0.8547 |
| M6-8 | 0.010 | 0.000 | 0.7587 | 0.128 | 0.021 | 0.2711 | 0.159 | 0.000 | 0.5609 | 0.369 | 0.003 | 0.0053* | 0.108 | 0.012 | 0.2135 |
| M7-1 | 0.115 | 0.019 | 0.4971 | 0.080 | 0.001 | 0.0009* | 0.234 | 0.002 | 0.0087* | 0.042 | 0.014 | 0.3110 | −0.108 | 0.000 | 1.0000 |
| Total | 0.006 | 0.065 | 0.131 | 0.093 | 0.096 | ||||||||||
| P = 0.2199 | P = 0.0007* | P = 0.0004* | P = 0.00000* | P = 0.00000* | |||||||||||
P = probability; SD = standard deviation.
Significant value.
Linkage disequilibrium.
Linkage-disequilibrium analysis applied to all the populations taken together, and it showed one case of significant disequilibrium of the M9-2 and M5-7 pair (χ2 = infinite, P = 0.0000) after the Bonferroni correction. The same analysis by population showed that only one population (Meta) had two significant linkage disequilibrium at two loci: at M9-2 and M5-7 (P = 0.0000) and M9-2 and M1-31 (P = 0.00212). With the Bonferroni correction, the latter disappeared. With this one exception, we conclude that the population dynamics of all STRP markers are independent.
Genetic heterogeneity.
The exact test with Markov chains for genetic heterogeneity using gene frequencies showed that eight of nine STRP markers used presented significant differences across the populations analyzed. Only M1-31 did not yield significant heterogeneity (P = 0.899); meanwhile, the global set of nine markers showed a striking statistically significant difference with the Fisher's test (χ2 = infinite, 18 df, P = 0.0000). The markers that showed most significant differences between population pairs by means of exact tests with Markov chains were M9-2 (8 cases), M11-4 (7 cases), M5-7 (7 cases), and M7-1 (7 cases), whereas M1-31 (1 case) and M8-2-2 (2 cases) presented least significant discriminatory capacity among the populations studied. The highest number of significant differences between population pairs were Caquetá-Magdalena (8 of 9 STRP markers) and Caquetá-Antioquia (7 of 9 STRP). Therefore, the more geographically distant populations were those with the highest number of significant comparison pairs. Antioquia-Magdalena (1 of 9 STRP), Caquetá-Norte de Santander (1 of 9 STRP), and Meta-Norte Santander (3 of 9 STRP) were the populations that presented the least number of significant comparison pairs. Antioquia and Magdalena are situated in the western area of the eastern Andean Mountains; meanwhile, Caquetá, Meta, and Norte de Santander are in the eastern area of this cordillera. This is the first evidence that the eastern Andean Cordillera may play an important role in genetically separating, in a moderate way, the populations of An. marajora in Colombia.
The application of the Wright's F statistics with the Weir and Cokerham22 procedure yielded the following results (Table 3). The overall value of FIT, 0.145 [confidence interval (CI) with 10,000 bootstrapping at 99% = 0.064–0.221), showed a significant homozygote excess at the total population level. Individually, all the loci studied, except M8-2-2 (P = 0.77), presented significant homozygote excess at this population level. The average value of FIS was slightly lower than the previous case, although it also showed homozygote excess at the subpopulation level (0.082 with a 99% CI of 0.007–0.155). M9-2, M1-31, M6-8, and M7-1 showed significant homozygote excess. These results corroborated the homozygote excess detected with the individual HWE tests. The genetic heterogeneity measured by FST was moderately significant (0.069; CI = 0.025–0.148). This means that each population had, on average, 93.1% of the genetic diversity found in the total Colombian An. marajora population. All the loci, except M1-31, showed significant heterogeneity independently to consider, or not, random breeding (G tests and log-likelihood G test). The three markers with the highest levels of genetic heterogeneity were M11-4, M5-7, and M10-1. Nei's20 genetic diversity analysis confirmed similar genetic heterogeneity values as the GST and GST ¢ statistics (0.057–0.07). When the RST statistic was used, the average values ranged from 0.042 to 0.047, depending on the procedures used. This statistic again showed a moderate divergence among the populations, with the highest genetic heterogeneity levels for M11-4 (RST = 0.152), M7-1 (RST = 0.117), and M5-7 (RST = 0.061). As in other analyses, the most similar pairs were Antioquia-Magdalena, Meta-Norte de Santander, and Caquetá-Meta. That is, the most similar populations were those at the western area from the eastern Andean Cordillera and two pairs of populations east of the mountains. The overall gene-flow estimates from an infinite island model were 3.37 (FST) and 5.07–5.70 (RST); meanwhile, for an n-dimensional island model, these values were 2.16 and 3.24–3.65, respectively. The gene-flow estimate from the private allele model of Slatkin36 was 3.6, very similar to estimates obtained with other methods. These gene-flow values could be considered moderate, although the populations are clearly related by gene flow. If we observe the gene-flow estimates by population pair with the unbiased estimator Rho, they agree quite well with previous analyses. Antioquia and Magdalena are really one unique population (Nm = infinite); meanwhile, Meta and Norte de Santander were highly homogenized by gene flow (Nm = 22.86–28.79) as well as Caquetá and Meta (Nm = 13.31–25.66) and Caquetá-Norte de Santander (Nm = 10.15–10.44). The population pairs most isolated by lower gene flow were Caquetá-Antioquia (Nm = 2.36–2.80) and Caquetá-Magdalena (Nm = 2.68–3.51).
Table 3.
Wright's F statistics values per locus according to Weir and Cokerham22
| Locus | FIT | FST | FIS | ||
|---|---|---|---|---|---|
| P1 | P2 | ||||
| M9-2 | 0.258; P = 0.0002* | 0.047 | 0.0002* | 0.0002* | 0.221; P = 0.0002* |
| M2 | 0.104; P = 0.0148* | 0.022 | 0.0002* | 0.0002* | 0.084; P = 0.0550 |
| M1-31 | 0.185; P = 0.0006* | 0.021 | 0.5214 | 0.2198 | 0.167; P = 0.0016* |
| M8-2-2 | −0.035; P = 0.7704 | 0.023 | 0.0002* | 0.0004* | −0.059; P = 0.8914 |
| M10-1 | 0.122; P = 0.0080* | 0.057 | 0.0002* | 0.0002* | 0.069; P = 0.0962 |
| M11-4 | 0.320; P = 0.0002* | 0.330 | 0.0002* | 0.0002* | −0.015; P = 0.6226 |
| M5-71 | 0.146; P = 0.0002* | 0.135 | 0.0002* | 0.0002* | 0.014; P = 0.3976 |
| M6-8 | 0.176; P = 0.0006* | 0.019 | 0.0002* | 0.0002* | 0.160; P = 0.0012* |
| M7-1 | 0.127; P = 0.0044* | 0.043 | 0.0002* | 0.0002* | 0.088; P = 0.0404* |
| Total | 0.145 | 0.069 | 0.069 | 0.069 | 0.082 |
| CI | 0.064–0.221 | 0.025–0.148 | 0.025–0.148 | 0.025–0.148 | 0.007–0.155 |
CI = 99% confidence interval.
Significant probabilities (P). P1 = assuming non-random breeding; P2 = assuming random breeding.
Assignment analysis.
The assignment analysis with the Structure 2.1 software showed the same results assuming different hypotheses. The least negative likelihood value was with K = 2 in all the cases. Therefore, independent of the conditions chosen, two gene pools seem to be present in the Colombian area sampled. One gene pool was composed of individuals from Antioquia and Magdalena (this gene pool was named Western pool); the other was composed of individuals from Norte de Santander, Meta, and Caquetá (this gene pool was named Eastern pool). These gene pools are separated by the eastern Andean Cordillera. With no geographic information, admixture model, correlated frequencies among populations, FST = 0.069, and migration rate of 0.05 in the Western pool, all the Antioquia and Magdalena individuals appeared, with the exception of three individuals of Antioquia and five individuals of Magdalena that were assigned with higher probability to the Eastern gene pool, plus one individual from Caquetá, four individuals from Meta, and eight individuals from Norte de Santander (these last 13 specimens sampled in the Eastern gene pool but showed more genetic resemblance with the Western gene pool). In the Eastern gene pool, the vast majority of the individuals of Caquetá, Meta, and Norte de Santander were assigned, with the aforementioned exceptions and the three and five individuals from Antioquia and Magdalena previously mentioned. One hybrid individual between the Eastern and Western gene pool was detected in Caquetá, and two hybrid individuals between the Eastern and Western gene pool were found in Meta and Norte de Santander (Figure 3A). With other conditions, such as a priori geographic information, admixture model, and independent gene frequencies among the populations, the results were similar. In the Eastern gene pool, all the individuals from Caquetá and the major part of the individuals from Meta and Norte de Santander were assigned, plus five individuals from Magdalena. In the Western gene pool, all the Antioquia individuals and the major fraction of the Magdalena individuals, together with four individuals from Meta and eight individuals from Norte de Santander, were included. Only one hybrid individual between the Eastern and Western gene pool in Meta and four hybrid individuals between these two gene pools in Norte de Santander were detected (Figure 3B). Therefore, the final picture was as follows: two gene pools (Eastern and Western ones), separated by the Eastern Andean Cordillera, were detected but with incomplete isolation, because mosquitoes from one gene pool were found in the other gene pool and vice versa. The two populations that showed the highest number of hybrids between both Eastern and Western gene pools were Norte de Santander and Meta. Therefore, the two most differentiated populations were those occupying the most extreme sampling geographic positions: Caquetá representing the Eastern gene pool and Antioquia representing the Western gene pool.
Figure 3.
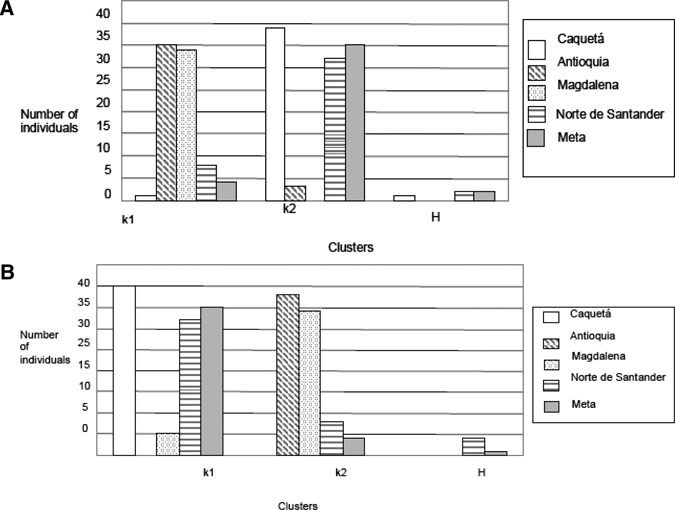
Assignment analysis of the An. marajoara individuals studied by means of two Structure 2.1 v software procedures. (A) Assignment analysis with no geographic information, admixture model, correlated frequencies among populations, FST = 0.069, and migration rate of 0.05. (B) a priori geographic information, admixture model, and independent gene frequencies among the population. K1 = western Colombian gene pool; K2 = eastern Colombian gene pool; H = hybrid individuals found.
Discussion
The overall genetic diversity found in Colombian An. marajoara populations (H = 0.578) was relatively modest with regard to that determined for Brazilian populations of An. marajoara, H = 0.715 (J. Conn and others, unpublished data). However, the populations from Trinidad (H = 0.538) and Boa Vista (H = 0.486), also studied by the same authors, presented values very similar to those reported here. These values suggest that central Brazilian and eastern Amazon populations may represent the central range of the species, whereas the Colombian populations were probably founded in a subsequent expansion with a certain loss of gene diversity because of founder effect. In fact, Lehr and others39 determined that the An. marajoara populations underwent a significant fragmentation event during the Pleistocene. All the Colombian populations analyzed showed very similar levels of expected heterozygosity (H = 0.547–0.587), with only Caquetá slightly lower (H = 0.481), as we quoted in Results, which could mean that this population has undergone more genetic drift than the other Colombian populations or suffered a slightly greater founder effect than the remainder populations analyzed. Nevertheless, some bottleneck analyses (the theory generated by Cornuet and others40 and Luikart and others41; the M ratio of Garza and Williamson42; the within locus kurtosis (k) test and the interlocus (g) test proposed by Reich and Goldstein43 and Reich and others44; Kimmel and others'45 test; Zhivotovsky and others'46 test; and Beaumont's theory with the MSVAR software47) were performed (shown elsewhere), and no evidence of this phenomenon was observed. Therefore, this loss of gene diversity was more likely because of the founder effect in the origin of the Colombian populations than by posterior, or recent, bottlenecks.
A comparison of gene diversity with other species of mosquitoes reveals the moderate gene diversity found in Colombian An. marajoara. For example, Conn and others48 estimated a value of H = 0.834 for An. darlingi, the main malaria vector in the Americas. Molina-Cruz and others49 determined an average H = 0.78 for An. albimanus in Central America and northern South America. In Africa, An. gambiae also yielded higher gene diversity than the Colombian populations of An. marajoara. Lehmann and others50,51 estimated values for different groups of populations for African malaria vectors, and the average values were, respectively, 0.62 and 0.66. In an identical sense, other studies with An. gambiae using microsatellites showed values ranging from 0.60 to 0.85.52,53 The same is apparent with An. maculatus in Thailand (H = 0.83–0.92).54
The mosquitoes sampled in the current study were obtained in urban and rural areas, representing diverse ecological conditions. For instance, the Norte de Santander sample was collected in rice fields, whereas the Magdalena, Antioquia, and Meta samples came from more urban environments and the Caquetá sample was obtained in an Amazonian habitat. In other studies, populations sampled in diverse ecological situations showed significantly different gene diversity levels. Onyabe and Conn55 found significant heterozygosity differences for populations of An. arabiensis among a savannah ecosystem (H = 0.606–0.743), a transition area of savannah–forest (H = 0.642), and a rain forest (H = 0.506). However, in the current study, no heterozygosity differences were found among the Colombian populations analyzed. Thus, natural history and demographic events, more than ecological traits, could be the main facts determining the gene diversity levels found.
With a few exceptions,56 HWE disequilibrium by homozygote excess is typically found in studies of malaria vector mosquitoes independently of the molecular markers used.51,55–58 In the present study, some homozygote excess causes could be discarded. (1) Null alleles. Although it is never totally impossible to discard the presence of null alleles using PCR procedures, the deviations from HWE for each one of the markers studied in each population was very different. If null alleles were frequent, we could expect similar homozygote excess trends in those loci affected by this phenomenon in different populations if the genetic heterogeneity among these populations is not very high (as it is in the current case). However, with the exceptions of M9-2 and M6-8, which presented homozygote excess in all the populations analyzed, the markers studied showed a very different dynamic. Therefore, we assumed a weak influence of null alleles on the homozygote excess found. (2) Hitchhiking and synteny. The linkage equilibrium analysis only revealed one case of significant disequilibrium between M9-2 and M5-7 in Meta and the total population. However, none of other loci pairs showed any evidence of significant disequilibrium. Possibly, this STRP pair could be placed within, or near, a chromosome rearrangement and therefore, is subjected to adaptative selection.59,60 Nevertheless, with the exception of the aforementioned case, the behavior of each marker used was independent from the other loci. This absence of significant gametic disequilibrium also indicated a small influence of genetic drift in the populations studied. (3) Endogamy. This affects the whole genome identically. Thus, if endogamy was important in our case, we would expect a similar homozygote excess for all the markers within each population studied, which we did not detect. Furthermore, the reproductive biology of the species does not favor endogamy. The ethological features, such as mate recognition, multiple oviposition sites, blood-meal location, and non-uniform distribution of breeding places, make the mating of mosquitoes of the same breed unlikely. Therefore, endogamy can effectively be discarded as a possible explanation. (4) Selection in favor of homozygotes. Selection could occur in one locus but not systematically in the majority of loci studied or in all the populations. Thus, the overall homozygote excess detected cannot be attributed to selection. In summary, microgeographic Wahlund effect (subdivision process within each one of the populations studied, with the exception of Caquetá) seems to be the most probable cause for this phenomenon. The existence of other Anopheles taxa in our sample, which could produce positive homozygote excess, could be discarded because, as we previously commented, all the exemplars were sequenced for ITS2 and white genes and were classified such as An. marajoara without any doubt (the three specimens from Magdalena that did not belong to this taxa were eliminated from the analyses). Thus, the picture could be as follows. We have observed that two gene pools (Eastern and Western) were detected in Colombia partially because of the eastern Andean cordillera. Additionally, within each population, we found a fraction of individuals with typical multi-locus genotypes of other populations. Only a small fraction of these migrants were hybrids between these Eastern and Western gene pools, which means that the majority of foreign mosquitoes in a given population consisted of migrants with no genetic interchange with the original mosquitoes. This produces homozygote excess by the Wahlund effect, even in very restricted local areas. This has also been detected in other malaria vector mosquitoes.51,61–63 Significant genetic heterogeneity (FST = 0.106) between two districts of Hermosillo (México) was discovered for Aedes aegypti.64 Bosio and others65 showed that significant genetic heterogeneity was present in Ae. aegypti populations from Thailand separated by 25 km. Similarly, Herrera and others66 found very restricted gene flow among Venezuelan Ae. aegypti populations at distances of 15 km, although, in these areas, there is intensive terrestrial and aerial traffic.
Although two gene pools were detected and the genetic heterogeneity was significant, it was only moderate (FST = 0.069). Other vector insect populations have presented similar genetic differentiation levels as follows: An. gambiae showed, in two Kenyan populations (Jego and Asembo), a value of FST = 0.072–0.10, although the genetic differentiation was conspicuously lower in other African areas separated by 6,000 km (FST = 0.016).67 Additionally, Lehmann and others50 showed that the genetic heterogeneity among 16 locations in 10 African countries for An. gambiae ranged from FST = 0–0.40, with a mean value around FST = 0.10. Ruiz-García and others68,69 showed similar genetic heterogeneity (FST = 0.067) among several Colombian populations of Psorophora columbiae in a geographical area (Tolima, Meta, and Cordoba Departments) that overlapped with our study area. De Souza and others70 showed a similar population differentiation in Ae. aegypti from Argentina (FST = 0.065) as well as An. albimanus,49 with FST values from 0.018 to 0.02 between Central American populations and from 0.057 to 0.082 between South American populations, similar to those shown here. Nevertheless, other species, like several An. nuñeztovari populations in Colombia and Brazil, presented substantially greater genetic heterogeneity (FST = 0.324),71 and several other species showed an underlying lower genetic heterogeneity, such as Ae. aegypti from Brazil (FST = 0.018)72 or Ae. albifasciatus from Argentina (FST = 0.003),73 than the mosquito populations analyzed here. Therefore, the genetic heterogeneity found in our study is only intermediate with respect to values found in other mosquitoes species. It is possible that human activity could enhance the gene-flow levels between the populations of An. marajoara studied, which has been reported for other mosquitoes (Ravel and others74 for Ae. aegypti in México; Vazeille and others75 for Ae. albopictus in Madagascar; García-Franco and others76 for Ae. aegypti in other Mexican populations; Bosio and others65 for Ae. aegypti in Thailand).
The migration of individuals and the gene-flow levels detected were elevated, despite the fact that some important geographical barriers separated the populations studied. Only the Eastern Andean cordillera was decisive in the partial differentiation of the two gene pools determined. Some other barriers such as the Cauca and Magdalena rivers in the western area or the Macarena Mountains and the Tepuyes formations in the eastern region studied did not represent any geographical barrier for the migration of An. marajoara throughout Colombia. The very different ecological conditions in the geographical areas analyzed also did not represent any problem for the dispersion of this potential malaria vector. Malaria has recently reemerged as a public-health burden in Colombia. From 18 to 24 million people are living in areas with high risk of exposure to malaria,2 which represents about 60% of the total Colombian human population. In 2008 alone, around 100,000 cases were officially reported in Colombia,77 although the real number is probably higher. Thus, the control of the malaria vectors such as An. marajoara is very important, because it shows considerable dispersal and a flexible adaptative capacity to anthropogenic environments. An. marajoara could probably be playing an important role either as a primary or secondary malaria vector in some Colombian areas. The occurrence of this species sympatrically with An. darlingi adds additional complexity to the study of the dynamics of malaria transmission in Colombia. From a population genetics point of view, the present results seem to suggest that the vector-control programs in Colombia do not affect the genetic structure of this species that is characterized by elevated gene-flow estimates throughout extensive areas of this country.
Acknowledgments
The authors thank technical personnel from Health Department of Magdalena (Katiuska Ariza), Antioquia (Armando Galeano), Caquetá (Alberto Polanía), Norte de Santander (Eulides Pabón and Pánfilo Lobo), and Meta (Luz Stella Buitrago). This research was performed with institutional support provided by both Walter Reed Army Institute of Research and the Colombian National Health Institute. Partial support was provided by National Institutes of Health Grant AI R0154139 to J.E.C. and Colciencias to H.B. Logistical support was obtained from the Pontificia Universidad Javeriana at Bogotá DC, Colombia, to M.R.-G. to carry out the population genetics analyses and write the present manuscript.
Footnotes
Authors' addresses: Helena Brochero, Facultad de Agronomía, Universidad Nacional de Colombia, Bogotá DC, Colombia, E-mail: embrochero@unal.edu.co. Cong Li and Richard Wilkerson, Division of Entomology, Walter Reed Army Institute of Research, Silver Spring, MD, E-mails: lic@si.edu and wilkersonr@si.edu. Jan E. Conn, Griffin Laboratory, Wadsworth Center, NYSDOH, Slingerlands, NY, E-mail: jconn@wadsworth.org. Manuel Ruiz-García, Laboratorio de Genética de Poblaciones Molecular-Biología Evolutiva, Unidad de Genética, Departamento de Biología, Facultad de Ciencias, Pontificia Universidad Javeriana, Cra 7ª. No 43-82, Bogotá DC, Colombia, E-mails: mruiz@javeriana.edu.co or mruizgar@yahoo.es.
References
- 1.Cortés J, Caraballo A, Contreras C, Plowe C. Origin and dissemination of Plasmodium falciparum drug-resistance mutations in South America. J Infect Dis. 2002;186:999–1006. doi: 10.1086/342946. [DOI] [PubMed] [Google Scholar]
- 2.Mendoza M, Nicholls R, Olano V, Cortés L. Manual de Manejo integral de la malaria. Instituto Nacional de Salud Ed. Bogotá, Columbia: Instituto Nacional de Salud; 2000. (Situación de la malaria en Colombia). [Google Scholar]
- 3.Herrera S, Suárez M, Sánchez G, Quiñones M, Herrera M. Uso de la técnica inmunoradiometrica (IRMA) en Anopheles de Colombia para la identificación de esporozoitos de Plasmodium. Colomb Med. 1987;18:57–60. [Google Scholar]
- 4.Collins W, Warren W, Skinner J, Sutton B. Infectivity of Plasmodium vivax to Anopheles albitarsis mosquitoes from Colombia. J Parasitol. 1985;71:771–773. [PubMed] [Google Scholar]
- 5.Quiñones M, Suárez M. Indoor resting heights of some anophelines in Colombia. J Am Mosq Control Assoc. 1990;6:602–604. [PubMed] [Google Scholar]
- 6.Brochero H, Rey G, Buitrago L, Olano V. Biting activity and breeding places of Anopheles species in the municipality Villavicencio, Meta, Colombia. J Am Mosq Control Assoc. 2005;21:182–186. doi: 10.2987/8756-971X(2005)21[182:BAABSO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 7.Servicio de Erradicación de la Malaria (SEM) Plan de erradicación de la malaria en Colombia. Volumen I y II. Ministerio de Salud Nacional; Bogotá, Columbia: Ministerio Nacional de Salud: 1957. [Google Scholar]
- 8.Rubio-Palis Y, Zimmermann R. Ecoregional classification of malaria vectors in the Neotropics. J Med Entomol. 1997;34:499–510. doi: 10.1093/jmedent/34.5.499. [DOI] [PubMed] [Google Scholar]
- 9.Conn J, Wilkerson R, Nazaré M, Segura O, Raimundo T, De Souza L. Emergence of a new neotropical malaria vector facilitated by human migration and changes in land use. Am J Trop Med Hyg. 2002;66:18–22. doi: 10.4269/ajtmh.2002.66.18. [DOI] [PubMed] [Google Scholar]
- 10.Brochero H, Li C, Wilkerson R. A previously unrecognized species in the Anopheles (Nyssorhynchus) albitarsis complex (Diptera: Culicidae) from Puerto Carreño, Colombia. Am J Trop Med Hyg. 2007;76:1113–1117. [PubMed] [Google Scholar]
- 11.Motoki M, Wilkerson R, Sallum M. The Anopheles albitarsis complex with the recognition of Anopheles oryzalimnetes Wilkerson and Motoki, n. sp. and Anopheles janconnae Wilkerson and Sallum, n. sp. (Diptera: Culicidae) Mem Inst Oswaldo Cruz. 2009;104:823–850. doi: 10.1590/s0074-02762009000600004. [DOI] [PubMed] [Google Scholar]
- 12.Narang S, Klein T, Perera O, Lima J, Tang A. Genetic evidence for the existence of cryptic species in the Anopheles albitarsis complex in Brazil: allozymes and mitochondrial DNA restriction fragment length polymorphism. Biochem Genet. 1993;1:97–112. doi: 10.1007/BF02399823. [DOI] [PubMed] [Google Scholar]
- 13.Conn J, Mitchell S, Cockburn A. Mitochondrial DNA variation within and between two species of neotropical Anopheline mosquitoes (Diptera: Culicidae) J Hered. 1997;88:98–107. doi: 10.1093/oxfordjournals.jhered.a023084. [DOI] [PubMed] [Google Scholar]
- 14.Posso C, González R, Cárdenas H, Tascón R. Estructura genética de Anopheles darlingi Root, An. nuneztovari Gabaldon y An. marajoara Galvão & Damasceno de Colombia mediante RAPD-PCR. Revista Colombiana de Entomología. 2006;32:49–56. [Google Scholar]
- 15.Faran M, Linthicum K. A handbook of the Amazonian species of Anopheles (Nyssorhynchus) Mosq Syst. 1981;13:1–85. [Google Scholar]
- 16.Li C, Wilkerson R. Identification of Anopheles (Nyssorhynchus) Albitarsis complex species (Diptera: Culicidae) using rDNA ITS2-based PCR primers. Mem Inst Oswaldo Cruz. 2005;100:495–500. doi: 10.1590/s0074-02762005000500009. [DOI] [PubMed] [Google Scholar]
- 17.Besansky N, Fahey T. Utility of the white gene in estimating phylogenetic relationships among mosquitoes (Diptera:Culicidae) Mol Biol Evol. 1997;14:442–444. doi: 10.1093/oxfordjournals.molbev.a025780. [DOI] [PubMed] [Google Scholar]
- 18.Wilkerson R, Parson T, Albringht T, Klein T, Braun M. Random amplified polymorphic DNA (RAPD) markers readily distinguish cryptic mosquito species (Diptera: Culicidae: Anopheles) Insect Mol Biol. 1993;1:205–211. doi: 10.1111/j.1365-2583.1993.tb00093.x. [DOI] [PubMed] [Google Scholar]
- 19.Li C, Wilkerson R, Fonseca D. Isolation of polmorphic microsatellite markers from the malaria vector Anopheles (Nyssorhynchus) marajoara. (Diptera: Culicidae) Mol Ecol Notes. 2005;5:65–67. [Google Scholar]
- 20.Nei M. Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA. 1973;70:3321–3323. doi: 10.1073/pnas.70.12.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Archie JW. Statistical analysis of heterozygosity data: independent sample comparisons. Evolution. 1985;39:623–637. doi: 10.1111/j.1558-5646.1985.tb00399.x. [DOI] [PubMed] [Google Scholar]
- 22.Weir B, Cokerham C. Estimating F statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 23.Raymond M, Rousset F. GenePop version 3.1 population genetics software for exact tests and ecumenicism. J Hered. 1995;86:248–249. [Google Scholar]
- 24.Raymond M, Rousset F. Updated version of GenePop version 3.1 described in: Raymond M, Rousset F. 1995. Population genetics software for exact tests and ecumenicism. J Hered. 2003;86:248–249. [Google Scholar]
- 25.Wright S. The genetical structure of populations. Ann Eugen. 1951;15:323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 26.Goudet J, Raymond M, de Meeus T, Rousset F. Testing differentiation in diploid populations. Genetics. 1996;144:1933–1940. doi: 10.1093/genetics/144.4.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goudet J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices (version 2.9.1) Switzerland: Institute of Ecology, University of Laussane; 2002. [Google Scholar]
- 28.Slatkin M. A measure of population subdivision based on microsatellite allele frequencies. Genetics. 1995;139:457–462. doi: 10.1093/genetics/139.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rouset F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics. 1997;145:1219–1228. doi: 10.1093/genetics/145.4.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodman S. RST CALC: a collection of computer programs for calculating unbiased estimates of genetic differentiation and determining their significance for microsatellite data. Mol Ecol. 1997;6:881–885. [Google Scholar]
- 31.Ruiz-García M. Analysis of the evolution and genetic diversity within and between Balearic and Iberian cat populations. J Hered. 1993;84:173–180. doi: 10.1093/oxfordjournals.jhered.a111312. [DOI] [PubMed] [Google Scholar]
- 32.Ruiz-García M. Genetic structure and evolution of different cat populations (Felis catus) in Spain, Italy, Argentina at microgeographical level. Acta Theriol (Warsz) 1998;43:39–66. [Google Scholar]
- 33.Ruiz-García M, Alvarez D. Genetic microstructure in two Spanish cat populations. I: genetic diversity, gene flow and selection. Genes Genet Syst. 2000;75:269–280. doi: 10.1266/ggs.75.269. [DOI] [PubMed] [Google Scholar]
- 34.Takahata N. Gene identity and genetic differentiation of populations in the finite island model. Genetics. 1983;104:497–512. doi: 10.1093/genetics/104.3.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crow J, Aoki K. Group selection for a polygenic behavioural trait. Proc Natl Acad Sci USA. 1984;81:6073–6077. doi: 10.1073/pnas.81.19.6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slatkin M. Rare alleles as indicators of gene flow. Evolution. 1985;39:53–65. doi: 10.1111/j.1558-5646.1985.tb04079.x. [DOI] [PubMed] [Google Scholar]
- 37.Barton N, Slatkin M. A quasi-equilibrium theory of the distribution of rare alleles in a subdivided population. Heredity. 1986;56:409–415. doi: 10.1038/hdy.1986.63. [DOI] [PubMed] [Google Scholar]
- 38.Pritchard J, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lehr M, Kilpatrick C, Wilkerson R, Conn J. Cryptic species in the Anopheles (Nyssorhynchus) albitarsis (Diptera: Culicidae) complex: incongruence between RAPD-PCR identification and analysis of mtDNA COI gene sequences. Ann Entomol Soc Am. 2005;98:908–917. doi: 10.1603/0013-8746(2005)098[0908:CSITAN]2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cornuet J, Piry D, Luikart G, Estoup A, Solignac M. New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics. 1999;153:1989–2000. doi: 10.1093/genetics/153.4.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luikart G, Sherwin W, Steele B, Allendorf F. Usefulness of molecular markers for detecting population bottlenecks via monitoring genetic changes. Mol Ecol. 1998;7:963–974. doi: 10.1046/j.1365-294x.1998.00414.x. [DOI] [PubMed] [Google Scholar]
- 42.Garza JC, Williamson EG. Detection of reduction in population size using data from microsatellite loci. Mol Ecol. 2001;10:305–318. doi: 10.1046/j.1365-294x.2001.01190.x. [DOI] [PubMed] [Google Scholar]
- 43.Reich DE, Goldstein DB. Genetic evidence for a Paleolitic human population expansion in Africa. Proc Natl Acad Sci USA. 1998;95:8119–8123. doi: 10.1073/pnas.95.14.8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reich DE, Feldman MW, Goldstein DB. Statistical properties of two tests that use multilocus data sets to detect population expansions. Mol Biol Evol. 1999;16:453–466. [Google Scholar]
- 45.Kimmel M, Chakravorty R, King JP, Bamshad M, Watkins WS, Jorde LB. Signatures of population expansion in microsatellite repeat data. Genetics. 1998;148:1921–1930. doi: 10.1093/genetics/148.4.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhivotovsky LA, Bennett L, Bowcock AM, Feldman MW. Human population expansion and microsatellite variation. Mol Biol Evol. 2000;17:757–767. doi: 10.1093/oxfordjournals.molbev.a026354. [DOI] [PubMed] [Google Scholar]
- 47.Beumont M. Detecting population expansions and decline using microsatellites. Genetics. 1999;153:2013–2029. doi: 10.1093/genetics/153.4.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conn J, Vineis J, Bollback J, Onyabe D, Wilkerson R, Povoa M. Population structure of the malaria vector Anopheles darlingi in a malaria-endemic region of eastern Amazonian Brazil. Am J Trop Med Hyg. 2006;74:798–806. [PubMed] [Google Scholar]
- 49.Molina-Cruz A, De Mérida A, Mills K, Rodriguez F, Schoua C, Yurrita M, Molina E, Palmieri M, Black W IV. Gene flow among Anopheles albimanus populations in Central America, South America and the Caribbean assessed by microsatellites and mitochondrial DNA. Am J Trop Med Hyg. 2004;71:350–359. [PubMed] [Google Scholar]
- 50.Lehmann T, Hawley W, Grebert H, Danga M, Atieli F, Collins F. The Rift Valley complex as a barrier to gene flow for Anopheles gambiae in Kenya. J Hered. 1999;90:613–621. doi: 10.1093/jhered/90.6.613. [DOI] [PubMed] [Google Scholar]
- 51.Lehmann T, Licht M, Elissa N, Maega T, Chimumbwa J, Watsenga T, Wondji C, Simard F, Hawley W. Population structure of Anopheles gambiae in Africa. J Hered. 2003;94:133–147. doi: 10.1093/jhered/esg024. [DOI] [PubMed] [Google Scholar]
- 52.Norris D, Shurtleff A, Touré Y, Lanzaro G. Microsatellite DNA polymorphism and heterozygosity among field and laboratory populations of Anopheles gambiae (Diptera: Culicidae) J Med Entomol. 2001;38:336–340. doi: 10.1603/0022-2585-38.2.336. [DOI] [PubMed] [Google Scholar]
- 53.Wondji C, Simard F, Fontenille D. Evidence for genetic differentiation between the molecular forms M and S within the Forest chromosomal form of Anopheles gambiae in an area of sympatry. Insect Mol Biol. 2002;11:11–19. doi: 10.1046/j.0962-1075.2001.00306.x. [DOI] [PubMed] [Google Scholar]
- 54.Rongnoparut R, Sirichotpakorn N, Rattanarithikul R, Yaicharoen S, Lithincum K. Estimates of gene flow among Anopheles maculatus populations in Thailand using microsatellite analysis. Am J Trop Med Hyg. 1999;60:508–515. doi: 10.4269/ajtmh.1999.60.508. [DOI] [PubMed] [Google Scholar]
- 55.Onyabe D, Conn J. Genetic differentiation of the malaria vector Anopheles gambiae across Nigeria suggests that selection limits gene flow. Heredity. 2001;87:647–658. doi: 10.1046/j.1365-2540.2001.00957.x. [DOI] [PubMed] [Google Scholar]
- 56.Estrada-Franco J, Lanzaro G, Ma M, Walker-Abbey A, Romans P, Galvan-Sanchez C, Céspedes J, Vargas-Sagarnaga R, Laughinghouse A, Columbus I, Gwadz R. Characterization of Anopheles pseudopunctipennis sensu lato from three countries of neotropical America from variation in allozymes and ribosomal DNA. Am J Trop Med Hyg. 1993;49:735–745. doi: 10.4269/ajtmh.1993.49.735. [DOI] [PubMed] [Google Scholar]
- 57.Goldstein B, Ruiz-Linares A, Cavalli-Sforza L, Feldman W. Genetic absolute dating on microsatellites and the origin of modern humans. Proc Natl Acad Sci USA. 1995;92:6723–6727. doi: 10.1073/pnas.92.15.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kamau L, Mukabana W, Hawley W, Lehmann T, Irungu L, Orago A, Collins F. Analysis of genetic variability in Anopheles arabiensis and Anopheles gambiae using microsatellite loci. Insect Mol Biol. 1999;8:287–297. doi: 10.1046/j.1365-2583.1999.820287.x. [DOI] [PubMed] [Google Scholar]
- 59.Kamau L, Hunt R, Coetzee M. Analysis of the population structure of Anopheles funestus Giles (Diptera: Culicidae) from Kenya using paracentric chromosomal inversion frequencies. J Med Entomol. 2002;39:78–83. doi: 10.1603/0022-2585-39.1.78. [DOI] [PubMed] [Google Scholar]
- 60.Mendes dos Santos J, Freitas Maia J, Tadeo P, Diaz G. Isoenzymatic variability among five Anopheles species belonging to the Nyssorhynchus and Anopheles subgenera of the Amazon Region, Brazil. Mem Inst Oswaldo Cruz. 2003;98:247–253. doi: 10.1590/s0074-02762003000200014. [DOI] [PubMed] [Google Scholar]
- 61.Coluzzi M, Sabatini A, Petrarca V, Di Deco M. Chromosomal differentiation and adaptation to human environments in the Anopheles gambiae complex. Trans R Soc Trop Med Hyg. 1979;73:483–497. doi: 10.1016/0035-9203(79)90036-1. [DOI] [PubMed] [Google Scholar]
- 62.Lanzaro G, Toure Y, Carnahans J, Zheng L, Dolo G, Traore S, Petrarca V, Vernic K, Taylor C. Complexities in the genetic structure of Anopheles gambiae populations in West Africa as revealed by microsatellite DNA analysis. Proc Natl Acad Sci USA. 1998;95:14260–14265. doi: 10.1073/pnas.95.24.14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mirabello L, Conn J. Molecular population genetics of the malaria vector Anopheles darlingi in Central and South America. Heredity. 2006;96:311–321. doi: 10.1038/sj.hdy.6800805. [DOI] [PubMed] [Google Scholar]
- 64.Ravel S, Monteny N, Velasco Olmos D, Escalante Verdugo J, Cuny G. A preliminary study of the population genetics of Aedes aegypti (Diptera: Culicidae) from Mexico using microsatellite and AFLP markers. Acta Trop. 2001;78:241–250. doi: 10.1016/s0001-706x(01)00083-3. [DOI] [PubMed] [Google Scholar]
- 65.Bosio CF, Harrington LC, Jones JW, Sithiprasasna R, Norris DE, Scott TW. Genetic structure of Aedes aegypti populations in Thailand using mitochondrial DNA. Am J Trop Med Hyg. 2005;72:434–442. [PubMed] [Google Scholar]
- 66.Herrera F, Urdaneta L, Rivero J, Zoghbi N, Ruiz J, Carrasquel G, Martinez JA, Pernalete M, Villegas P, Montoya A, Rubio-Palis Y, Rojas E. Population genetic structure of the dengue mosquito Aedes aegypti in Venezuela. Mem Inst Oswaldo Cruz. 2006;10:625–633. doi: 10.1590/s0074-02762006000600008. [DOI] [PubMed] [Google Scholar]
- 67.Lehmann T, Hawley W, Grebert H, Collins F. The effective population size of Anopheles gambiae in Kenya: implications for population structure. Mol Biol Evol. 1998;15:264–276. doi: 10.1093/oxfordjournals.molbev.a025923. [DOI] [PubMed] [Google Scholar]
- 68.Ruiz-García M, Ramírez D, Bello F, Alvarez D. Psorophora columbiae and Psorophora toltecum (Diptera: Culicidae) Colombian populations cannot be differentiated by isoenzymes. Genet Mol Res. 2003;2:229–259. [PubMed] [Google Scholar]
- 69.Ruiz-García M, Bello F, Ramírez D, Alvarez D. Genetic structure of the genera Psorophora (Diptera: Culicidae) in Columbian and North American populations using isoenzymes and ITS2 sequences. Russ J Genet. 2006;42:752–765. [PubMed] [Google Scholar]
- 70.De Souza G, De Dutari GP, Gardenal C. Genetic structure of Aedes albifasciatus (Diptera Culicidae) populations in Central Argentina determined by random amplified polymorphic DNA-polymerase chain reaction markers. J Med Entomol. 2000;86:400–404. doi: 10.1093/jmedent/36.3.400. [DOI] [PubMed] [Google Scholar]
- 71.Scarpassa V, Geurgas S, Azeredo-Espin A, Tadei W. Genetic divergence in mitochondrial DNA of Anopheles nuneztovari (Diptera: Culicidae) from Brazil and Colombia. Genet Mol Biol. 2000;23:71–78. [Google Scholar]
- 72.Dinardo-Miranda L, Contel E. Enzymatic variability in natural populations of Aedes aegypti (Diptera: Culicidae) from Brazil. J Med Entomol. 1996;33:726–733. doi: 10.1093/jmedent/33.5.726. [DOI] [PubMed] [Google Scholar]
- 73.De Souza G, Jimenez A, Blanco A, Gardenal C. Gene flow in Aedes albifasciatus (Diptera: Culicidae) from Central Argentina. J Med Entomol. 1996;33:894–900. doi: 10.1093/jmedent/33.6.894. [DOI] [PubMed] [Google Scholar]
- 74.Ravel S, Monteny N, Velasco Olmos D, Escalante Verdugo J, Cuny G. A preliminary study of the population genetics of Aedes aegypti (Diptera: Culicidae) from Mexico using microsatellites and AFLP markers. Acta Trop. 2001;78:241–250. doi: 10.1016/s0001-706x(01)00083-3. [DOI] [PubMed] [Google Scholar]
- 75.Vazeille M, Mousson L, Rakatoarivony I, Villeret R, Rodhain F, Duchemin JB, Failloux AB. Population genetic structure and competence as a vector for dengue type 2 virus of Aedes aegypti and Aedes albopictus from Madagascar. Am J Trop Med Hyg. 2001;65:491–497. doi: 10.4269/ajtmh.2001.65.491. [DOI] [PubMed] [Google Scholar]
- 76.García-Franco F, Lourdes Muñoz M, Lozano-Fuentes S, Fernandez-Salas I, García-Rejon J, Beaty BJ, Black WC IV. Large genetic distances among Aedes aegypti populations along the South Pacific coast of Mexico. Am J Trop Med Hyg. 2002;64:594–598. doi: 10.4269/ajtmh.2002.66.594. [DOI] [PubMed] [Google Scholar]
- 77.Ministerio de la Protección Social, Instituto Nacional de Saud Sistema nacional de vigilancia SIVIGILA, 2009. Informe Quincenal Epidemiológico Nacional. 2009;14:47–69. [Google Scholar]