

Abstract
During the past two decades, Dengue virus-3 (DENV-3) has re-emerged in the Western Hemisphere causing significant epidemics of dengue fever (DF) and dengue hemorrhagic fever (DHF). In an effort to understand the molecular evolution of DENV-3 and their relationships to other DENV-3 circulating in the western hemisphere, we conducted a phylogenetic study on DENV-3 isolates made between 2002 and 2007 in the metropolitan area of Medellín, Colombia. An unexpected co-circulation of two different variants of DENV-3 subtype III during at least 5 years in Medellín was found. In addition, a more complete analysis of DENV-3 viruses isolated in other South American regions revealed the existence of three different subtype III lineages, all derived from independent introductions. This study documents significant genetic diversity of circulating viruses within the same subtype and an unusual capacity of the population of this city to support continuous circulation of multiple variants of dengue virus.
Introduction
Dengue viruses (DENV) (genus Flavivirirus, family Flaviviridae) include four serotypes (DENV-1 to DENV-4) that cause the most important arthropod-borne viral infection of humans with about 100 million cases and 25,000 deaths annually.1,2 The DENV cause classic dengue (“break-bone”) fever (DF) and more severe and sometimes fatal dengue hemorrhagic fever (DHF)/dengue shock syndrome (DSS). In tropical and subtropical regions of the Western Hemisphere, DF and DHF are considered two of the most serious public health problems.2–4
The pathogenesis of DHF is incompletely understood. Several factors including age and previous exposure can increase the risk for DHF.1,5 Recent studies also suggest that more virulent strains, and in particular the Asian subtypes (or genotypes), might also play an important role in the increase of DHF in American countries. Rico-Hesse and others6 showed that introduction of a DENV-2 subtype arriving from Southeast Asia produced an increase in proportion of DHF/DSS cases. Lanciotti and others7 and Messer and others,8 have postulated that a genetic shift between two subtype III lineages of DENV-3, which occurred in 1989, may be responsible for the increase in severity in Sri Lanka.
Of particular interest is the reemergence of DENV-3 in the Western Hemisphere. In 1994, after an absence of 17 years, DENV-3 was detected in Nicaragua and Panama.9,10 Soon thereafter, the virus was isolated in Mexico and other Central American countries causing DF and DHF.8,11–13 From 1998 to 2003, the virus spread across the Caribbean islands.8,14–16 In 1999 and 2000, DENV-3 was detected in Ecuador, Venezuela, and Brazil and later in other South American countries.17–19
The molecular evolution of DENV-3 has been studied in numerous American countries, including Nicaragua, Venezuela, Mexico, Brazil, Paraguay, Cuba, Peru, and Argentina.16–18,20–24 All of these studies point to subtype III as the only DENV-3 circulating in the Western Hemisphere. However, recent works by three independent groups reported the presence of DENV-3 isolates in Brazil and Colombia that belong to a branch of subtype I, also designated as subtype V.25–27
In Colombia, DENV-3 was reported to cause an epidemic in 1975–1977 but disappeared soon after.28 The DENV-3 was isolated again in 2001 in the eastern province of Santander.29 It then spread to other provinces in the following years, including Antioquia and its capital, Medellín, where DENV has been regularly diagnosed since 1992 with peak incidence in 1998, 2003, and 2007 (Figure 1). Most cases detected have been of DF, but since 1995, DHF cases have occurred. Between 2002 and 2007, 14,539 cases of DF and 182 cases of DHF were reported. Until 2001, only DENV-1, -2, and -4 were isolated. Although DENV-3 was first detected in Medellín in 2002, it was not the predominant serotype until 2005, while DENV-1, -2, and -4 continued circulating.30 DENV-3 is highly prevalent now in Medellín and in other parts of Colombia and other South American countries.8
Figure 1.
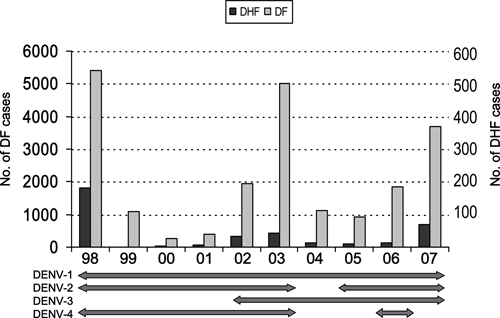
Dengue fever (DF) and dengue hemorrhagic fever (DHF) cases reported in Medellín metropolitan area during 1998–2007. Both events are plotted in different scales. Horizontal bars in the bottom shows the serotypes isolated each year in the same time scale.
In an effort to understand the origin and evolution of DENV-3 circulating in Medellín and any possible relation to the severity or epidemic behavior of the disease, a phylogenetic analysis was conducted of DENV-3 isolated from clinical cases between 2002 and 2007 in the area. This analysis was further expanded using DENV-3 isolated in other South American countries to determine potential circulation patterns and origin of viruses found in the Medellín area.
Materials and Methods
Geographic location.
The Medellín metropolitan area is a conurbation of 10 municipalities in the province of Antioquia, northwestern Colombia. It is located at latitude: 6°09′52²N, longitude: 75°25′23²W, in a valley 1,500–1,600 meters above the sea level (Figure 2). The mean temperature is 24°C. The total population is about three million people.
Figure 2.

Location of the Medellín metropolitan area in the province of Antioquia and in Colombia. The numbers correspond to the following municipalities where the viruses were isolated: 1. Copacabana, 2. Bello, 3. Medellín, 4. Itagüi, 5. Envigado.
Viruses.
All the DENV-3 isolates sequenced in this study were obtained from the virus collection of the Laboratorio Departamental de Salud Publica (LDSP, Table 1), during the 6-year period from 2002 to 2007. Eighteen viruses obtained from DF or DHF cases occurring in Medellín and surrounding municipalities were selected to represent all the years in the period. These cases were classified according to the World Health Organization (WHO) criteria for DHF.31 All viruses had been isolated in Aedes albopictus C6/36 cells and stored at −70°C until testing. Only primary isolates or first passage viruses were used in this study.
Table 1.
DENV-3 isolates from Medellín metropolitan area included in this study
| Isolate identification | Clinical presentation* | Patient age (years) | Municipality | Date of isolation | GenBank accession no. |
|---|---|---|---|---|---|
| 3419 | DHF | 58 | MEDELLÍN | 2007 | FJ389906 |
| 5292 | DF | ND† | MEDELLÍN | 2003 | FJ389907 |
| 15859 | DF | 31 | BELLO | 2002 | FJ389908 |
| 16407 | DF | ND | MEDELLÍN | 2002 | FJ389909 |
| 22379 | DF | 47 | MEDELLÍN | 2004 | FJ389910 |
| 11049 | DF | 25 | MEDELLÍN | 2003 | FJ389911 |
| 31233 | DF | 27 | BELLO | 2004 | FJ389912 |
| 3453 | DF | 46 | MEDELLÍN | 2007 | FJ389914 |
| 3832 | DF | 22 | MEDELLÍN | 2006 | FJ389915 |
| 18206 | DF | 27 | MEDELLÍN | 2005 | FJ389916 |
| 2836 | DF | 9 | ENVIGADO | 2003 | HM030549 |
| 2754 | DF | 59 | ITAGUI | 2007 | HM030550 |
| 28888 | DF | 15 | COPACABANA | 2006 | HM030551 |
| 5379 | DF | 57 | ITAGUI | 2003 | HM030552 |
| 5952 | DF | 40 | BELLO | 2003 | HM030553 |
| 6003 | DF | 69 | COPACABANA | 2007 | HM030554 |
| 7370 | DF | 9 | MEDELLÍN | 2006 | HM030555 |
| 12363 | DF | 18 | MEDELLÍN | 2005 | HM030556 |
DF = dengue fever; DHF = dengue hemorrhagic fever.
ND = no data.
Molecular procedures.
Viral RNA (vRNA) was extracted from cell culture supernatant using a commercial silica-based method (QIAamp viral RNA, Qiagen, Valencia, CA). The vRNAs were copied into complementary DNAs (cDNAs) using random hexamers and RAV-2 reverse transcriptase (Amersham, Piscataway NJ). A cDNA segment of 1,806 nucleotides encompassing the M and E genes was amplified by polymerase chain reaction (PCR). The PCR mix included 1.75 mM MgCl, 0.25 mM dNTPs, 0.5 μM primers, and 0.05 U/μL Taq DNApol (Promega, Madison WI). Amplification was performed in 40 cycles of 94°C for 15 seconds, 55°C for 30 seconds, and 72°C for 2 minutes. Products were verified by 0.8% agarose gel electrophoresis and then purified using the QIAquick PCR purification kit (Qiagen). Both strands of the amplicons were sequenced using appropriate primers and the Big Dye Terminator Cycle Sequencing System (ABI, Foster City, CA) following the protocols recommended by the manufacturer. The sequences of the primers used in the PCR and sequencing are available from the authors upon request. Sequences obtained in this study were deposited in GenBank (accession nos. FJ389906 to FJ389916 and HM030549 to HM030556).
Sequence datasets.
The sequences of the complete M and E genes (1,704 nucleotides) of viruses isolated in this study were manually aligned with other DENV-3 sequences obtained in other studies7,8,16,18,22,23 and available in GenBank. These included three other Colombian sequences of viruses isolated in the province of Santander. Sequences were selected to maximize the representation by country, year, and subtype. Sequences of viruses isolated in the Western Hemisphere since 1994 were included in higher number than those of Asia and Africa. We did not include the subtype IV Puerto Rican sequences of the 1960–70s, because they are thought to be recombinants.32 We also excluded sequences of subtype V other than the prototype strain H87/Philippines/56, because all of them are very similar and probably derived from the same prototype virus that has been maintained in many laboratories in the world for reference and control purposes.
Phylogenetic and other analyses.
Phylogenetic analyses were conducted with Bayesian approaches using MrBayes v3.1.2 and BEAST v1.4.8 software packages and related utility programs.33 Model settings used in both programs were the Hasegawa–Kishino–Yano substitution model, with gamma distribution of among-site rate variation (HKY + Γ). The transition/transversion ratio and alpha shape parameter of the gamma distribution were estimated separately for the third codon position; this corresponds to the model suggested by Shapiro and others,34 which had a good fit for protein-coding nucleotide data with relatively few parameters. By MrBayes, two independent Markov chain Monte Carlo (MCMC) analyses of four chains each were run simultaneously sampling one in every 100 trees. The analyses were halted when they converged onto a stationary distribution as assessed by an average standard deviation of split frequencies lower than 0.02. A 50% majority-rule consensus tree was computed excluding the initial 20% of each run. To estimate the times of the most recent common ancestors (TMRCA) and other evolutionary parameters, three independent analyses of 10 million generations each were performed using BEAST, and then merged using Log Combiner v1.4.8. The evolutionary model was the same used in MrBayes with an uncorrelated-lognormal relaxed molecular clock and a Bayesian Skyline coalescent prior. The TRACER v1.4 software package33 was used to visualize and verify the convergence of estimated evolutionary parameters of both, MrBayes and BEAST outputs. The FigTree v1.1.2 package was used to draw the phylogenetic trees. To explore for selective forces, the Nei and Gojobori (NG86) method was used for estimating the ratio of nonsynonymous substitutions per nonsynonymous site to synonymous substitutions per synonymous site (dN/dS) as implemented in MEGA v3.1.35
Results
In this study, M and E genes of 18 DENV-3 viruses isolated between 2002 and 2007 in the Medellín area were sequenced, aligned to other DENV-3 isolates, and analyzed phylogenetically. A preliminary analysis of 109 DENV sequences, including the other three DENV serotypes as outgroup, was performed to correctly locate the root of the tree. Sequences in the outgroup and those that were identical or closely related to others, were excluded from the final analysis for space reasons. The final data set was composed of 90 sequences from DENV-3 isolated in different countries and years. The phylogenetic analysis using MrBayes was run for 1,000,000 generations with a 20% burn-in, producing a consensus tree where the previously described subtypes I, II, and III appeared as monophyletic groups (Figure 3). The prototype strain H87/Philippines/56 appeared in a separate branch, corresponding to the subtype V described by Wittke and others.36 Most isolates of Southeast Asia and Western Pacific islands grouped in subtypes I and II, as previously reported.7,8,24 Isolates from India, Sri Lanka, Africa, and the Americas, including all the Colombian viruses sequenced in this study, clustered in subtype III.
Figure 3.

Majority-rule consensus tree of DENV-3 sequences resulting from the Bayesian phylogenetic analysis done with MrBayes v3.1.2. Isolates sequenced in this study are bolded. Numbers close to nodes are posterior probabilities for the corresponding clade. The names of the subtypes (or genotypes), defined according to Wittke and others,36 are indicated below the corresponding branches. The lineages defined in this study are pointed by arrows. See the text for details about the substitution model used and the placing of the root. The tree was drawn with the FigTree v1.1.2 package.
American DENV-3 viruses formed a highly supported monophyletic group inside subtype III. Isolates from South America were distributed in three different and well-supported clades: the first was composed of viruses from Venezuela and Colombia (2000–2008), including nine of our isolates and the three from the province of Santander (Colombia). The second clade is composed of viruses from Brazil, Paraguay, and Bolivia (2000–2008), and some isolates from the Caribbean (Martinique 2000–2001, Cuba 2000, and Puerto Rico 2003). The third clade, which shares a common ancestor with the previous one, included all the viruses isolated in Ecuador and Peru (2000–2005), the remainder of our Colombian isolates (2003–2007), and two isolates from Cuba (2001–2002), and two from Nicaragua (2008–2009). These three clades represent separate introductions of DENV-3 into South America,17,24 and here they will be named after the country in which they were first detected as the Venezuelan, Brazilian, and Ecuadorian lineages, respectively.
Thus, Colombian isolates from the same years and cities belong to two distinct lineages, the Venezuelan and the Ecuadorian. Differences among Colombian isolates grouping in the same lineage were of 0 to 15 nucleotides (99.1% to 100.0% identity), and between isolates in different lineages, differences were of 21 to 34 nucleotides (98.0% to 98.8% identity). Colombian viruses in the Venezuelan lineage grouped in three separate subclades suggesting repeated importation of viruses from Venezuela. Colombian viruses in the Ecuadorian lineage grouped in a single clade but they are paraphyletic, because two recent Nicaraguan isolates emerged from the same clade, suggesting exportation of that lineage from Colombia to Central America.
We estimated TMRCA and mean evolutionary rates of the lineages defined here using BEAST and dN/dS using the method of Nei and Gojobori.37 The estimated TMRCA for the Brazilian, Ecuadorian, and Venezuelan lineages were between 1997 and 1999 (Table 2). The TMRCA estimated for all the American subtype III, and for viruses isolated in Medellín was 1991. Evolutionary rates estimated for the different lineages and other groups were between 3.6 × 10−4 and 8.6 × 10−4 substitutions per site per year (s/s/y). These estimates were not significantly different as revealed by the overlapping 95% highest probability density (HPD) values. All dN/dS ratios were lower than 0.1, indicating that purifying selection was the predominant type of selective force.
Table 2.
Estimated time of the most recent common ancestor (TMRCA) and other evolutionary parameters estimated for lineages or groups of viruses in the dataset used in this study
| Lineage or group | TMRCA (95% HPDs*) | Mean evolutionary rate in s/s/y† (95% HPDs) | dN/dS‡ |
|---|---|---|---|
| American Subtype III | 1991 (1989–1992) | 7.4 (5.4–9.1) × 10−4 | 0.044 |
| Venezuelan | 1999 (1997–2000) | 4.7 (1.4–7.9) × 10−4 | 0.053 |
| Ecuadorian | 1997 (1996–1999) | 8.6 (5.7–11.5) × 10−4 | 0.052 |
| Brazilian | 1997 (1996–1998) | 7.2 (2.1–11.8) × 10−4 | 0.099 |
| Medellín viruses | 1991 (1989–1993) | 3.6 (1.7–7.7) × 10−4 | 0.033 |
95% HPDs: 95% highest probability density.
s/s/y: substitutions per site per year.
dN/dS: average ratio of nonsynonymous substitutions per nonsynonymous site to synonymous substitutions per synonymous site.
To determine the amino acid changes in the dataset, viral sequences were translated, aligned, and the substitutions were mapped to the branches in the phylogenetic tree using conventional Fitch optimization. Changes were observed in two and 64 positions of the deduced sequences of M and E proteins, respectively. Most of the substitutions mapped either to the long basal branches connecting different subtypes or to the terminal branches. There were only three amino acid substitutions in the branches of the viruses sequenced in this study: V → A in position E-81, A → V in E-329, and G → E in E-340. The first one is unique to all the Colombian viruses in the Ecuadorian lineage, whereas the last two appeared in most Colombian viruses of Venezuelan lineage. The change in E-329 is also present in all viruses of the Brazilian lineage and Puerto Rican viruses of 2005 and 2007, indicating parallel substitutions. These positions are localized in loops of the structural domain II (E-81) or III (E-329, E-340) of E-glycoprotein.38 In summary, there were 0–2 amino acid differences within and 1–3 between lineages.
Discussion
The primary purpose of this study was to describe the origin and evolution of DENV-3 circulating in Medellín, Colombia. We found that the viruses isolated in this city during 2002–2007 were polyphyletic in origin and belonged to two different lineages, Venezuelan and Ecuadorian, derived from separate introductions of DENV-3 into South America. The most unusual finding is that these two lineages have remained co-circulating in the same city for at least 5 years (2003–2007).
Although co-circulation of different serotypes in a single place, referred to as hyperendemicity, has been reported from multiple countries, simultaneous circulation of different lineages of the same serotype is usually a short-lived event. Previous works have shown that the introduction of a second DENV variant of the same serotype in a region is soon followed by the disappearance of one of them.6,8,36,39 In the most complete phylogenetic work to date on DENV-3, Araujo and others concluded that “co-circulation of different DENV-3 in a single location is a rare event.” After exhaustively reviewing the phylogenetic literature on DENV we found only one case similar to what we describe here: the co-circulation of two lineages (B and C) of DENV-1 between 1998 and 2002 in Yangon, Myanmar, which replaced the previously circulating lineage A.40 However, it is possible that other cases of co-circulation could have passed unnoticed because of limited sampling.
The paucity of co-circulation of different variants in a single place seems to be the result of stochastic events occurring during interepidemic periods.36,40 Dengue has been diagnosed in the Medellín area every single month since 2002, as reported in the laboratory-based surveillance system of this illness carried out in LDSP (Ospina MC, personal observation, data not shown). In this way the homogenizing effect of population bottlenecks could have been minimized and different lineages could have co-existed in a large and previously non-immune population. We are currently monitoring DENV-3, and other DENV serotypes, to study the long-term outcome of this viral co-circulation in Medellín.
The clinical and epidemiological significance of the co-existence of these two lineages described here is still not clear. At the continental level there have been severe cases associated with viruses in all three lineages.16,18,41 In our sample of isolates there was only one DHF case that grouped in the Venezuelan lineage but this is not statistically significant. We did not detect any major difference in evolutionary rates or in overall dN/dS ratios that could point to differences in adaptive processes among lineages. The consequences of the described amino acid substitutions are also uncertain. More research is needed to explore potential differences in virulence or in fitness among the three lineages of DENV-III that circulates in South America, including full genome sequencing and phenotypic studies.
The origin and dispersion of DENV-3 subtype III in the Western Hemisphere has been studied in other works.8,16–18,22,24 Although its origin, either Asiatic or African remains obscure, it is now clear that the virus could have been introduced in the late 1980s or early 1990s, several years before its first detection in Nicaragua and Panama in 1994. Our estimate of the most recent common ancestor (TMRCA) suggests that subtype III could have been introduced between 1989 and 1993, which is close to previous estimates.24 The South American isolates segregated in three separated branches (Figure 3), which were previously identified by Kochel and others17 and Araujo and others.24 These branches are here designated as Venezuelan, Brazilian, and Ecuadorian lineages, according to their more likely site of entrance into the continent. All these lineages probably originated between 1996 and 1999 according to their estimated TMRCA (Table 2). The Brazilian lineage includes isolates from Cuba 2000, Martinique 2000–2001, and Puerto Rico 2003, which suggest a Caribbean origin for this lineage.22 Araujo and others suggested Mexico as the most likely origin of the Venezuelan lineage, but we show here that they are more closely related to viruses from Puerto Rico 1999–2007, which also points to the Caribbean as its possible source. The origin of the Ecuadorian lineage is more obscure. Cuban isolates of 2001 and 2002 are included in it. Because DENV-3 has been circulating in Ecuador since 1999,17 a viral exportation event from South America to Cuba for this lineage looks more likely, as proposed by Araujo and others.24 However, the existence of a common ancestor for Ecuadorian and Brazilian lineages that we dated around 1996 could indicate that the Ecuadorian lineage also originated in the Caribbean. A conclusive inference of the origin of these three variants must wait until more isolates from Central America and the Caribbean from the late nineties are available for analysis.
The dispersion of the three lineages into South America has already been described by Araujo and others.24 By using a balanced sample of isolates from different countries, our analysis also provided a comprehensive view of the dispersion of DENV-3 in South America (Figure 3). The Brazilian lineage, first detected in 2000 near Rio de Janeiro, seems to have reached a wide dispersion in that country, as shown by the Aquino and others22 study. That variant then passed to Paraguay (2002), eastern Bolivia (2003), and Argentina (2007).17,22,23 The Venezuelan lineage, also detected in 2000,18 passed to Colombia where it was detected in 2001 in the eastern province of Santander but does not seems to have extended to other countries.29 The Ecuadorian lineage, first detected in 1999 in that country, was isolated in Peru in 200017 and in Colombia in the southern province of Putumayo in 2002.27 Although other studies did not find overlaps in the areas of dispersion of the three lineages, we show here that two of them have met in Medellín, at least since 2003. It is foreseeable that in the next years, other places could report similar findings.
Excluding the old Puerto Rican isolates of the sixties and seventies (not included in our analysis), all other American DENV-3 isolates analyzed belong to subtype III, as previously reported.8,9,16–18,22 This subtype exhibited a virulent phenotype in Sri Lanka and Mozambique in the 1980s.7,36 We did not detect circulation of a branch of subtype I (or subtype V in the classification of Wittke and others36) as it was recently reported.25–27 Our findings are in disagreement with those of the study of Usme-Ciro and others,27 who supposedly isolated two DENV-3 subtype I in 2005 in Medellín and Bello, the same area studied by us. On the basis of our limited sampling, we cannot rule out that subtype I and III had co-circulated there and that they were segregated in the two studies by chance. However, this possibility looks unlikely. Future studies could shed some light on this intriguing point.
In summary, Colombia seems to be the first South American country maintaining simultaneous transmission of two distinct DENV-3 subtype III lineages. The prolonged co-circulation of these two lineages in the Medellín metropolitan area exemplifies the potential for DENV lineages to persist in a single place when the environmental conditions are appropriate.
Acknowledgments
We thank Yenny Goez and Andres Higuita for their technical assistance, and Thomas Yuill and Tonie Rocke for critical review of this manuscript.
Footnotes
Financial support: This work was supported by a career development Research Award from The Gorgas Memorial Foundation of Tropical and Preventive Medicine.
Disclosure: J. Osorio is a co-founder of Invivagen, Inc., a company working on the development of a dengue vaccine. This statement is made in the interest of full disclosure and not because the author considers this to be a conflict of interest.
Authors' addresses: Marta C. Ospina, Dirección Seccional de Salud y Protección Social de Antioquia, Laboratorio Departamental de Salud Pública de Antioquia, Medellín, Colombia, E-mail: franciscodiaz314@gmail.com. Francisco J. Diaz, Grupo Inmunovirología, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia, E-mail: mospinao@antioquia.gov.co. Jorge E. Osorio, Department of Pathobiological Sciences, School of Veterinary Medicine, University of Wisconsin, Madison, WI, E-mail: osorio@svm.vetmed.wisc.edu.
References
- 1.Guzman MG, Kouri G. Dengue and dengue hemorrhagic fever in the Americas: lessons and challenges. J Clin Virol. 2003;27:1–13. doi: 10.1016/s1386-6532(03)00010-6. [DOI] [PubMed] [Google Scholar]
- 2.Monath TP. Dengue: the risk to developed and developing countries. Proc Natl Acad Sci USA. 1994;91:2395–2400. doi: 10.1073/pnas.91.7.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gubler DJ, Clark GG. Dengue/dengue hemorrhagic fever: the emergence of a global health problem. Emerg Infect Dis. 1995;1:55–57. doi: 10.3201/eid0102.952004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kyle JL, Harris E. Global spread and persistence of dengue. Annu Rev Microbiol. 2008;62:71–92. doi: 10.1146/annurev.micro.62.081307.163005. [DOI] [PubMed] [Google Scholar]
- 5.Clyde K, Kyle JL, Harris E. Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J Virol. 2006;80:11418–11431. doi: 10.1128/JVI.01257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rico-Hesse R, Harrison LM, Salas RA, Tovar D, Nisalak A, Ramos C, Boshell J, de Mesa MT, Nogueira RM, da Rosa AT. Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology. 1997;230:244–251. doi: 10.1006/viro.1997.8504. [DOI] [PubMed] [Google Scholar]
- 7.Lanciotti RS, Lewis JG, Gubler DJ, Trent DW. Molecular evolution and epidemiology of dengue-3 viruses. J Gen Virol. 1994;75:65–75. doi: 10.1099/0022-1317-75-1-65. [DOI] [PubMed] [Google Scholar]
- 8.Messer WB, Gubler DJ, Harris E, Sivananthan K, de Silva AM. Emergence and global spread of a dengue serotype 3, subtype III virus. Emerg Infect Dis. 2003;9:800–809. doi: 10.3201/eid0907.030038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.CDC Dengue type 3 infection–Nicaragua and Panama, October–November 1994. MMWR Morb Mortal Wkly Rep. 1995;44:21–24. [PubMed] [Google Scholar]
- 10.Guzman MG, Vazquez S, Martinez E, Alvarez M, Rodriguez R, Kouri G, de los Reyes J, Acevedo F. Dengue in Nicaragua, 1994: reintroduction of serotype 3 in the Americas. Bol Oficina Sanit Panam. 1996;121:102–110. [PubMed] [Google Scholar]
- 11.Balmaseda A, Sandoval E, Perez L, Gutierrez CM, Harris E. Application of molecular typing techniques in the 1998 dengue epidemic in Nicaragua. Am J Trop Med Hyg. 1999;61:893–897. doi: 10.4269/ajtmh.1999.61.893. [DOI] [PubMed] [Google Scholar]
- 12.Usuku S, Castillo L, Sugimoto C, Noguchi Y, Yogo Y, Kobayashi N. Phylogenetic analysis of dengue-3 viruses prevalent in Guatemala during 1996–1998. Arch Virol. 2001;146:1381–1390. doi: 10.1007/s007050170098. [DOI] [PubMed] [Google Scholar]
- 13.Diaz FJ, Black WC, 4th, Farfan-Ale JA, Lorono-Pino MA, Olson KE, Beaty BJ. Dengue virus circulation and evolution in Mexico: a phylogenetic perspective. Arch Med Res. 2006;37:760–773. doi: 10.1016/j.arcmed.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 14.Peyrefitte CN, Couissinier-Paris P, Mercier-Perennec V, Bessaud M, Martial J, Kenane N, Durand JP, Tolou HJ. Genetic characterization of newly reintroduced dengue virus type 3 in Martinique (French West Indies) J Clin Microbiol. 2003;41:5195–5198. doi: 10.1128/JCM.41.11.5195-5198.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peyrefitte CN, Pastorino BA, Bessaud M, Gravier P, Tock F, Couissinier-Paris P, Martial J, Huc-Anais P, Cesaire R, Grandadam M, Tolou HJ. Dengue type 3 virus, Saint Martin, 2003–2004. Emerg Infect Dis. 2005;11:757–761. doi: 10.3201/eid1105.040959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez-Roche R, Alvarez M, Holmes EC, Bernardo L, Kouri G, Gould EA, Halstead S, Guzman MG. Dengue virus type 3, Cuba, 2000–2002. Emerg Infect Dis. 2005;11:773–774. doi: 10.3201/eid1105.040916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kochel T, Aguilar P, Felices V, Comach G, Cruz C, Alava A, Vargas J, Olson J, Blair P. Molecular epidemiology of dengue virus type 3 in northern South America: 2000–2005. Infect Genet Evol. 2008;8:682–688. doi: 10.1016/j.meegid.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Uzcategui NY, Comach G, Camacho D, Salcedo M, Cabello de Quintana M, Jimenez M, Sierra G, Cuello de Uzcategui R, James WS, Turner S, Holmes EC, Gould EA. Molecular epidemiology of dengue virus type 3 in Venezuela. J Gen Virol. 2003;84:1569–1575. doi: 10.1099/vir.0.18807-0. [DOI] [PubMed] [Google Scholar]
- 19.Miagostovich MP, dos Santos FB, de Simone TS, Costa EV, Filippis AM, Schatzmayr HG, Nogueira RM. Genetic characterization of dengue virus type 3 isolates in the State of Rio de Janeiro, 2001. Braz J Med Biol Res. 2002;35:869–872. doi: 10.1590/s0100-879x2002000800002. [DOI] [PubMed] [Google Scholar]
- 20.Nogueira RM, Schatzmayr HG, de Filippis AM, dos Santos FB, da Cunha RV, Coelho JO, de Souza LJ, Guimaraes FR, de Araujo ES, De Simone TS, Baran M, Teixeira G, Jr, Miagostovich MP. Dengue virus type 3, Brazil, 2002. Emerg Infect Dis. 2005;11:1376–1381. doi: 10.3201/eid1109.041043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz FJ, Farfan-Ale JA, Olson KE, Lorono-Pino MA, Gubler DJ, Blair CD, Black WC, 4th, Beaty BJ. Genetic variation within the premembrane coding region of dengue viruses from the Yucatan peninsula of Mexico. Am J Trop Med Hyg. 2002;67:93–101. doi: 10.4269/ajtmh.2002.67.93. [DOI] [PubMed] [Google Scholar]
- 22.Aquino VH, Anatriello E, Goncalves PF, DA Silva EV, Vasconcelos PF, Vieira DS, Batista WC, Bobadilla ML, Vazquez C, Moran M, Figueiredo LT. Molecular epidemiology of dengue type 3 virus in Brazil and Paraguay, 2002–2004. Am J Trop Med Hyg. 2006;75:710–715. [PubMed] [Google Scholar]
- 23.Barrero PR, Mistchenko AS. Genetic analysis of dengue virus type 3 isolated in Buenos Aires, Argentina. Virus Res. 2008;135:83–88. doi: 10.1016/j.virusres.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 24.Araujo JM, Nogueira RM, Schatzmayr HG, Zanotto PM, Bello G. Phylogeography and evolutionary history of dengue virus type 3. Infect Genet Evol. 2009;9:716–725. doi: 10.1016/j.meegid.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Bordignon-Nogueira M, Stella V, Bordignon J, Batista W, de Borba L, Pereira da Silva L, Hoffmann F, Probst C, Duarte dos Santos C. Evidence for the co-circulation of dengue virus type 3 genotypes III and V in the northern region of Brazil during the 2002–2004 epidemics. Mem Inst Oswaldo Cruz. 2008;103:483–488. doi: 10.1590/s0074-02762008000500013. [DOI] [PubMed] [Google Scholar]
- 26.Figueiredo L, Cecílio A, Portela Ferreira G, Drumond B, Germano de Oliveira J, Bonjardim C, Peregrino Ferreira P, Kroon E. Dengue virus 3 genotype 1 associated with dengue fever and dengue hemorrhagic fever, Brazil. Emerg Infect Dis. 2008;14:314–316. doi: 10.3201/eid1402.070278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Usme-Ciro JA, Mendez JA, Tenorio A, Rey GJ, Domingo C, Gallego-Gomez JC. Simultaneous circulation of genotypes I and III of dengue virus 3 in Colombia. Virol J. 2008;5:101. doi: 10.1186/1743-422X-5-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groot H. The reinvasion of Colombia by Aedes aegypti: aspects to remember. Am J Trop Med Hyg. 1980;29:330–338. doi: 10.4269/ajtmh.1980.29.330. [DOI] [PubMed] [Google Scholar]
- 29.Ocazionez RE, Cortes FM, Villar LA, Gomez SY. Temporal distribution of dengue virus serotypes in Colombian endemic area and dengue incidence. Re-introduction of dengue-3 associated to mild febrile illness and primary infection. Mem Inst Oswaldo Cruz. 2006;101:725–731. doi: 10.1590/s0074-02762006000700004. [DOI] [PubMed] [Google Scholar]
- 30.Diaz F, Ospina M, Higuita E, Osorio J. Molecular characterization of dengue viruses isolated in Medellín, Colombia. Meeting of the American Society of Tropical Medicine and Hygiene; Philadelphia, PA: 2007. [Google Scholar]
- 31.WHO . Dengue Haemorrhagic Fever: Diagnosis, Treatment, Prevention and Control. Geneva: WHO; 1999. [Google Scholar]
- 32.Worobey M, Rambaut A, Holmes EC. Widespread intra-serotype recombination in natural populations of dengue virus. Proc Natl Acad Sci USA. 1999;96:7352–7357. doi: 10.1073/pnas.96.13.7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shapiro B, Rambaut A, Drummond AJ. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol Biol Evol. 2006;23:7–9. doi: 10.1093/molbev/msj021. [DOI] [PubMed] [Google Scholar]
- 35.Kumar S, Tamura K, Nei M. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- 36.Wittke V, Robb TE, Thu HM, Nisalak A, Nimmannitya S, Kalayanrooj S, Vaughn DW, Endy TP, Holmes EC, Aaskov JG. Extinction and rapid emergence of strains of dengue 3 virus during an interepidemic period. Virology. 2002;301:148–156. doi: 10.1006/viro.2002.1549. [DOI] [PubMed] [Google Scholar]
- 37.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 38.Modis Y, Ogata S, Clements D, Harrison SC. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J Virol. 2005;79:1223–1231. doi: 10.1128/JVI.79.2.1223-1231.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Myat Thu H, Lowry K, Jiang L, Hlaing T, Holmes EC, Aaskov J. Lineage extinction and replacement in dengue type 1 virus populations are due to stochastic events rather than to natural selection. Virology. 2005;336:163–172. doi: 10.1016/j.virol.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 40.Thu HM, Lowry K, Myint TT, Shwe TN, Han AM, Khin KK, Thant KZ, Thein S, Aaskov J. Myanmar dengue outbreak associated with displacement of serotypes 2, 3, and 4 by dengue 1. Emerg Infect Dis. 2004;10:593–597. doi: 10.3201/eid1004.030216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miagostovich MP, dos Santos FB, Fumian TM, Guimaraes FR, da Costa EV, Tavares FN, Coelho JO, Nogueira RM. Complete genetic characterization of a Brazilian dengue virus type 3 strain isolated from a fatal outcome. Mem Inst Oswaldo Cruz. 2006;101:307–313. doi: 10.1590/s0074-02762006000300015. [DOI] [PubMed] [Google Scholar]