

Abstract
Changes in regional O2 tension that occur during fracture and skeletal unloading may stimulate local bone cell activity and ultimately regulate bone maintenance and repair. The mechanisms by which bone cells sense and respond to changes in O2 tension are unclear. In this study we investigated the effects of low O2 on activation of the hypoxia response element (HRE), prostaglandin E2 (PGE2) production, PGE2 receptor (EP) expression and proliferation in MC3T3-E1 osteoblastic cells. Cells were cultured for up to 72 hours in 2% O2 (considered hypoxic), 5% O2 (in the range of normal O2 tension in vivo) or 21% O2 (commonly used for cell culture). Cells cultured in 2% O2 showed activation of the HRE, increased PGE2 release, increased EP1 expression, and reduced cell proliferation compared to cells grown at 21% O2. Similarly, cells cultured in 5% O2 showed increased expression of EP1 and a trend toward a decrease in proliferation, but no activation of the HRE or increase in PGE2 levels. Expression of EP2, EP3 and EP4 were not affected by O2 tension. The differences in EP receptor profile observed in cells grown at 5% compared to 21% O2 suggest that bone cell phenotype may be altered under routine cell culture conditions. Furthermore, our data suggest that hypoxia-dependent PGE2 production and EP1 expression in bone cells may play a role in bone remodeling and repair in regions of compromised or damaged bone, where O2 tension is low.
Keywords: Osteoblast, Oxygen, Prostaglandin, Prostanoid receptor, Proliferation, Hypoxia, Hypoxia response element
Introduction
O2 tension in the environment surrounding bone cells can vary under certain circumstances. For example, skeletal unloading has been shown to induce osteocyte hypoxia (Dodd et al., 1999). In addition, bone cells in the vicinity of a fracture may experience a significant decrease in O2 tension due to disruption of the vasculature (Brighton and Krebs, 1972; Heppenstall et al., 1975). Regions of low O2 tension may also exist in the areas surrounding a microcrack due to a disruption of the lacunar-canalicular network and at the junction of graft and host bone tissue. Hypoxia can activate specific genes whose protein products mediate adaptive responses to O2 deprivation as well as those that potentiate O2 delivery (Semenza, 2002). For example, some cells up-regulate the expression of glycolytic enzymes and glucose transporters to facilitate a switch from oxidative phosphorylation to glycolysis (Semenza, 2000). In bone cells, hypoxia has been shown to up-regulate the expression of vascular endothelial growth factor (VEGF), a potent angiogenic factor (Steinbrech et al., 1999; Steinbrech et al., 2000), which facilitates new blood vessel formation and may potentiate O2 delivery. Hypoxia-dependent regulation of gene expression is believed to take place at the transcriptional level and to be mediated by hypoxia-sensitive transcription factors such as hypoxia-inducible factor-1α (HIF-1α) (Semenza, 1998). Under normoxic conditions, HIF-1α undergoes rapid ubiquitination and proteosomal degradation. Under hypoxic conditions, this degradation is inhibited, HIF-1α translocates to the nucleus, dimerizes with HIF-1β and binds to its consensus sequence within the hypoxia responsive element (HRE) (D’Angio and Finkelstein, 2000).
Hypoxia in most tissues is generally defined as between 1 and 2% O2, a condition under which HIF-1 is activated (Semenza, 2002). Less well defined is the normal range of O2 tension bone cells experience in vivo. O2 tension is around 13% (100mmHg) in arterial blood, approximately 6% (45mmHg) in mixed venous blood and considerably lower than 6% in most tissues (Vanderkooi et al., 1991; Vaupel et al., 1991). It is possible, therefore, that bone cells reside in O2 tensions of 6% or lower as their distance from the vasculature increases.
Although the data are contradictory, reduced O2 tension has been shown to have significant effects on bone cell physiology. For example, some studies have shown that reduced O2 tension leads to bone cell proliferation (Tuncay et al., 1994), while others have demonstrated decreased cell proliferation (Steinbrech et al., 1999; Utting et al., 2006), apoptosis (Chae et al., 2001) and decreased Runx2 expression (Park et al., 2002). In addition, little is known about the signaling pathways induced under hypoxic conditions that elicit such changes in bone cell activity. To gain insight into the signaling pathways involved in hypoxic regulation of bone cell activity, we chose to look at the relationship between hypoxia and prostaglandin E2 (PGE2) signaling. PGE2 is an important modulator of bone cell physiology and has been shown to be upregulated in response to hypoxia in other cells types (Michiels et al., 1993; Roszinski and Jelkmann, 1987). Interestingly, PGE2 levels have been shown to be elevated at a fracture site (Dekel et al., 1981) an area where O2 tension is significantly lowered (Brighton and Krebs, 1972; Heppenstall et al., 1975). Evidence suggests that under these circumstances, PGE2 and cyclooxygenase-2 are crucial for fracture repair (Simon et al., 2002; Zhang et al., 2002).
PGE2 has diverse effects on bone cell physiology, one of which includes functioning as an autocrine factor to regulate osteoblast proliferation and differentiation (Feyen et al., 1985; Fujimori et al., 1989; Igarashi et al., 1994). In addition, PGE2 has been shown to regulate osteoclastic bone resorption (Chambers, 1980; Kobayashi et al., 2005; Okuda et al., 1989). The specific effect of PGE2 on cellular functions is dependent on both the concentration of PGE2 available (Hakeda et al., 1985; Hakeda et al., 1986) and the type of prostanoid receptor expressed on the surface of bone cells. Prostanoid receptors include EP1, EP2, EP3 and EP4 (Coleman et al., 1994; Regan et al., 1994). MC3T3-E1 cells have been shown to express EP1, EP2 and EP4, while all 4 receptors were identified in an osteocytic cell line (MLO-Y4) (Cherian et al., 2003). In addition, EP1, EP2 and EP3 were expressed in rat calvariae (Kasugai et al., 1995 ). EP1 receptor activation has been shown to increase proliferation and reduce differentiation in MC3T3-E1 cells (Suda et al., 1996) and increase bone formation in rat tibiae (Tang et al., 2005). In contrast, EP4 receptor activation reduces proliferation and increases differentiation in osteoblast cells (Ito et al., 2006; Suda et al., 1996). In addition, the EP4 receptor may be critical during bone formation and resorption (Miyaura et al., 2000; Sakuma et al., 2000; Suzawa et al., 2000; Yoshida et al., 2002). The EP2 receptor has been shown to enhance osteoclastogenesis and bone resorption in vitro (Suzawa et al., 2000, Li et al., 2000). Although the EP3 receptor is expressed in bone tissue, its function in osteoblast cells remains to be determined.
To examine the relationship between hypoxia and PGE2 signaling in bone cells we looked at the effects of reducing O2 from an ambient tension of 21% (at which most cell culture is performed) to 5% and 2%, on PGE2 release, EP1 and EP4 expression, as well as cell proliferation. To confirm cellular hypoxia, we used pimonidazole hydrochloride, commercially available as Hypoxyprobe™-1. Hypoxyprobe™-1 forms protein adducts in cells at pO2<10mmHg (1.4% O2) that can be detected and visualized using immunocytochemistry. In addition, we also looked at the activity of HIF-1α, which activates gene transcription in hypoxic cells by binding to a specific HRE contained in those genes. We constructed an HRE-luciferase reporter plasmid and recorded luciferase activity in cells cultured at 2%, 5% and 21% O2.
Methods
Cell culture
MC3T3-E1, mouse osteoblastic cells (clone 14) were cultured at a seeding density of 10,000 cells/cm2 in 100mm Petri dishes with α-minimum essential medium (α-MEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (P/S). 20 hours after seeding, media was replaced with α-MEM containing 0.1% FBS and 1% P/S, supplemented with or without 100μM indomethacin treatement (a nonselective inhibitor of COX-1 and COX-2), pre-conditioned in 2%, 5% or 21% O2 tension for 24 hours. For ambient (21%) O2 tension experiments, cells were cultured in a standard humidified incubator at 37°C with a 95% air and 5% CO2 atmosphere. For reduced O2 tension experiments, cells were cultured in humidified incubators at 37°C with 5% CO2 and with O2 tension reduced to either 2% or 5% using supplemental N2 (HERAcell® 150, Kendro, USA.). Cells were removed from the incubators at specified time points, immediately placed on ice and both media and whole cell lysates were collected.
Detection of cellular hypoxia
Pimonidazole hydrochloride (Hypoxyprobe™-1, Chemicon International, Inc, Temecula, CA) was used to determine cellular hypoxia. At pO2<10mmHg (1.4% O2) Hypoxyprobe™-1 forms protein adducts in cells which can be detected and then visualized using a monoclonal antibody, Hypoxyprobe™-1-Mab1, covalently bound to FITC. To detect cellular hypoxia, cells were seeded onto 24mm glass coverslips placed within the wells of a 6-well plate, at a density of 100,000 cells per well and treated with media supplemented with or without 200μM Hypoxyprobe™-1, which had been pre-conditioned in 2%, 5% or 21% O2 for 24 hours. Cells were then placed into incubators set at 2%, 5% or 21% O2 respectively, for 3 hours after which they were removed and placed on ice. Each coverslip was washed 2 times with ice cold PBS, fixed in 90% methanol for 10 min at −20°C, and then blocked with 3% bovine serum albumin (BSA) in PBS for 2 hours. The coverslips were incubated with Hypoxyprobe™-1-Mab1 at a concentration of 1:200 in PBS for 45 min, washed in PBS, mounted onto glass slides and imaged using a Nikon E600 epifluorescence microscope with exposure times fixed for all slides. Cells not treated with pimonidazole were fixed and stained in the same manner and served as negative controls.
Hypoxia response element (HRE) reporter plasmid construct
A pGL3-HRE reporter plasmid vector was created by ligating the promoter region of human vascular endothelial growth factor (hVEGF) from −1005 to 379, which contains the HRE (AGCGTG), into the Kpn1-Mlu1 sites of pGL3-Basic vector (Promega, Madison, WI) that contains no promoter but a firefly luciferase (luc) coding sequence (Forsythe et al., 1996). The promoter sequence was confirmed by DNA sequencing.
Transient HRE expression assays
Cells were seeded into 24 well plates at a density of 85,000 cells/well and grown overnight. Cells were then co-transfected with pGL3-HRE DNA and a Renilla luciferase vector, pRL-TK (Promega, Madison, WI) using Lipofectamine™ 2000 and Optimem (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol, which has been previously used (You et al., 2002). The pRL-TK Vector was used as an internal control to normalize the difference in transfection efficiency. After 19 hours, media were replaced with α-MEM containing 0.1% FBS and 1% P&S that had been pre-conditioned in 2%, 5% or 21% O2 for 24 hours. Cells were then cultured at 2%, 5% or 21% O2, respectively for 24 hours. Cell lysates were collected and luciferase activity levels were measured using the Dual-Luciferase® Reporter Assay System (Promega Corp., Madison, WI) and a Turner Designs Model 20/20 luminometer. 100μM CoCl2, a known activator of HIF-1α (Wang and Semenza, 1993), was used as a positive control for HRE activation.
PGE2 release
Conditioned media were collected from cells cultured at 2%, 5% or 21% O2 for up to 72 hours. Media were centrifuged at 2000 RPM for 15 mins and PGE2 concentration in the supernatant was determined using a commercially available enzyme immunoassay system (GE Healthcare, Piscataway, NJ). PGE2 levels were normalized to total cell protein for each sample, and reported as a fold increase as compared to cells cultured in 21% O2 for 1 hour.
Western blotting
Up to 72 hours after exposing cells to different O2 tensions, total cell lysates were collected in RIPA buffer [1% Igepal CA-630, 0.5% sodium deoxycholate (Sigma –Aldrich, St. Louis, MO) and 0.1% SDS (Bio-Rad, Hercules CA) in PBS]. Protein concentration was determined by the Lowry method using the DC Protein Assay (Bio-Rad, Hercules, CA). Equal amounts of protein (20μg) were loaded into SDS-polyacrylamide gels, resolved by electrophoresis and transferred to nitrocellulose membranes that were blocked in 5% non-fat milk in Tris-buffered saline with 0.05% Tween-20 (TBST) prior to antibody incubation. Ram seminal vesicle microsomes were used as a positive control for EP2 detection, as suggested by the manufacturer (Cayman Chemicals, Ann Arbor, MI). Whole cell lysate from a kidney cell line (293) was used as a positive control for EP3 detection since high levels of EP3 receptor have been detected in kidney cells(Kotani et al., 1995).
For detection of EP receptors, membranes were incubated overnight at 4 °C with polyclonal antibodies against the respective proteins (Cayman Chemicals, Ann Arbor, MI). Following three washes in TBST, the membranes were incubated with appropriate secondary antibodies linked to horseradish peroxidase (HRP) for 1 hour (Jackson ImmunoResearch Laboratories, West Grove, PA) at 1:10,000 dilution. After washing in TBST and then PBS, the membranes were soaked in ECL detection reagents for 1 min (Amersham, UK) and then exposed to X-ray film. To confirm equal loading of protein, the membranes were stripped and probed for expression of β-actin (anti-β-actin mAb, 1:5,000, Abcam, Cambridge, MA), the cellular levels of which have been shown to be unaffected by O2 tension (Zhong and Simons, 1999).
Proliferation assay
Cellular proliferation was determined using a bromodeoxyuridine (BrdU) flow kit (BD Biosciences, San Diego, CA). Cells were treated with reduced serum media (0.1% FBS), or reduced serum media supplemented with 5μM 17-phenyl trinor PGE2 (an EP1 agonist), or 10μM AH6809 (an EP1 antagonist) (Cayman Chemicals, Ann Arbor, MI), or DMSO as a vehicle control and exposed to 2%, 5% and 21% O2. After 21 hours, cells were treated with 10μM BrdU for 3 hours. Cells were collected and stained for BrdU incorporation using an anti-BrdU FITC-conjugated antibody, and total DNA content was stained with 5μl 7-AAD (as supplied) followed by analysis using FACScan according to the manufacturer’s instructions.
Statistical analysis
To compare luciferase activity between O2 tensions, a one-way analysis of variance (ANOVA) was used with a Dunn’s multiple comparisons post-hoc test. To compare PGE2 levels and proliferation rates between O2 tensions, unpaired t-tests were performed.
Results
Detection and confirmation of cellular hypoxia
In order to determine whether MC3T3-E1 cells were experiencing and responding to cellular hypoxia (<1.4% O2), we used an immunofluorescent staining technique, Hypoxyprobe™-1, and a luciferase reporter assay, respectively. Immunofluorescent staining was evident in cells cultured at 2% O2 for 3 hours (Figure 1), which indicates the presence of Hypoxyprobe™-1 adducts and therefore a pO2<10mmHg (<1.4% O2). Fluorescent staining was not different from control (no Hypoxyprobe™-1) in cells cultured at 5% and 21% O2. HRE-luciferase activity levels in MC3T3-E1 cells cultured at 2% O2 were significantly increased compared to those measured at both 5% and 21% O2 (Figure 2). These data confirm that MC3T3-E1 cells cultured at 2% O2 were experiencing hypoxia during the course of the experiments. Our data suggest that at 2% O2, HIF-1α is stabilized in MC3T3-E1 cells, translocates to the nucleus and binds to the HRE in target genes. In addition, treatment of cells with CoCl2 at 21% O2 also increased luciferase activity levels when compared to cells grown at 2%, 5% and 21% O2. This validated our experimental system, as CoCl2 is a known activator of HIF-1α (Wang and Semenza, 1993).
Figure 1.
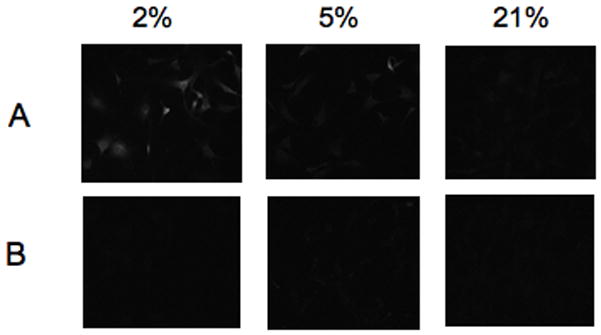
Fluorescent immunocytochemical staining for hypoxia (Hypoxyprobe™-1 adducts) in MC3T3- E1 cells cultured for 3 hours in 2%, 5% or 21% O2. Cells were cultured with (A) or without (B) 200 μM Hypoxyprobe™-1. Fluorescent staining indicates the presence of Hypoxyprobe™-1 adducts which form at pO2<10mmHg (1.4% O2).
Figure 2.

Effect of hypoxia on expression of pGL3-HRE vector. MC3T3-E1 cells were cultured for 24 hours at 2%, 5% or 21% O2 after which luciferase activity levels were measured. Some cells were treated with 100μM CoCl2 as a positive control for HRE activation. Bars represent mean luciferase activity ± SE, expressed in relative luciferase units (RLU) and normalized to a Renilla luciferase internal control. * Represents a statistically significant difference from 2% O2, p < 0.05, n=4.
Effects of hypoxia on PGE2 levels in MC3T3-E1 cells
PGE2, an important modulator of bone cell physiology, has been shown to be upregulated in response to hypoxia in other cell types and is elevated in the reduced O2 environment of a fracture site. In this study, as predicted, PGE2 levels in media collected from cells grown at 2% O2 were significantly elevated over those from cells cultured at 5% and 21% O2 at 24 hours (Figure 3). PGE2 levels remained significantly elevated for 48 hours. There was no detectable difference in PGE2 levels between cells cultured at 5% and 21% O2 at any time point. Treatment of cells with 100μM indomethacin (a nonselective inhibitor of COX-1 and COX-2) inhibited PGE2 release at all O2 tensions and all time points. Our data show that PGE2 levels are elevated in bone cells in response to hypoxia. We propose that PGE2 may act locally to modulate bone cell physiology and aid in remodeling and repair in regions of damaged bone.
Figure 3.

Effects of hypoxia on PGE2 levels in MC3T3- E1 cells cultured for up to 72 hours in 2%, 5% or 21% O2. PGE2 levels were normalized to total cell protein for each sample. Bars represent mean fold increase in PGE2 levels ± SE, as compared to cells cultured in 21% O2 for 1hour. * Represents a statistically significant difference from 2% O2 at the same time point, p < 0.05, n=4.
Effects of hypoxia on expression of prostanoid receptors in MC3T3-E1 cells
PGE2 exerts its effects through stimulation of specific cell surface EP receptors, EP1–4. Our data show that the expression of EP1 was similar at all O2 tensions at 6 hours (Figure 4A). However, after 24 hours, EP1 expression increased in cells cultured at 2% and 5% O2 and remained elevated for up to 72 hours. EP1 expression at 2% O2 was consistently higher than at 5% between 24 and 72 hours. EP1 expression in cells cultured at 21% O2 did not change over time. This response is not specific to the MC3T3-E1 cell line. Similar increases in EP1 expression were observed in human osteoblastic cells (MG63) cultured at 2% and 5% O2, at 24 and 48 hours. Expression of EP4 was similar at all O2 tensions and did not change with time in culture (Figure 5A). Treatment with indomethacin had no effect at any O2 tension or time point on either EP1 or EP4 expression (Figure 4B and 5B). Interestingly, we did detect expression of EP2 and EP3 in MC3T3-E1 cells, but levels were similar at all O2 tensions, and also did not change with time in culture (Figure 6). EP3 protein was detected at 65 and 52 kDa which indicates a variation in the degree of post translational modification of the receptor. Changes in the expression level of EP1 which has been shown to be involved in auto-amplification of PGE2 production, may play a role in enhancing or prolonging the action of PGE2 under hypoxic conditions.
Figure 4.

Representative western blots for EP1 protein levels in MC3T3-E1 cells. A. EP1 protein in cells cultured for up to 72 hours in 2%, 5% or 21% O2. B. EP1 protein levels in cells cultured for up to 72 hours in 2%, 5% or 21% O2 in the presence of 100μM indomethacin. β-actin was used as a loading control, n=3.
Figure 5.

Representative western blots for EP4 protein levels in MC3T3-E1 cells. A. EP4 protein in cells cultured for up to 72 hours in 2%, 5% or 21% O2. B. EP4 protein levels in cells cultured for up to 72 hours in 2%, 5% or 21% O2 in the presence of 100μM indomethacin. β-actin was used as a loading control, n=3.
Figure 6.
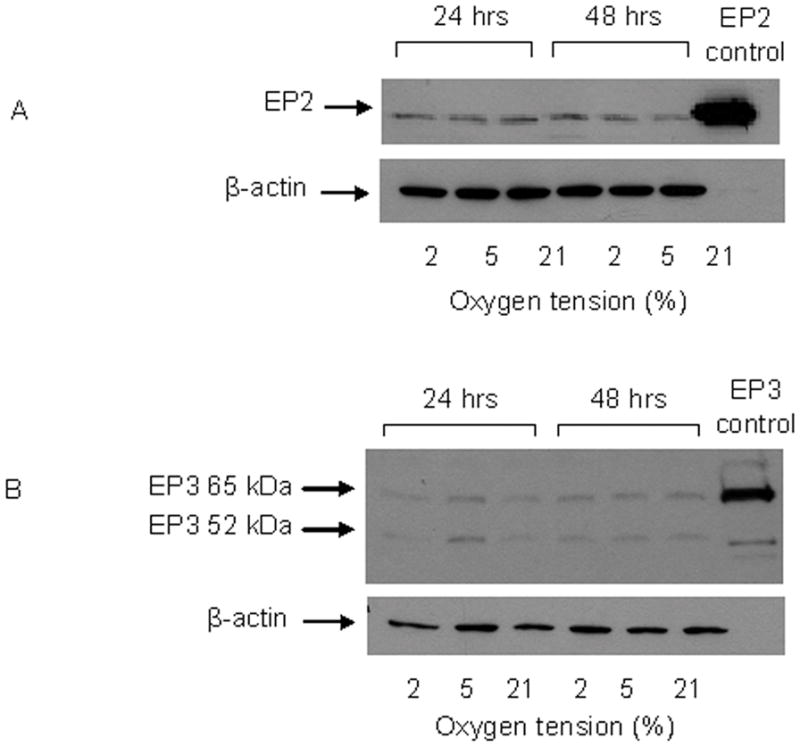
Representative western blots for EP2 and EP3 protein levels in MC3T3-E1 cells. A. EP2 protein levels in cells cultured for 24 and 48 hours in 2%, 5% or 21% O2. Ram seminal vesicle microsomes were used as a positive control for EP2 as suggested by the manufacturer. B. EP3 protein levels in cells cultured for 24 and 48 hours in 2%, 5% or 21% O2. Whole cell lysate from kidney 293 cells was used as a positive control for EP3 detection. β-actin was used as a loading control, n=2.
Effects of hypoxia on MC3T3-E1 proliferation
PGE2, acting through EP receptors has diverse effects on bone cell physiology including modulation of osteoblast proliferation and differentiation. Specifically, in MC3T3-E1 cells, activation of EP1 has been shown to lead to increased proliferation. Interestingly, in our studies, proliferation was significantly inhibited by over 40% in MC3T3-E1 cells cultured at 2% O2 when compared to those cultured at 21% O2 (Figure 7). There was also a trend for a decreased proliferation rate in 5% O2. Neither 17-phenyl trinorPGE2 (an EP1 agonist) nor AH6809 (an EP1 antagonist) had an effect on proliferation rate at any O2 tension. These data suggest that lowering O2 tension inhibits proliferation in osteoblasts. Furthermore, PGE2 acting through EP1 does not seem to be responsible for this decrease in proliferation.
Figure 7.

Effects of hypoxia on cell proliferation. MC3T3-E1 cells were cultured in 2, 5 or 21% O2 for 24 hours with or without 5μM 17-phenyl trinor PGE2, 10μM AH6809, or DMSO vehicle control. BrdU was allowed to incorporate into the DNA of proliferating cells for 3 hours and then detected using FACScan. Bars represent mean percent of cells with BrdU incorporation normalized to the percent incorporation in 21% O2 ± SE. *represents statistically significant difference compared to 2%, p<0.05, n=4.
Discussion
Our data demonstrate that osteoblasts can sense and respond to decreases in O2 tension. In this study we looked at the effect of lowering O2 tension from 21%, at which most cell culture is performed, to 2%, which is generally considered hypoxic for cells, and 5%, which is within the range of normal O2 tensions measured in a variety of body tissues in vivo (Vanderkooi et al., 1991). We confirmed that osteoblastic cells experienced and responded to hypoxia by examining the effects of hypoxia on expression of an HRE-luciferase reporter construct. Our data showed that luciferase activity levels in cells cultured at 2% O2 were significantly increased compared to those measured at both 5% and 21% O2. This suggests that at 2% O2 HIF-1α translocates to the nucleus and binds to the HRE. Cellular hypoxia was further confirmed by visualizing Hypoxyprobe™-1 adducts that have been shown to form at cellular O2 tensions of <1.4%.
Our data suggest that PGE2 levels are elevated in bone cells in response to hypoxia. Similar increases in PGE2 have been observed in renal mesangial cells exposed to approximately 1–2% O2 (Kurtz et al., 1985; Roszinski and Jelkmann, 1987). In human endothelial cells exposed to anoxia there was a 5-fold increase in PGE2 levels after 2 hours and a concomitant increase in activation of PLA2 (Michiels et al., 1993). While it has been shown in vivo that PGE2 levels are increased in the tibiae and surrounding tissues 1–14 days after fracture (Dekel et al., 1981), a situation in which hypoxia ensues due to disruption of the vasculature (Brighton and Krebs, 1972), ours may be the first study to show hypoxia-induced PGE2 production in bone cells in vitro. Interestingly, fracture healing has been shown to fail in COX-2 knockout mice (Simon et al., 2002). Furthermore, both specific and non-specific COX enzyme inhibitors have been shown to have deleterious effects on fracture healing in animal models (Seidenberg and An, 2004). Taken together, these data suggest that under certain circumstances, hypoxia may stimulate the production of PGE2 to aid in bone repair and remodeling.
Ultimately, PGE2 exerts its effects through stimulation of specific cell surface EP receptors. Our studies show that all EP receptors are expressed by MC3T3-E1 cells, and that expression levels of EP2, EP3 and EP4 were insensitive to changes in O2 tension. In contrast, EP1 receptor expression was strongly elevated in cells cultured at 2% and 5% O2 compared to 21% O2, with expression consistently highest in 2% O2 and occurring in parallel with the hypoxia-stimulated increase in PGE2. To our knowledge this is the first study to show that hypoxia can modulate EP receptor expression. The role of the EP1 receptor under hypoxic conditions and in bone biology in general is unclear. An EP1 agonist was shown to enhance expression of fibronectin and α5β1 integrin in rat primary osteoblasts (Tang et al., 2005). In the same study local administration of PGE2 or the EP1 agonist into the metaphysis of the tibia increased fibronectin andα5β1 integrin immunostaining and promoted bone formation (Tang et al., 2005). This contradicts evidence to suggest that stimulation of the EP1 receptor in MC3T3-E1 cells enhances proliferation, but suppresses differentiation (Suda et al., 1996). In our study we observed a significant decrease in cell proliferation in MC3T3-E1 cells cultured at 2% O2 when compared to those cultured at 21%. Furthermore, neither the EP1 antagonist or agonist had any effect on cell proliferation at any O2 tension. Importantly, the EP1 receptor has been shown to be involved in auto-amplification of PGE2 production (Suda et al., 1998). The EP1 receptor may, therefore, play a role in enhancing and prolonging the action of PGE2 under hypoxic conditions, for example during fracture healing.
In this study we show that lowering O2 tension significantly inhibits osteoblast proliferation. This supports previous studies which showed that lowering O2 tension to around 5% inhibited proliferation in MC3T3-E1 cells for up to 5 days (Steinbrech et al., 1999). In addition, in primary rat osteoblasts a decrease in proliferation was noted in cells cultured for between 6 and 18 days in 2% O2 (Utting et al., 2006). We also showed that proliferation rates were not affected by an EP1 receptor agonist and antagonist. These data suggest that PGE2 acting through EP1 is not responsible for the decrease in osteoblast proliferation. Indeed, PGE2 has been shown in many studies to stimulate osteoblast proliferation (Conconi et al., 2002; Frost et al., 1997). Furthermore an EP1 receptor agonist has been shown to increase proliferation in MC3T3-E1 cells (Suda et al., 1996). It is possible then, that the increase in PGE2 levels and EP1 receptor expression in osteoblasts in response to hypoxia might be an attempt by the cells to enhance impaired proliferation rates.
The O2 concentration in bone tissue in vivo is unknown, however, mean tissue levels of O2 generally fall between 1 and 9%, most being at the lower end of this range (Vanderkooi et al., 1991). It is interesting to note that lowering O2 tension to a more physiologically relevant level of 5% also had effects on bone cell phenotype. While there was no induction of PGE2 levels in 5% O2, EP1 receptor expression was increased and there was a trend for a decrease in cell proliferation rates. The fact that PGE2 levels were not affected suggests that 5% O2 is not adequate to induce hypoxic stress. This is confirmed by HRE-luciferase data in Figure 2 and the absence of Hypoxyprobe™ staining in Figure 1. However, the up-regulation of EP1 receptor levels and the decrease in proliferation do seem to suggest that under these conditions that the phenotype of bone cells may be altered. O2 is a powerful regulator and there are many examples of how cell physiology is significantly altered by changing the O2 tension from a potentially hyperoxic level of O2 (21%) which is routinely used in cell culture incubators, to a lower but more physiologic O2 tensions. For example, it has been shown that O2 levels are critical for stem cell proliferation, differentiation, apoptosis and migration (Csete, 2005). The reasons for this are unclear, however, it is possible that the regulation of cell processes observed at different O2 tensions may be due to generation of more or less reactive oxygen species (ROS) (Csete, 2005). ROS can harm cells by causing oxidative damage to DNA, protein and lipids, and as such, have been implicated in an array of disease states including osteoporosis, rheumatoid arthritis and aging studies. It has been shown that there is a reduction in ROS generated when O2 levels are lowered in culture (Csete, 2005). Using dichlorofluorescein, a fluorescent probe for ROS, we have observed that ROS levels in MC3T3-E1 cells exposed to 2% O2 for 24 hrs are reduced compared to cells cultured in 21% O2 (Lee and Yellowley unpublished data). Taken together, the change in the receptor profile and cell proliferation in MC3T3-E1 cells grown at 5% O2 compared to 21% O2 emphasizes a need for further investigation of normal cell behavior at more physiologic levels of O2.
Acknowledgments
Contract grant sponsor: National Institute of Aging
Contract grant number: R01 AG022305-0551
Contract grant sponsor: National Institute of Arthritis, Musculoskeletal and Skin Diseases
Contract grant number: F31 AR053467-01
We would like to thank Dr. Julie Baumber and Dr. Stuart Meyers for their assistance with flow cytometry for this study. This work was supported by the National Institute of Aging R01 AG022305-0551 (CEY) and a National Institute of Arthritis, Musculoskeletal and Skin Diseases National Research Service Award F31 AR053467-01 (CML).
References
- Brighton CT, Krebs AG. Oxygen tension of healing fractures in the rabbit. J Bone Joint Surg Am. 1972;54(2):323–332. [PubMed] [Google Scholar]
- Chae HJ, Kim SC, Han KS, Chae SW, An NH, Kim HM, Kim HH, Lee ZH, Kim HR. Hypoxia induces apoptosis by caspase activation accompanying cytochrome C release from mitochondria in MC3T3E1 osteoblasts. p38 MAPK is related in hypoxia-induced apoptosis. Immunopharmacol Immunotoxicol. 2001;23(2):133–152. doi: 10.1081/iph-100103855. [DOI] [PubMed] [Google Scholar]
- Chambers TJ. The cellular basis of bone resorption. Clin Orthop Relat Res. 1980;(151):283–293. [PubMed] [Google Scholar]
- Cherian PP, Cheng B, Gu S, Sprague E, Bonewald LF, Jiang JX. Effects of mechanical strain on the function of Gap junctions in osteocytes are mediated through the prostaglandin EP2 receptor. J Biol Chem. 2003;278(44):43146–43156. doi: 10.1074/jbc.M302993200. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46(2):205–229. [PubMed] [Google Scholar]
- Conconi MT, Tommasini M, Baiguera S, De Coppi P, Parnigotto PP, Nussdorfer GG. Effects of prostaglandins E1 and E2 on the growth and differentiation of osteoblast-like cells cultured in vitro. Int J Mol Med. 2002;10(4):451–456. [PubMed] [Google Scholar]
- Csete M. Oxygen in the cultivation of stem cells. Ann N Y Acad Sci. 2005;1049:1–8. doi: 10.1196/annals.1334.001. [DOI] [PubMed] [Google Scholar]
- D’Angio CT, Finkelstein JN. Oxygen regulation of gene expression: a study in opposites. Mol Genet Metab. 2000;71(1–2):371–380. doi: 10.1006/mgme.2000.3074. [DOI] [PubMed] [Google Scholar]
- Dekel S, Lenthall G, Francis MJ. Release of prostaglandins from bone and muscle after tibial fracture. An experimental study in rabbits. J Bone Joint Surg Br. 1981;63-B(2):185–189. doi: 10.1302/0301-620X.63B2.7217139. [DOI] [PubMed] [Google Scholar]
- Dodd JS, Raleigh JA, Gross TS. Osteocyte hypoxia: a novel mechanotransduction pathway. Am J Physiol. 1999;277(3 Pt 1):C598–602. doi: 10.1152/ajpcell.1999.277.3.C598. [DOI] [PubMed] [Google Scholar]
- Feyen JH, Di Bon A, van der Plas A, Lowik CW, Nijweide PJ. Effects of exogenous prostanoids on the proliferation of osteoblast-like cells in vitro. Prostaglandins. 1985;30(5):827–840. doi: 10.1016/0090-6980(85)90011-5. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16(9):4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost A, Jonsson KB, Nilsson O, Ljunggren O. Inflammatory cytokines regulate proliferation of cultured human osteoblasts. Acta Orthop Scand. 1997;68(2):91–96. doi: 10.3109/17453679709003987. [DOI] [PubMed] [Google Scholar]
- Fujimori A, Tsutsumi M, Fukase M, Fujita T. Cyclooxygenase inhibitors enhance cell growth in an osteoblastic cell line, MC3T3-E1. J Bone Miner Res. 1989;4(5):697–704. doi: 10.1002/jbmr.5650040508. [DOI] [PubMed] [Google Scholar]
- Hakeda Y, Ikeda E, Kurihara N, Nakatani Y, Maeda N, Kumegawa M. Induction of osteoblastic cell differentiation by forskolin. Stimulation of cyclic AMP production and alkaline phosphatase activity. Biochim Biophys Acta. 1985;838(1):49–53. doi: 10.1016/0304-4165(85)90248-x. [DOI] [PubMed] [Google Scholar]
- Hakeda Y, Yoshino T, Natakani Y, Kurihara N, Maeda N, Kumegawa M. Prostaglandin E2 stimulates DNA synthesis by a cyclic AMP-independent pathway in osteoblastic clone MC3T3-E1 cells. J Cell Physiol. 1986;128(2):155–161. doi: 10.1002/jcp.1041280204. [DOI] [PubMed] [Google Scholar]
- Heppenstall RB, Grislis G, Hunt TK. Tissue gas tensions and oxygen consumption in healing bone defects. Clin Orthop Relat Res. 1975;(106):357–365. doi: 10.1097/00003086-197501000-00048. [DOI] [PubMed] [Google Scholar]
- Igarashi K, Hirafuji M, Adachi H, Shinoda H, Mitani H. Role of endogenous PGE2 in osteoblastic functions of a clonal osteoblast-like cell, MC3T3-E1. Prostaglandins Leukot Essent Fatty Acids. 1994;50(4):169–172. doi: 10.1016/0952-3278(94)90140-6. [DOI] [PubMed] [Google Scholar]
- Ito M, Nakayama K, Konaka A, Sakata K, Ikeda K, Maruyama T. Effects of a prostaglandin EP4 agonist, ONO-4819, and risedronate on trabecular microstructure and bone strength in mature ovariectomized rats. Bone. 2006 doi: 10.1016/j.bone.2006.02.054. [DOI] [PubMed] [Google Scholar]
- Kasugai S, Oida S, Iimura T, Arai N, Takeda K, Ohya K, Sasaki S. Expression of prostaglandin E receptor subtypes in bone: expression of EP2 in bone development. Bone. 1995;17(1):1–4. doi: 10.1016/8756-3282(95)00134-y. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Mizoguchi T, Take I, Kurihara S, Udagawa N, Takahashi N. Prostaglandin E2 enhances osteoclastic differentiation of precursor cells through protein kinase A-dependent phosphorylation of TAK1. J Biol Chem. 2005;280(12):11395–11403. doi: 10.1074/jbc.M411189200. [DOI] [PubMed] [Google Scholar]
- Kotani M, Tanaka I, Ogawa Y, Usui T, Mori K, Ichikawa A, Narumiya S, Yoshimi T, Nakao K. Molecular cloning and expression of multiple isoforms of human prostaglandin E receptor EP3 subtype generated by alternative messenger RNA splicing: multiple second messenger systems and tissue-specific distributions. Mol Pharmacol. 1995;48(5):869–879. [PubMed] [Google Scholar]
- Kurtz A, Jelkmann W, Pfeilschifter J, Bauer C. Role of prostaglandins in hypoxia-stimulated erythropoietin production. Am J Physiol. 1985;249(1 Pt 1):C3–8. doi: 10.1152/ajpcell.1985.249.1.C3. [DOI] [PubMed] [Google Scholar]
- Li X, Okada Y, Pilbeam CC, Lorenzo JA, Kennedy CR, Breyer RM, Raisz LG. Knockout of the murine prostaglandin EP2 receptor impairs osteoclastogenesis in vitro. Endocrinology. 2000;141(6):2054–2061. doi: 10.1210/endo.141.6.7518. [DOI] [PubMed] [Google Scholar]
- Michiels C, Arnould T, Knott I, Dieu M, Remacle J. Stimulation of prostaglandin synthesis by human endothelial cells exposed to hypoxia. Am J Physiol. 1993;264(4 Pt 1):C866–874. doi: 10.1152/ajpcell.1993.264.4.C866. [DOI] [PubMed] [Google Scholar]
- Miyaura C, Inada M, Suzawa T, Sugimoto Y, Ushikubi F, Ichikawa A, Narumiya S, Suda T. Impaired bone resorption to prostaglandin E2 in prostaglandin E receptor EP4-knockout mice. J Biol Chem. 2000;275(26):19819–19823. doi: 10.1074/jbc.M002079200. [DOI] [PubMed] [Google Scholar]
- Okuda A, Taylor LM, Heersche JN. Prostaglandin E2 initially inhibits and then stimulates bone resorption in isolated rabbit osteoclast cultures. Bone Miner. 1989;7(3):255–266. doi: 10.1016/0169-6009(89)90082-2. [DOI] [PubMed] [Google Scholar]
- Park JH, Park BH, Kim HK, Park TS, Baek HS. Hypoxia decreases Runx2/Cbfa1 expression in human osteoblast-like cells. Mol Cell Endocrinol. 2002;192(1–2):197–203. doi: 10.1016/s0303-7207(02)00036-9. [DOI] [PubMed] [Google Scholar]
- Regan JW, Bailey TJ, Pepperl DJ, Pierce KL, Bogardus AM, Donello JE, Fairbairn CE, Kedzie KM, Woodward DF, Gil DW. Cloning of a novel human prostaglandin receptor with characteristics of the pharmacologically defined EP2 subtype. Mol Pharmacol. 1994;46(2):213–220. [PubMed] [Google Scholar]
- Roszinski S, Jelkmann W. Effect of PO2 on prostaglandin E2 production in renal cell cultures. Respir Physiol. 1987;70(2):131–141. doi: 10.1016/0034-5687(87)90045-4. [DOI] [PubMed] [Google Scholar]
- Sakuma Y, Tanaka K, Suda M, Komatsu Y, Yasoda A, Miura M, Ozasa A, Narumiya S, Sugimoto Y, Ichikawa A, Ushikubi F, Nakao K. Impaired bone resorption by lipopolysaccharide in vivo in mice deficient in the prostaglandin E receptor EP4 subtype. Infect Immun. 2000;68(12):6819–6825. doi: 10.1128/iai.68.12.6819-6825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidenberg AB, An YH. Is there an inhibitory effect of COX-2 inhibitors on bone healing? Pharmacol Res. 2004;50(2):151–156. doi: 10.1016/j.phrs.2003.12.017. [DOI] [PubMed] [Google Scholar]
- Semenza G. Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol. 2002;64(5–6):993–998. doi: 10.1016/s0006-2952(02)01168-1. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev. 1998;8(5):588–594. doi: 10.1016/s0959-437x(98)80016-6. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88(4):1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- Simon AM, Manigrasso MB, O’Connor JP. Cyclo-oxygenase 2 function is essential for bone fracture healing. J Bone Miner Res. 2002;17(6):963–976. doi: 10.1359/jbmr.2002.17.6.963. [DOI] [PubMed] [Google Scholar]
- Steinbrech DS, Mehrara BJ, Saadeh PB, Chin G, Dudziak ME, Gerrets RP, Gittes GK, Longaker MT. Hypoxia regulates VEGF expression and cellular proliferation by osteoblasts in vitro. Plast Reconstr Surg. 1999;104(3):738–747. doi: 10.1097/00006534-199909030-00019. [DOI] [PubMed] [Google Scholar]
- Steinbrech DS, Mehrara BJ, Saadeh PB, Greenwald JA, Spector JA, Gittes GK, Longaker MT. VEGF expression in an osteoblast-like cell line is regulated by a hypoxia response mechanism. Am J Physiol Cell Physiol. 2000;278(4):C853–860. doi: 10.1152/ajpcell.2000.278.4.C853. [DOI] [PubMed] [Google Scholar]
- Suda M, Tanaka K, Natsui K, Usui T, Tanaka I, Fukushima M, Shigeno C, Konishi J, Narumiya S, Ichikawa A, Nakao N. Prostaglandin E receptor subtypes in mouse osteoblastic cell line. Endocrinology. 1996;137(5):1698–1705. doi: 10.1210/endo.137.5.8612504. [DOI] [PubMed] [Google Scholar]
- Suda M, Tanaka K, Yasoda A, Natsui K, Sakuma Y, Tanaka I, Ushikubi F, Narumiya S, Nakao K. Prostaglandin E2 (PGE2) autoamplifies its production through EP1 subtype of PGE receptor in mouse osteoblastic MC3T3-E1 cells. Calcif Tissue Int. 1998;62(4):327–331. doi: 10.1007/s002239900440. [DOI] [PubMed] [Google Scholar]
- Suzawa T, Miyaura C, Inada M, Maruyama T, Sugimoto Y, Ushikubi F, Ichikawa A, Narumiya S, Suda T. The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology. 2000;141(4):1554–1559. doi: 10.1210/endo.141.4.7405. [DOI] [PubMed] [Google Scholar]
- Tang CH, Yang RS, Fu WM. Prostaglandin E2 stimulates fibronectin expression through EP1 receptor, phospholipase C, protein kinase Calpha, and c-Src pathway in primary cultured rat osteoblasts. J Biol Chem. 2005;280(24):22907–22916. doi: 10.1074/jbc.M500130200. [DOI] [PubMed] [Google Scholar]
- Tuncay OC, Ho D, Barker MK. Oxygen tension regulates osteoblast function. Am J Orthod Dentofacial Orthop. 1994;105(5):457–463. doi: 10.1016/S0889-5406(94)70006-0. [DOI] [PubMed] [Google Scholar]
- Utting JC, Robins SP, Brandao-Burch A, Orriss IR, Behar J, Arnett TR. Hypoxia inhibits the growth, differentiation and bone-forming capacity of rat osteoblasts. Exp Cell Res. 2006;312(10):1693–1702. doi: 10.1016/j.yexcr.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Vanderkooi JM, Erecinska M, Silver IA. Oxygen in mammalian tissue: methods of measurement and affinities of various reactions. Am J Physiol. 1991;260(6 Pt 1):C1131–1150. doi: 10.1152/ajpcell.1991.260.6.C1131. [DOI] [PubMed] [Google Scholar]
- Vaupel P, Schlenger K, Knoop C, Hockel M. Oxygenation of human tumors: evaluation of tissue oxygen distribution in breast cancers by computerized O2 tension measurements. Cancer Res. 1991;51(12):3316–3322. [PubMed] [Google Scholar]
- Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993;90(9):4304–4308. doi: 10.1073/pnas.90.9.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Oida H, Kobayashi T, Maruyama T, Tanaka M, Katayama T, Yamaguchi K, Segi E, Tsuboyama T, Matsushita M, Ito K, Ito Y, Sugimoto Y, Ushikubi F, Ohuchida S, Kondo K, Nakamura T, Narumiya S. Stimulation of bone formation and prevention of bone loss by prostaglandin E EP4 receptor activation. Proc Natl Acad Sci U S A. 2002;99(7):4580–4585. doi: 10.1073/pnas.062053399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Madrid LV, Saims D, Sedivy J, Wang CY. c-Myc sensitizes cells to tumor necrosis factor-mediated apoptosis by inhibiting nuclear factor kappa B transactivation. J Biol Chem. 2002;277(39):36671–36677. doi: 10.1074/jbc.M203213200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Schwarz EM, Young DA, Puzas JE, Rosier RN, O’Keefe RJ. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J Clin Invest. 2002;109(11):1405–1415. doi: 10.1172/JCI15681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Simons JW. Direct comparison of GAPDH, beta-actin, cyclophilin, and 28S rRNA as internal standards for quantifying RNA levels under hypoxia. Biochem Biophys Res Commun. 1999;259(3):523–526. doi: 10.1006/bbrc.1999.0815. [DOI] [PubMed] [Google Scholar]