

Abstract
Substitution mutations in the BRAF serine/threonine kinase are found in a variety of human cancers. Such mutations occur in ∼70% of human malignant melanomas, and a single hyperactivating V600E mutation is found in the activation segment of the kinase domain and accounts for more than 90% of these mutations. Given this correlation, the molecular mechanism for BRAF regulation as well as oncogenic activation has attracted considerable interest, and activated forms of BRAF, such as BRAFV600E, have become attractive targets for small molecule inhibition. Here we report on the identification and subsequent optimization of a potent BRAF inhibitor, CS292, based on an organometallic kinase inhibitor scaffold. A cocrystal structure of CS292 in complex with the BRAF kinase domain reveals that CS292 binds to the ATP binding pocket of the kinase and is an ATP competitive inhibitor. The structure of the kinase–inhibitor complex also demonstrates that CS292 binds to BRAF in an active conformation and suggests a mechanism for regulation of BRAF by phosphorylation and BRAFV600E oncogene-induced activation. The structure of CS292 bound to the active form of the BRAF kinase also provides a novel scaffold for the design of BRAFV600E oncogene selective BRAF inhibitors for therapeutic application.
RAF1 kinases were originally identified as cellular homologues of v-raf oncogenes acquired by retroviruses and contain three members: CRAF (RAF-1 or c-RAF-1), BRAF, and ARAF (1–3). RAF family kinases are central players in the highly conserved mitogen-activated protein kinase (MAPK) signaling pathway (RAS-RAF-MEK-ERK) which relays signals from the extracellular space through receptor tyrosine kinases (RTKs) to the nucleus to promote the expression of genes involved in cell proliferation and survival. RAF kinases function by specifically phosphorylating MEK1/2 within the kinase activation loop leading to the subsequent activation of MEK1/2, which in turn activates ERK1/2. Activated ERK1/2 translocates into the nucleus and activates transcription factors to promote cellular outcomes, including survival, growth, proliferation, and differentiation (4). RAF family kinases are subject to very complex mechanisms of regulation from a variety of different protein kinases and scaffolding proteins (4). Among the RAF isoforms, BRAF differs significantly from CRAF and ARAF because it requires fewer regulatory events for activation (4). BRAF has also been shown to be the major activator of MEK1/2, and it possesses a significantly higher level of basal activity compared to the other RAF isoforms (5–10). The importance of BRAF as the major RAF effector of the MAPK signaling pathway is highlighted by the finding that BRAF mutations are found in a variety of cancers, including 67% of melanomas, 30–50% of thyroid cancers, 30% of ovarian cancers, 5–20% of colorectal cancers, and 1–3% of other cancer types (11). The vast majority of these mutations render the BRAF kinase constitutively active. Notably, a single valine to glutamate substitution at position 600 (V600E) within the activation segment of BRAF accounts for ∼90% of cancer-associated BRAF mutations, and in vitro BRAFV600E shows greatly elevated kinase activity and an increased level of transformation in fibroblasts and melanocytes (11–15). An extensive mapping of cancer genome mutations has also shown that BRAF is mutated in as much as 7% of human cancers, and this fact alone points to BRAF as one of the most important oncogenes, particularly in human melanoma. Taken together, the BRAF kinase represents an excellent target for anticancer drug development.
Since the discovery of BRAF as an important oncogene, significant effort has been directed toward the discovery of small molecule BRAF inhibitors. Sorafenib (BAY43-9006), initially identified as a CRAF inhibitor, was shown to be a potent BRAF inhibitor, and it was also found to potently inhibit several other kinases, including c-KIT, VEGFR, and PDGFR (16). Sorafenib was recently approved by the FDA for treatment of renal carcinoma despite its inefficacy in melanoma patients (17). A number of other BRAF inhibitor leads are being developed and are in various stages of development (18–21), with some compounds showing very promising preclinical results (22–26).
X-ray crystal structures of BRAF and BRAFV600E bound to Sorafenib revealed that the inhibitor binds within the ATP binding site and to an inactive conformation of the BRAF kinase. Although this structure provided important clues for the structure-based design of second-generation BRAF inhibitors, it did not provide information about an active conformation of the BRAF kinase, which is likely to be similar to the conformation that is adopted by activating oncogenic BRAF mutants such as BRAFV600E. In addition, the activation loop spanning residue 600 was largely disordered in both structures. Nonetheless, the authors proposed an indirect mechanism for BRAF oncogenic activation whereby a Val to Glu substitution at position 600 might disrupt an inactive conformation of BRAF kinase (13). The structural similarity between BRAFWT and BRAFV600E bound to Sorafenib also reflects the lack of selectivity for Sorafenib inhibiton of BRAF and BRAFV600E in vitro (13).
The use of organometallic compounds as scaffolds for developing protein kinase inhibitors has been previously described (27) and involves mimicking the nonselective kinase inhibitor staurosporine by replacing the alkaloid portion with a metal atom that can coordinate up to six ligands (Figure 1a). This approach provides several advantages over purely organic kinase inhibitors, most notably, the exploration of chemical space not available to traditional organic molecules (28–30). The preparation of such compounds is highly efficient, and the metal coordination bonds have been shown to be highly inert within a biological environment with no metal-related cytotoxi-cities (31–33). This approach has been applied to the development of potent and specific inhibitors for a number of protein kinases, including GSK3 and PIM1 (29, 32) and most recently PI3K (34).
Figure 1.
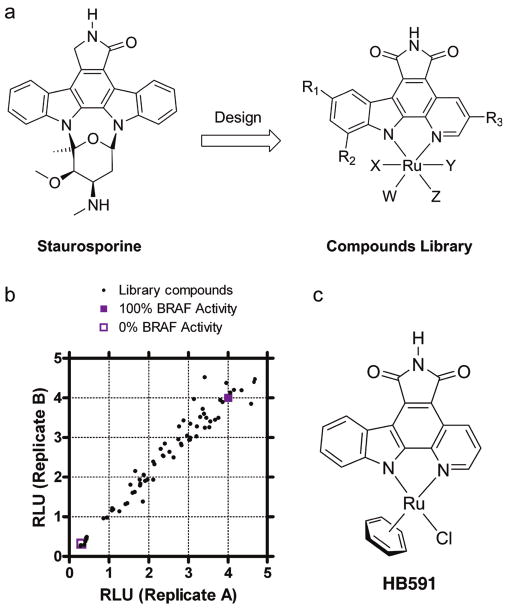
Initial lead organometallic inhibitor identification for BRAF kinase. (a) Chemical structures of staurosporine and the organoruthenium scaffold library. (b) Initial screen of the organometallic compound library using the ELISA-based BRAF activity. Relative light units (RLU) are plotted for duplicate measurements along the abscissa and ordinate, and inhibitors that yield the lowest RLU values along the diagonal represent the most potent and reproducible inhibitors. (c) Chemical structure of the racemic BRAF lead inhibitor HB591 identified from the organometallic compound library.
Here we report on the preparation of a potent and BRAFV600E selective kinase inhibitor, CS292, based on a novel organoruthenium scaffold. We also present an X-ray crystal structure of BRAF bound to CS292. The cocrystal structure reveals that the inhibitor is bound to an active conformation of BRAF in which a large portion of the activation segment, including Val 600, is well resolved in the electron density map. This structure suggests a molecular mechanism for oncogenic activation of BRAF and provides a scaffold for V600E-specific BRAF inhibitors.
Materials And Methods
Protein Expression and Purification
Human BRAF kinase domain, BRAF-KD (residues 433–726) with an N-terminal purification tag (MDRGSH6GS), and full-length mouse p50cdc37 were cloned into a pFastBac Dual vector. BRAF wild-type and V600E mutant kinase domains were expressed and purified essentially as previously described (13). Briefly, Sf9 cells infected with the BRAF kinase domain harboring baculovirus were resuspended in sonication buffer for sonication, and the lysate was cleared by high-speed centrifugation. Equilibrated Talon resin was added into the cleared lysate; the resin was washed with 10 column volumes of wash buffer [25 mM Tris (pH 8.0), 250 mM NaCl, 5 mM imidazole, and 10% glycerol] and then eluted with addition of buffer [25 mM Tris (pH 7.0), 250 mM NaCl, 160 mM imidazole, and 10% glycerol]. The eluant from the Talon resin was diluted 3-fold and loaded on a SP resin column. SP resin was extensively washed [25 mM Tris (pH 8.0), 50 mM NaCl, 1 mM dithiothreitol, and 10% glycerol] and eluted with high-salt buffer [25 mM Tris (pH 8.0), 500 mM NaCl, 1 mM dithiothreitol, 1 mM EDTA, and 10% glycerol]. Recombinantly expressed λ phosphatase was then added to the SP eluant and incubated for 2 h at room temperature followed by 7-fold dilution in buffer [25 mM Tris (pH 8.0), 1 mM dithiothreitol, 1 mM EDTA, and 10% glycerol]. This dilution was reloaded on an SP resin column to remove the λ phosphatase. The final SP eluant was concentrated and loaded onto a Superdex 200 gel filtration column equilibrated in buffer [25 mM Tris (pH 8.0), 300 mM NaCl, 1 mM dithiothreitol, 1 mM EDTA, and 5% glycerol]. Gel filtration fractions were concentrated with a final glycerol concentration of 15% to a protein concentration of 1.5 mg/mL. BRAF-KD protein was immediately used for crystallization.
GST-MEK-His protein was overexpressed at 37 °C in Escherichia coli BL21 (Gold) cells (Invitrogen) in LB medium until the OD value reached 0.4–0.6. Isopropyl 1-thio-β-d-galactopyrano-side (IPTG, 0.5 mM) was then added to the culture, which was grown at 15 °C for an additional 16 h. The cell pellet was resuspended and sonicated in sonication buffer [20 mM HEPES (pH 7.5), 500 mM NaCl, 10 mM 2-mercaptoethanol (BME), 10 mM imidazole, 0.1 mg/mL phenylmethanesulfonyl fluoride (PMSF), and 5% glycerol]. The lysate was cleared by highspeed centrifugation before it was loaded onto a Ni-NTA resin column pre-equilibrated in sonication buffer. The protein-bound Ni-NTA resin was then extensively washed with 20 column volumes of wash buffer, and the GST-MEK-His protein was eluted with an imidazole gradient from 10 to 150 mM in wash buffer. Fractions corresponding to GST-MEK-His were pooled and applied to a Superdex 200 column equilibrated in buffer [20 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM BME, and 5% glycerol]. Gel filtration fractions were pooled and concentrated to 10 mg/mL before being flash-frozen in liquid nitrogen and stored at −80 °C until use.
Crystallization, Data Collection, and Structure Determination
BRAF-KD crystals were obtained by mixing 1.35 μL of BRAF-KD with 1.35 μL of crystallization reservoir [200 mM magnesium acetate tetrahydrate, 100 mM sodium cacodylate trihydrate (pH 6.5), and 20% polyethylene glycol 8000] (Hampton Research) supplemented with 0.3 μL of 100 mM nicotinamide adenine dinucleotide (Hampton Research) using the microbatch method underneath light mineral oil (Sigma). Crystals reached the maximum size of ∼30 μm × ∼30 μm × ∼200 μm after 1 week. Increasing concentrations of a racemic mixture of CS292 in cryoprotectant solution [200 mM magnesium acetate tetrahydrate, 100 mM sodium cacodylate trihydrate (pH 6.5), 25% polyethylene glycol 8000, and 15% glycerol] were then added in 15 min intervals to a final concentration of 1 mM and the mixtures incubated for 4 h to overnight. Soaked crystals were harvested in fresh cryoprotectant solution and flash-frozen in liquid propane. Data were collected at beamline GM/CA-CAT 23ID-B at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL). Diffraction images were collected using the 10 μm minibeam from a single crystal. Diffraction data were indexed, integrated, and scaled using the HKL2000 package (HKL Research) and further processed using the CCP4 program suite (39). The structure was determined by molecular replacement using Molrep (40) from the CCP4 suite with a previously reported BRAF-KD structure (PDB entry 1UWH) as a search model. The space group was determined to be P41212, and each asymmetric unit contained two molecules. Electron density corresponding to the organometallic BRAF inhibitor of CS292 was well resolved in one of the two molecules in the asymmetric unit, and the ligand model was placed into the electron density from the calculated Fo – Fc map and adjusted in Coot (41). The electron density suggested that only one of the two enantiomers of the CS292 inhibitor was bound. Parameter and topology files for CS292 used in the refinements were generated from the HIC-UP XDICT server (http://alpha2.bmc.uu.se/hicup/xdict.html). This was followed by additional refinement using CNS (42), and the final model was checked for errors by using the CNS composite omit map for protein models and simulated annealing omit map for the organoruthenium inhibitors. The final model was refined with excellent geometrical and refinement statistics (Table 1).
Table 1.
Crystallographic Data Collection and Structural Refinement Statistics
| data collection | APS 23ID-B with 10 μm minibeam |
| complex | BRAF–CS292 |
| space group | P41212 |
| cell dimensions | |
| a, c (Å) | 94.91, 163.35 |
| resolution (Å) | 50−3.2 |
| Rmergea,b | 0.277 (0.735) |
| I/σ(I)a | 9.9 (2.2) |
| completenessa (%) | 99.9 (99.0) |
| multiplicitya | 13.3 (6.4) |
| refinement | |
| Rworkc/Rfreed | 23.2%/28.0% |
| root-mean-square deviation | |
| bond lengths (Å) | 0.0099 |
| bond angles (deg) | 1.54 |
Data for the highest-resolution shell shown in parentheses.
Rmerge = Σhkl|I(hkl)−〈I(hkl)〉|/Σhkl|I(hkl), where 〈I(hkl)〉 is the mean of the sym-metry-equivalent reflections of I(hkl).
Rwork = Σ‖Fo| – |Fc‖/|Fo|.
Rfree=ΣT‖Fo|–|Fc‖/ΣT|Fo| (where T is a test data set of 9.3% of the total reflections randomly chosen and set aside before refinement).
All structural superpositions were generated by overlaying the Cα positions of previously determined kinase and kinase–ligand structures and by using secondary structure matching (SSM) in Coot (43). All structural graphics were generated in PyMol (DeLano Scientific LLC).
In Vitro Kinase Assay
Recombinantly expressed GST-MEK-His, diluted in TTBS buffer [20 mM Tris (pH 7.5), 150 mM NaCl, and 0.05% Tween 20] to 50 μg/mL in a volume of 100 μL, was bound to the wells of a 96-well glutathione-coated plate (Pierce Biotechnology). One microliter of compounds (as racemic mixtures) with 2× serial dilutions in a 100% DMSO stock solution was added to a mixture of 50 μL of a buffer containing 50 mM HEPES (pH 7.0) with 0.7 pmol of BRAF-KD kinase. This mixture was incubated at room temperature for 1 h before it was added to the GST-MEK-His-bound wells of the 96-well plate. An additional 50 μL of phosphorylation buffer [50 mM HEPES (pH 7.0), 200 mM NaCl, 10 mM MgCl2, and 200 μM ATP] was added to the well mixture to start the kinase reaction at 37 °C for 30 min with intermittent shaking. The kinase reaction was stopped by extensive washing with TTBS buffer, and a 1:5000 dilution of anti-phospho-MEK1 (Ser218/222)/ MEK2 (Ser222/226) monoclonal antibody (Millipore) in TTBS buffer was subsequently added to the wells and incubated for 1 h with shaking. Goat anti-rabbit IgG (H + L)−HRP conjugate (Bio-Rad Laboratories) in a 1:5000 dilution was added to the wells for incubation at room temperature with shaking. Finally, the SuperSignal ELISA Pico chemiluminescent substrate (Pierce Biotechnology) was added to the wells. The luminescence signal was recorded with a luminescence filter using a Wallac 1420 luminometer (PerkinElmer). Data were processed, and IC50 values were derived from fitting the data to a sigmoidal dose– response curve using GraphPad Prism.
Compounds and Chemical Synthesis
All reactions were carried out using oven-dried glassware and conducted under a positive pressure of argon unless otherwise specified. NMR spectra were recorded on a DMX-360 (360 MHz) or Bruker AM-500 (500 MHz) spectrometer. Infrared spectra were recorded on a Perkin-Elmer 1600 series FTIR spectrometer. Low-resolution mass spectra were obtained on an LC platform from Micromass using the ESI technique. ES-TOF spectra were measured by Waters Micromass MS Technologies. High-resolution mass spectra were obtained with a Micromass AutoSpec instrument using either CI or ES ionization. Compounds 1 (44), 2 (45), 3 (46), 5 (47), and 11 (48) were prepared according to reported literature procedures. Reagents and solvents were used as received from standard suppliers. HB591, NP580, and CS292 were synthesized according to the scheme below by splitting the appropriate benzene chloride dimer with the necessary pyridocarbazole fragment in acetonitrile with potassium carbonate (Scheme 1).
Scheme 1.
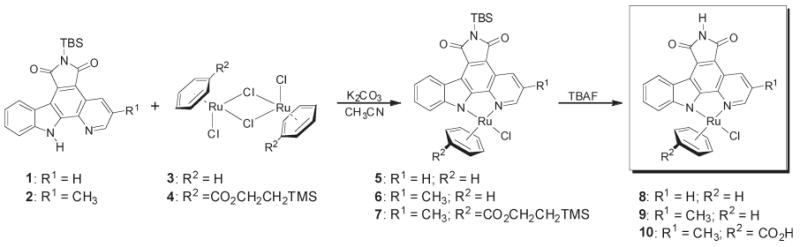
Synthesis of Pyridocarbazole Half-Sandwich Complexes
The TMS ethyl ester-substituted benzene chloride dimer (4) was synthesized from the corresponding cyclohexadiene (11) and commercially available ruthenium trichloride in trimethylsilylethanol. The complete sequence is shown in Scheme 2.
Scheme 2.
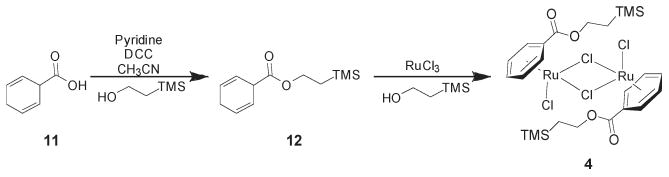
Synthesis of Modified Benzene Chloride Dimer 4
Isoquinoline complex CNSE030 was synthesized in a similar manner using analogous free ligand 13 (Scheme 3).
Scheme 3.
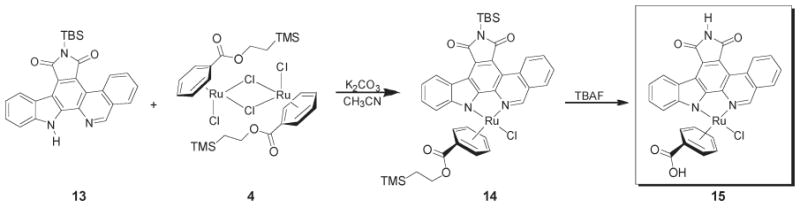
Synthesis of Isoquinoline-Based Half-Sandwich Complex 15
(i) Synthesis of Trimethylsilyl Benzoate Chloride Dimer (4)
A solution of 11 in acetonitrile (6.3 mL) was cooled to 0 °C. Pyridine (505 μL, 6.24 mmol), dicyclohexylcarbodiimide (DCC) (682 mg, 3.31 mmol), and trimethylsilylethanol (TMS-EtOH) (490 μL, 3.43 mmol) were added at once. A precipitate rapidly formed from the clear colorless solution as it was allowed to warm to room temperature overnight. After the mixture had been stirred overnight, the precipitate was removed by vacuum filtration. The supernatant was concentrated and subjected to silica gel chromatography with hexanes and EtOAc (15:1). The combined product eluents were dried in vacuo to provide compound 12 (500 mg, 72%) as a clear colorless oil.
Compound 12 (634 mg, 2.83 mmol) was then dissolved in TMSEtOH (4.0 mL), and RuCl3·3H2O (178 mg, 0.681 mmol) was added. The dark green reaction mixture was heated to 100 °C overnight during which time an orange precipitate formed. Complex 4 could be isolated as an orange solid (235 mg, 88%) by cold vacuum filtration followed by MeOH and Et2O washings: 1H NMR (500 MHz, CD3CN) δ 6.45 (d, J = 6.0 Hz, 4H), 6.02 (t, J = 5.6 Hz, 2H), 5.80 (t, J = 6.0 Hz, 4H), 4.45 (m, 4H), 1.16 (m, 4H), 0.10 (s, 18H); 13C NMR (125 MHz, CD3CN) δ 166.0, 90.9, 89.4, 83.0, 81.9, 65.9, 18.3, −1.4; IR (film) v (cm−1) 3079, 2952, 2927, 2897, 2849, 1726, 1624, 1399, 1288, 1270, 1251, 1175, 1107, 1043, 931, 861, 838, 771; HRMS calcd for C52H51NNaO2Ru (monomer) (M − Cl + 2CH3CN) + 441.0339, found (M − Cl + 2CH3CN)+ 441.0287.
(ii) Synthesis of Complex HB591
Compound HB591 (8)
Silyl-protected complex 5 (20 mg, 0.033 mmol) was solvated in THF (2.0 mL) under argon and cooled to 0 °C. A 1 M solution of TBAF in THF (36 μL, 0.036 mmol) was added at once. The reaction mixture was then allowed to stir for 10 min before the reaction was quenched with saturated NH4Cl(aq). The aqueous layer was extracted twice with EtOAc. The combined organics were dried with Na2SO4, concentrated, and subjected to silica gel chromatography with methylene chloride and methanol (35:1). The product eluted as a dark red band. Collecting only the purest fractions as determined by 1H NMR gave a red-purple solid (8.3 mg) in 50% yield: 1H NMR (360 MHz, DMSO-d6) δ 11.05 (s, 1H), 9.72 (d, J = 5.1 Hz, 1H), 9.09 (d, J = 8.4 Hz, 1H), 8.67 (d, J = 8.1 Hz, 1H), 8.10 (d, J = 8.2 Hz, 1H), 7.87 (dd, J = 8.2, 5.2 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.37 (t, J = 7.2 Hz, 1H), 6.23 (s, 6H); 13C NMR (90 MHz, DMSO-d6) δ 170.7, 170.5, 153.2, 152.6, 151.1, 141.8, 133.5, 130.6, 126.1, 123.9, 123.5, 122.2, 120.7, 119.5, 116.0, 113.7, 112.6, 85.5; IR (film) v (cm−1) 3202, 3061, 2958, 2918, 2851, 1748, 1702, 1637, 1582, 1524, 1497, 1473, 1419, 1343, 1267, 1229, 1023, 1009, 824, 796, 746; HRMS calcd for C25H17N4O2Ru (M − Cl + CH3CN) + 507.0395, found (M − Cl + CH3CN)+ 507.0399.
(iii) Synthesis of Complex NP580
(a) Compound 6
A suspension of ligand 2 (300 mg, 0.723 mmol), dimer 3 (271 mg, 0.542 mmol), and K2CO3 (110 mg, 0.795 mmol) in CH3CN (30 mL) was purged with argon and stirred at room temperature overnight. The reaction was then pushed to completion by heating the mixture to 55 °C for 4 h. The resulting thick red reaction mixture was cooled to room temperature and concentrated. The crude material was subjected to silica gel chromatography with methylene chloride and methanol (from 100:1 to 35:1). The combined product eluents were dried to provide the half-sandwich complex 6 (416 mg, 91 %) as a red solid: 1H NMR (360 MHz, CDCl3) δ 9.10 (d, J = 1.6 Hz, 1H), 9.05 (d, J = 1.6 Hz, 1H), 8.89 (d, J = 7.2 Hz, 1H), 7.81 (d, J = 8.2 Hz, 1H), 7.60 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 7.38 (ddd, J = 8.0, 7.1, 0.9 Hz, 1H), 5.96 (s, 6H), 2.67 (s, 3H), 1.03 (s, 9H), 0.61 (s, 6H); 13C NMR (90 MHz, CDCl3) δ 175.9, 174.9, 153.4, 152.1, 151.4, 141.3, 135.2, 133.4, 133.3, 126.6, 125.8, 124.9, 121.9, 120.3, 114.8, 114.8, 114.6, 83.6, 26.7, 19.6, 19.4, −3.7; IR (film) v (cm−1) 3062, 2929, 2857, 1743, 1686, 1567, 1497, 1468, 1417, 1340, 1301, 1265, 1233, 1169, 1050, 1010, 830, 746; HRMS calcd for C32H33N4O2RuSi (M − Cl + CH3CN)+ 635.0516, found (M − Cl + CH3CN)+ 635.1499.
(b) Compound NP580 (9)
Silyl-protected complex 6 (29 mg, 0.046 mmol) was solvated in CH2Cl2 (2.9 mL) and cooled to 0 °C. A 1 M solution of TBAF in THF (104 μL, 0.069 mmol) was added at once. The reaction mixture was then allowed to stir for 15 min before the reaction was quenched with saturated NH4Cl(aq). The aqueous layer was extracted twice with EtOAc. The combined organics were washed twice with 10% HCl, dried with Na2SO4, concentrated, and subjected to silica gel chromatography with methylene chloride and methanol (75:1). The product eluted as a dark red band that could be further purified by dissolving in acetone and MeOH and diluting into a 1:1 hexane/diethyl ether mixture, giving a purple precipitate (14 mg) in 58% yield: 1H NMR (500 MHz, DMSO-d6) δ 11.01 (s, 1H), 9.63 (s, 1H), 8.88 (s, 1H), 8.65 (d, J = 8.3 Hz, 1H), 8.07 (d, J = 8.3 Hz, 1H), 7.60 (ddd, J = 7.7, 7.6, 1.3 Hz, 1H), 7.34 (dd, J = 7.5, 0.7 Hz, 1H), 6.22 (s, 6H), 2.71 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.3, 176.0, 159.8, 158.4, 156.6, 145.6, 139.1, 138.0, 136.1, 131.4, 129.3, 129.1, 126.1, 124.8, 121.4, 118.7, 117.5, 89.0, 24.3; IR (film) v (cm−1) 3061, 2920, 2844, 1746, 1713, 1700, 1647, 1634, 1556, 1539, 1520, 1490, 1470, 1454, 1418, 1339, 1296, 1266, 1231, 1147, 1023, 1003, 745; HRMS calcd for C24H16N3O2Ru (M − Cl)+ 480.0286, found (M − Cl) + 480.0009.
(iv) Synthesis of Complex CS292
(a) Compound 7
Free ligand 2 (11.0 mg, 0.027 mmol), K2CO3 (4.0 mg, 0.029 mmol), and modified benzene chloride dimer 4 (13.5 mg, 0.017 mmol) were added together to a dry 10 mL two-neck round-bottomed flask and placed under argon. A 1:1 DCM/CH3CN mixture (2 mL) was then added to the reaction mixture. After the reaction mixture had been stirred overnight at room temperature, the mixture was concentrated and subjected to silica gel chromatography with toluene and acetone (6:1). The product (7) eluted as a dark purple band (15.5 mg) in 76% yield: 1H NMR (500 MHz, CDCl3) δ 9.09 (s, 2H), 8.87 (d, J = 7.7 Hz, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.59 (ddd, J = 8.1,7.1,1.1 Hz, 1H), 7.38 (t, J = 7.4 Hz, 1H), 6.96 (d, J = 6.1 Hz, 1H), 6.92 (d, J = 5.8 Hz, 1H), 6.44 (t, J = 5.6 Hz, 1H), 6.11 (t, J = 5.8 Hz, 1H), 5.48 (t, J = 5.7 Hz, 1H), 4.32 (m, 2H), 2.69 (s, 3H), 1.04 (s, 9H), 0.83 (m, 2H), 0.60 (d, J = 1.2 Hz, 6H), 0.02 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 175.8, 174.8, 165.1, 153.0, 152.1, 151.2, 141.1, 135.2, 133.6, 133.2, 126.6, 125.8, 124.8, 121.7, 120.4, 115.1, 114.7, 114.4, 94.1, 90.7, 90.0, 79.2, 76.7, 65.7, 26.7, 19.7, 19.3, 17.6, −1.2, −3.7; IR (film) v (cm−1) 3062, 2942, 2925, 2856, 1741, 1728, 1715, 1685, 1642, 1569, 1518, 1497, 1471, 1415, 1385, 1338, 1295, 1265, 1230, 1170, 1110, 1050, 1011, 926, 837, 748; HRMS calcd for C38H45N4O4RuSi2 [(M − Cl) + (CH3CN)]+ 779.2023, found [(M − Cl) + (CH3CN)]+ 779.2058.
(b) Compound CS292 (10)
Silyl-protected complex 7 (13.5 mg, 17.5 mmol) was solvated in THF (2 mL). A1M solution of TBAF in THF (45 μL, 45 mmol) was added at once. The reaction mixture was then allowed to stir at 32 °C for 10 min. The mixture was then cooled to room temperature, poured into saturated NH4OAc(aq), and extracted twice with EtOAc. The combined organics were washed with 10% HCl, dried with Na2SO4, and concentrated. The dark red solid could then be subjected to silica gel chromatography with a methylene chloride/ methanol/TFA mixture (75:1:0.001). The combined red fractions could be washed with 1 M HCl, dried with MgSO4, and concentrated to yield a dark red solid (6.9 mg) in 71% yield: 1H NMR (500 MHz, DMSO-d6) δ 11.04 (s, 1H), 9.51 (s, 1H), 8.91 (s, 1H), 8.64 (d, J = 7.8 Hz, 1H), 8.13 (d, J = 8.2 Hz, 1H), 7.60 (dd, J = 7.6,1.2 Hz, 1H), 7.35 (t, J = 7.5 Hz, 1H), 7.08 (d, J = 6.0 Hz, 1H), 7.03 (d, J = 6.0 Hz, 1H), 6.56 (t, J = 5.7 Hz, 1H), 6.41 (t, J = 5.9 Hz, 1H), 6.19 (t, J = 5.9 Hz, 1H), 2.71 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 170.7, 170.4, 166.3, 153.8, 152.5, 151.0, 139.9, 133.6, 132.8, 130.6, 126.0, 123.8, 123.5, 120.5, 119.5, 115.7, 113.2, 112.3, 91.2, 91.0, 90.2, 78.4, 78.0, 18.8; IR (film) v (cm−1) 3417, 3059, 2954, 2922, 2852, 1749, 1713, 1626, 1567, 1419, 1339, 1267, 1232, 1175, 1150, 1109, 1012, 940, 903, 819, 749; HRMS calcd for C25H16N3O4Ru (M − Cl)+ 524.0184, found (M − Cl)+ 524.0189.
(v) Synthesis of Complex CNSE030
(a) Compound 14
Free ligand 13 (23 mg, 0.050 mmol), K2CO3 (8.0 mg, 0.060 mmol), and modified benzene chloride dimer 4 (25 mg, 0.033 mmol) were added together to a dry 10 mL round-bottomed flask and placed under argon. A 1:1 DCM/CH3CN mixture (2 mL) was then added to the reaction mixture. After the mixture had been stirred overnight at room temperature, it had turned dark purple. The reaction mixture was concentrated and subjected to silica gel chromatography with a 6:1 toluene/acetone mixture ramped to a 3:1 toluene/acetone mixture. The product (14) eluted as a dark purple band (38 mg) in 98 % yield: 1H NMR (500 MHz, CDCl3) δ 10.49 (d, J = 8.5 Hz, 1H), 9.83 (s, 1H), 9.07 (d, J = 7.8 Hz, 1H), 8.07 (d, J = 7.7 Hz, 1H), 8.02 (t, J = 7.6 Hz, 1H), 7.76 (d, J = 8.1 Hz, 1H), 7.71 (t, J = 7.2 Hz, 1H), 7.60 (t, J = 7.5 Hz, 1H), 7.37 (t, J = 7.5 Hz, 1H), 6.85 (d, J = 5.0 Hz, 2H), 6.27 (m, 1H), 6.04 (m, 1H), 5.42 (m, 1H), 4.22 (ddd, J = 10.5, 7.2, 2.2 Hz, 2H), 1.08 (s, 9H), 0.72 (m, 8H), −0.04 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 175.2, 174.5, 165.1, 157.0, 153.9, 151.8, 137.8, 133.5, 133.5, 132.8, 130.1, 129.6, 128.3, 127.4, 126.6, 124.5, 120.9, 120.0, 117.2, 116.5, 114.0, 93.9, 91.1,90.2, 79.2, 77.9, 65.6, 26.8,19.4,17.5, −1.4, −3.4; IR (film) v (cm−1) 3065, 2952, 2925, 2850, 1730, 1686, 1621, 1576, 1536, 1480, 1464, 1438, 1377, 1359, 1326, 1297, 1270, 1230, 1148, 1109, 1080, 919, 828, 757; HRMS calcd for C28H28F3N3O2PRuSi (M − Cl + DMSO-d6) + 858.2273, found (M − Cl + DMSO-d6)+ 858.2291.
(b) Compound CS030 (15)
Silyl-protected complex 14 (37.0 mg, 0.046 mmol) was solvated in THF (2.5 mL). A 1 M solution of TBAF in THF (183 μL, 0.183 mmol) was added at once. The mixture was then allowed to stir at room temperature over 45 min and then heated to 45 °C for 1 h. The reaction mixture was poured into saturated NH4OAc(aq) and extracted twice with EtOAc. The combined organics were washed with 10% HCl, dried with Na2SO4, concentrated, and subjected to silica gel chromatography with a methylene chloride/methanol/ HOAc mixture (75:1:0.01). The product eluted as a dark red band (9.2 mg) in 60% yield: 1H NMR (500 MHz, DMSO-d6) δ 10.52 (d, J = 8.6 Hz, 1H), 10.34 (s, 1H), 8.87 (d, J = 7.8 Hz, 1H), 8.46 (d, J= 7.9 Hz,1H), 8.16 (m, 2H), 8.03 (t, J = 7.4 Hz, 1H), 7.67 (t, J = 7.6 Hz, 1H), 7.37 (t, J = 7.5 Hz, 1H), 7.12 (d, J = 6.0 Hz, 1H), 7.09 (d, J = 6.0 Hz, 1H), 6.59 (t, J = 5.7 Hz, 1H), 6.43 (t, J = 5.9 Hz, 1H), 6.28 (t, J = 5.8 Hz, 1H); 13C NMR (90 MHz, DMSO-d6) δ 170.8, 169.8, 166.3, 158.0, 153.3, 151.6, 137.0, 133.2, 132.8, 131.3, 129.9, 129.2, 128.5, 127.7, 127.0, 124.8, 123.2, 119.7, 119.2, 115.5, 115.3, 114.5, 91.7, 91.0, 90.6, 79.2, 78.5; IR (film) v (cm−1) 3202, 2954, 2922, 2850, 1743, 1719, 1699, 1657, 1621, 1578, 1537, 1459, 1438, 1378, 1344, 1326, 1294, 1265, 1229, 1021, 864, 824, 752; HRMS calcd for C28H15ClN3O4Ru (M − H)− 593.9795, found (M − H)− 593.9769.
Results and Discussion
Identification of BRAF Inhibitors through Organometallic Scaffold Design and in Vitro Enzymatic Screening
To identify an initial lead compound for BRAF kinase inhibition from an organometallic scaffold, we developed an ELISA-based assay system to detect the phosphorylation of BRAF substrate MEK1. Briefly, in the ELISA, full-length human MEK1 protein kinase with an N-terminal GST fusion affinity tag is immobilized on a glutathione-coated microtiter plate. BRAF kinase is added to the MEK1-immobilized plate, and MEK1 phosphorylation is detected with a phospho-specific antibody coupled with a HRP-linked IgG to generate a luminescence signal proportional to the amount of phosphorylated MEK1. This assay was utilized to screen a library of 68 organometallic compounds (Figure 1b), leading to the identification of compound HB591 as the most potent BRAF inhibitor (Figure 1c) with an IC50 in the sub-micromolar range. We prepared a series of HB591 analogues and found that the addition of a methyl group to the pyridine fragment (NP580) and a carboxylic acid to the η6-benzene (CS292) yielded more potent BRAF inhibitors (Figure 2a,b). The most potent BRAF inhibitor, CS292, was also shown tohave an ∼2-fold selectivity for BRAFV600E (IC50 = 0.21 μM) over the wild-type enzyme (IC50 = 0.37 μM) (Figure 2a,b). As a negative control, we demonstrated that the pyridocarbazole portion alone without the ruthenium complex is a poor inhibitor for both BRAFWT and BRAFV600E with IC50 values in the midmicromolar range (Figure 2c), demonstrating the importance of the ruthenium complex of the compound for potent inhibition.
Figure 2.
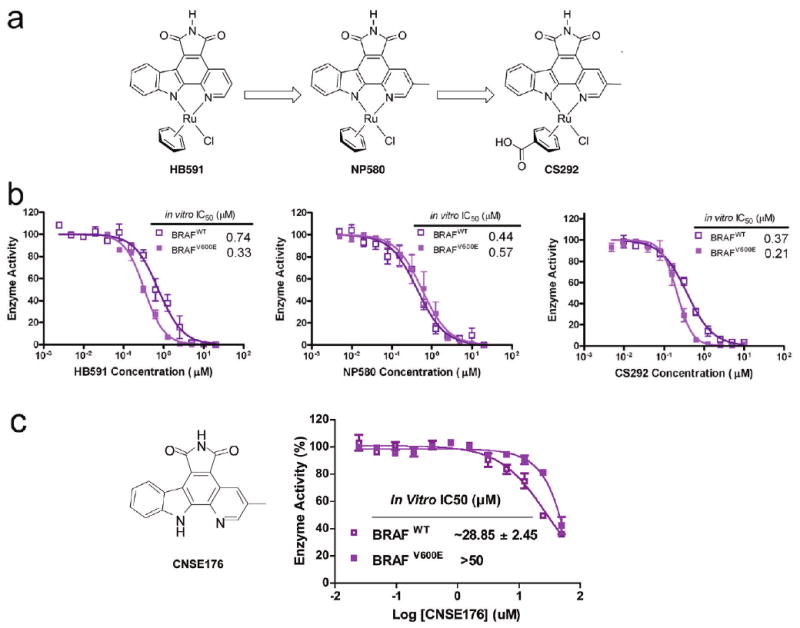
Modifications on BRAF lead inhibitor HB591. (a) Chemical structures of lead inhibitor HB591, precursor NP580, and CS292. (b) Dose–response curves of HB591, NP580, and CS292 from an ELISA-based BRAF kinase inhibition assay. (c) Chemical structure and dose– response curve of the pyridocarbazle compound CNSE176 without the ruthenium complex portion. The data represent an average of triplicate measurements.
Crystal Structure of CS292 in Complex with BRAF
To understand the molecular basis for BRAF inhibition by CS292, we carried out crystallization trials of the kinase domain (residues 433–726) of BRAF and BRAFV600E with a racemic mixture of CS292 and were able to obtain well-diffracting crystals of only the BRAF–CS292 complex for X-ray structure determination. The BRAF–CS292 crystals contained two copies of the BRAF kinase domain in one asymmetric unit cell, with one of the two copies containing well-resolved electron density that fit one of the two enantiomers of the CS292 inhibitor better than the other. CS292 binds proximal to the ATP binding cleft of BRAF located between the N-lobe and C-lobe of the kinase domain (Figure 3a). The overall conformation of the BRAF kinase domain in complex with CS292 is largely identical to that of the BRAF– Sorafenib complex with no significant perturbations introduced by the CS292 inhibitor. An overlay of the BRAF–CS292 complex structure with cAMP-dependent protein kinase (PKA) in complex with ATP shows a significant overlap of the pyrido-carbazole fragment of CS292 with the adenosine and ribose moieties of the ATP molecule (Figure 3a). The rest of the CS292 molecule largely occupies the hydrophobic space formed proximal to the hinge region of the N-lobe and C-lobe, orthogonal to the P-loop extension. CS292 is well-resolved from the analysis of the electron density around the ATP phosphate binding pocket (Figure 3b). Specifically, the pyridocarbazole moiety of CS292 is intercalated into the space between residues Trp531 and Phe583 forming π–π stacking interactions (Figure 3c). Residue His539 from the αD helix in the C-lobe of the kinase domain also makes π–π stacking interactions with the benzene ligand of inhibitor CS292. Indeed, this particular interaction appears to be responsible for the preferential binding of one CS292 enantiomer over the other to the BRAF kinase in the crystals. In addition, the maleimide moiety of CS292 forms hydrogen bonding interactions with the hinge region of the N-lobe and C-lobe. In particular, an oxygen atom from one of the maleimide carbonyls forms a hydrogen bond with the amide nitrogen of Cys532, and the maleimide nitrogen forms a potential water-mediated hydrogen bond with the main chain carbonyl of Glu530. There are also extensive van der Waals contacts between CS292 and other residues in the protein pocket such as Ser536, Ser535, Gly534, Cys532, Ile463, Val471, and Asn580. The chloride ligand of CS292 is pointed toward Ile463, which is located in the middle of the P-loop, leaving very limited space beyond the chloride ligand of CS292. The carboxylated benzene ligand occupies a solvent-exposed region of the ATP binding pocket and does not make specific interactions with the protein moiety. The hydro-philicity of this carboxylate group on the rather hydrophobic benzene ligand in this solvent-exposed region may explain the importance of the carboxylate group for inhibitor potency. Taken together, structural evidence reveals extensive and specific interactions between CS292 and the ATP binding pocket of the BRAF kinase domain, establishing CS292 as an ATP competitive inhibitor and confirming its potent inhibitory properties against both BRAFWT and BRAFV600E.
Figure 3.
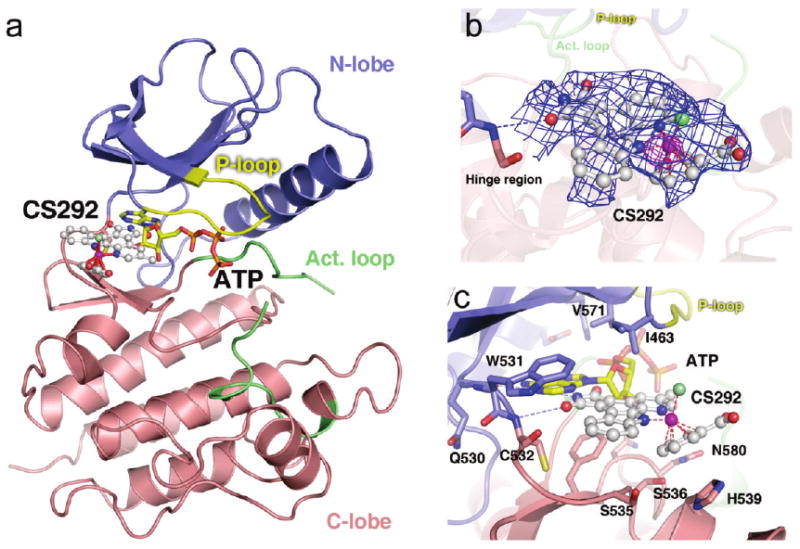
Crystal structure of BRAF in complex with CS292. (a) Overall structure of the BRAF kinase domain in complex with CS292. The N-lobe, C-lobe, P-loop, and activation loop (Act. loop) of the kinase domain are colored blue, red, yellow, and green, respectively. Compound CS292 is shown as a white ball and stick model with the ruthenium atom highlighted in magenta and coordination bonds displayed as red dashes. The PKA–ATP complex structure was overlaid with the BRAF–CS292 complex structure to show the binding of ATP to the BRAF kinase active site (43). The ATP model is colored yellow for the adenine and the ribose fragments and orange for the phosphates. The same color coding scheme is preserved in all panels of this figure. (b) Electron density map corresponding to CS292 of the BRAF–CS292 cocrystal structure. The blue map is contoured at 4σ from a simulated annealing Fo – Fc omit map without any contribution from the CS292 inhibitor model. The Fo – Fc omit map that is colored magenta is contoured at 8σ to indicate the position of the ruthenium metal atom. (c) Details of interactions of CS292 with the BRAF active site. Hydrogen bonding interactions are represented with blue dashed lines.
The BRAF Structure Is in an Active Conformation
A superposition of the BRAF–CS292 complex structure with the active conformation of PKA with bound ATP reveals a striking similarity between the conformations of these two structures (35, 36) (Figure 4a). An active conformation of residues centered around the catalytic core of protein kinases is divided into two groups of residues responsible for stabilizing ATP and facilitating the phospho-transfer reaction from ATP to the substrate. With regard to residues that participate in ATP stabilization, extensive studies on the structural basis for activation of PKA have established that two residues are responsible for positioning the α- and β-phosphates of the ATP molecule for hydrolysis, a lysine residue (Lys72PKA and Lys483BRAF) from the β3 strand which is stabilized by a salt bridge interaction with a glutamate residue (Glu91PKA and Glu501BRAF) on the αC helix (35, 36). An asparagine residue (Asn171PKA and Asn581BRAF) from the β7 strand also mediates this catalytic arrangement by forming hydrogen bonding interactions with an aspartic acid residue (Asp184PKA and Asp594BRAF) of the activation loop DFG motif which appropriately positions the terminal ATP γ-phosphate through Mg2+ ions and another aspartic acid residue (As-p166PKA and Asp576BRAF) from the catalytic loop. Residue Asp166PKA is also positioned by Asn171PKA to serve as a general base for phospho transfer. A structural superposition of the BRAF–CS292 complex with the active conformation of the PKA structure reveals that all of the homologous residues of BRAF (Lys483, Glu501, Asn581, Asp594, and Asp576) overlap well with the corresponding PKA residues with a root-mean-square deviation of 0.433 Å. In addition, the activation loop of BRAF also has a conformation similar to that of the active PKA loop with a root-mean-square deviation of 0.453 Å between the Cα atoms of the corresponding ordered activation loop residues (residues 593–600 in BRAF and residues 184–191 in PKA) (Figure 4a). Taken together, our structural evidence strongly suggests that BRAF adopts an active conformation in the presence of the organoruthenium inhibitor, CS292.
Figure 4.
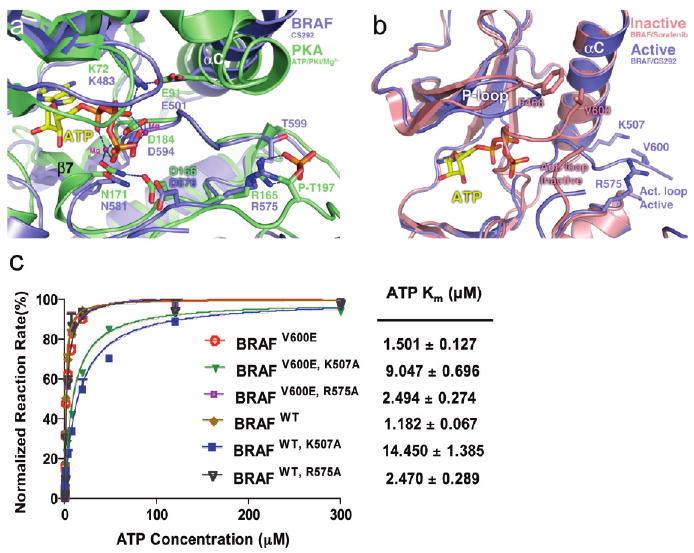
BRAF kinase in an active conformation. (a) Superposition of the BRAF–CS292 complex protein fragment with the PKA–ATP complex (PDBentry 1ATP). The BRAF kinase is colored blue and PKA green. Essential catalytic residues are shown in stick figure representation with color coding corresponding to each kinase. Magnesium ions are shown as magenta spheres, and hydrogen bonding interactions are shown as blue dashes. Part of the activation loop is truncated for better viewing of the PKA phospho-Thr197. (b) Superposition of the BRAF–CS292 complex with BRAF in an active conformation with the BRAF–Sorafenib complex in an inactive conformation. The BRAF–CS292 complex is colored blue and the BRAF–Sorafenib complex red. (c) ATP Km measurements of BRAFV600E and BRAFWT kinases and effect of K507A and R575A mutations on Km,ATP. The reaction rate is normalized against the Vmax of each kinase construct, and Km values are extrapolated from a sigmoidal curve fitting model and listed in units of micromolar. All compounds were used as racemic mixtures.
A Comparison of BRAF Bound to CS292 and Sorafenib and a Proposed Mechanism for BRAF Oncogenic Activation
Despite the overall structural similarity between the BRAF– CS292 and BRAF–Sorafenib complexes, the overlay of these two structures reveals a major difference in the conformation of the activation loop (Figure 4b), which is free of crystal contacts. In the inactive conformation from the BRAF–Sorafenib structure, the Val600 residue forms a hydrophobic contact with the hydrophobic portion of the P-loop, particularly with the side chain of Phe468. This hydrophobic interaction induces the activation loop of BRAF bound to Sorafenib to adopt a conformation that occupies the phosphate-binding pocket formed by the P-loop and the β7–β8 sheet. The specific conformation of the activation loop is largely reminiscent of the inactive conformation of protein kinases in which the activation segment is flipped toward the P-loop. For example, when the ordered residues of the BRAF activation loop (residues 589–599) of the BRAF–Sorafenib complex are superimposed with the corresponding loops of the active form of PKA (PDB entry 1ATP, residues 180–190) and the inactive form of the c-Abl tyrosine kinase bound to the STI-571 inhibitor (PDB entry 1FPU, residues 377–387), the root-mean-square deviation values for Cα atoms are 4.293 and 1.641 Å, respectively. In contrast, in the BRAF–CS292 complex structure, the activation loop is significantly more ordered and adopts a dramatically different conformation that corresponds more to an active kinase. In particular, starting from residue Asp594, the activation segment approaches the αC helix, placing residue Val600 in the proximity of the aliphatic region of Lys507 of the αC helix. This activation loop conformation mirrors the position of the activation segment of BRAF from a recently reported BRAF crystal structure in complex with inhibitor SB590885, although a large portion of the activation loop is missing from this structure (37). This more open conformation of the activation segment is proposed to be in a more accessible conformation for ATP and protein substrate binding.
With the determination of both inactive and active conformations of the BRAF structure in hand, a mechanism can be proposed to explain the mechanism for phospho-regulation and V600E oncogenic activation of BRAF kinase activity. When the two activating phosphorylation events occur on residues Thr599 and Ser602 of BRAF, which are immediately adjacent to the Val600 residue, the negatively charged phosphates will destabilize the inactive conformation by disrupting the hydrophobic interactions formed between Val600 of the activation segment and residues in the P-loop. At the same time, favorable salt bridge interactions could also be formed with either residue Lys507 or the highly conserved Arg575 residue from the catalytic loop, which in turn stabilizes the active conformation of the kinase (Figure 4b). This active conformation results in an alleviation of the blockage of the ATP binding site that was present in the inactive kinase conformation. We propose that when Val600 is mutated to Glu, as is the case with the oncogenic BRAFV600E mutant, the negatively charged glutamate residue mimics the effect of Thr599 and Ser602 phosphorylation by disrupting the inactive conformation and stabilizing an active conformation by forming a salt bridge interaction between Lys507 and/or Arg575 and the substituted Glu600 residue.
The scenario proposed above is consistent with an active phosphorylated or V600E substituted form of BRAF that is more accessible for ATP and substrate binding than the wild-type unmodified BRAF protein. On the basis of this, we hypothesize that the inactive unphosphorylated BRAFWT will have different ATP binding properties compared with the phosphorylated BRAFWT and BRAFV600E activating mutant, with the active conformation harboring higher ATP binding affinity. When we compare the ATP Km values of the BRAFWT and BRAFV600E proteins produced in Sf9 cells, no significant difference is observed (Figure 4c), consistent with the presence of the active phosphorylated form of BRAFWT.
To probe the roles of Lys507 and Arg575 in stabilizing the open active conformation of BRAF to facilitate ATP binding, K507A and R575A mutants were prepared in the context of BRAFWT and BRAFV600E. We hypothesized that the loss of the positive charge will undermine the association of Lys507 and/or Arg575 with Glu600, which in turn would disrupt the ability of the activation loop to adopt an active conformation to facilitate ATP binding. The results of the analysis of these mutants show that a lysine to alanine substitution at position 507 does indeed increase the ATP Km value by ∼5–10-fold for BRAFWT and BRAFV600E, while an arginine to alanine substitution at position 575 shows a more modest effect on ATP Km of ∼2-fold (Figure 4c). These results suggest that Lys507 and, to a lesser extent, Arg575 both contribute in forming salt bridges to stabilize the open active conformation.
Structural Implications for the Future Design of BRAFV600E Selective Organometallic BRAF Inhibitors
Close examination of the ATP binding pockets of the inactive and active forms of BRAF kinase reveals major differences in the ATP binding pockets. As shown in panels a and b of Figure 5, the active site pocket possesses a much larger ATP phosphate binding space and the active BRAF conformation also has a unique hydrophobic pocket only present in the active conformation of the BRAF kinase. This pocket, which is formed by residues Thr529, Leu514, Phe595, Gly593, and Leu505 (Figure 5a), has been proposed to be a “Raf selective pocket” in a more recent study (38). The CS292 inhibitor also binds in a unique orientation, pointing its O2 carbonyl toward this hydrophobic pocket (Figure 5c). A close examination of the path leading to this “Raf-specific pocket” reveals that this path is 11 Å long with the side chain of residue Asp594 and other main chain atoms outlining the interior. We hypothesize that the derivatization on the carbonyl or the adjacent methylpyridine moiety to extend further toward the Raf selectivity pocket would enhance the inhibitory potency. Since Asp594 adopts distinct conformations in the active BRAF conformation compared to the inactive BRAF conformation, designing a linker supporting the protrusion into the Raf-specific pocket while it still interacts with the active conformation of Asp594 will most likely improve the selectivity of the next generation of inhibitors against the BRAFV600E oncogenic mutant kinase over the wild-type enzyme. Indeed, replacement of pyridine with isoquinoline (compound CNSE030) enhances inhibition potency and selectivity toward the BRAFV600E oncogenic mutant by ∼3-fold relative to those of compound CS292 (Figure 5d).
Figure 5.
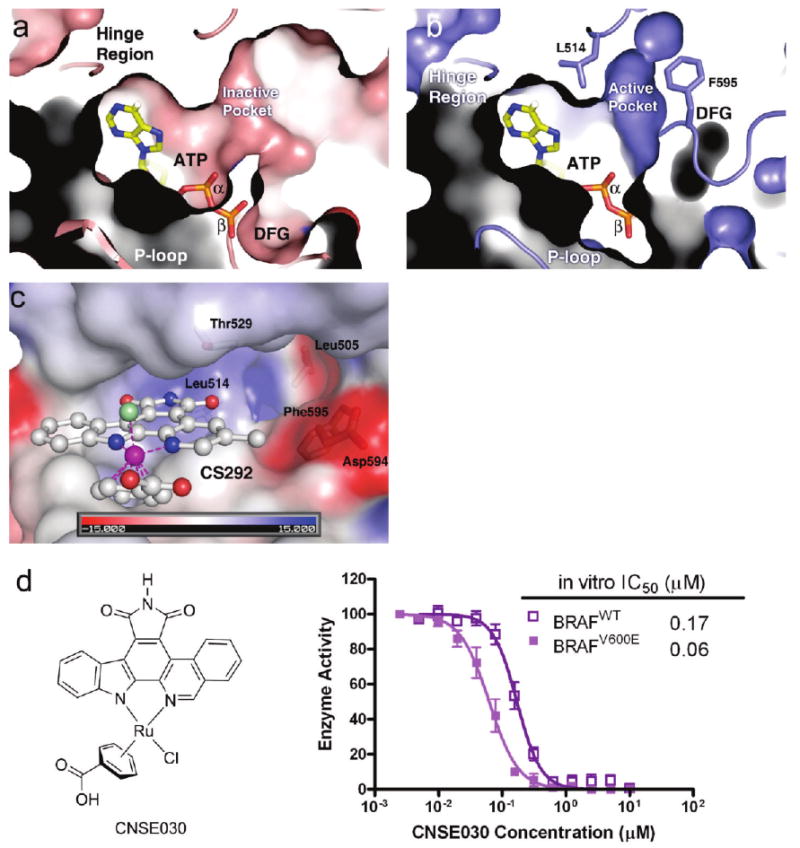
Details of the ATP binding site of the active BRAF conformation. (a) Superposition of ATP in the ATP binding site of BRAF in an inactive conformation (BRAF–Sorafenib). Note that the β- and γ-phosphates of ATP clash with the ATP binding pocket due to the inward activation loop. (b) Superposition of ATP in the ATP binding site of BRAF in an active conformation (BRAF–CS292). Phe595 and Leu514 are shown in stick model to indicate the active pocket. Note the different positions of the DFG motif indicating dramatic differences in activation loop conformation. (c) Electrostatic potential maps of the BRAF–CS292 complex structure highlighting the residues forming the “BRAF-specific pocket”. (d) Chemical structure of compound CNSE030 and its inhibition against BRAFWT and BRAFV600E. The data represent an average of triplicate measurements.
Taken together, the structural differences between the active and inactive conformations of the BRAF kinase, together with the structure of the organometallic CS292 inhibitor bound to the active form of the BRAF kinase, provide an entry point for the design of second-generation potent BRAFV600E oncogenic mutant-specific organometallic inhibitors.
Acknowledgments
We thank Dr. Richard Marais (The Institute of Cancer Research, London, U.K.) for providing the full-length human BRAF clone, Dr. Wade Harper (Harvard Medical School, Boston, MA) for providing the full-length mouse p50cdc37 clone, Michael Olson (Beatson Institute for Cancer Research, Glasgow, U.K.) for providing the full-length human MEK1 clone, and the Wistar Protein Expression facility for preparing recombinant BRAF and BRAFV600E proteins in insect cells. This paper is dedicated to the memory of Peng Xie.
Footnotes
This work was supported by Grant CA 114046 from the National Institutes of Health.
Abbreviations: CI, chemical ionization; c-KIT, proto-oncogene receptor tyrosine kinase; DCC, dicyclohexylcarbodiimide; DCM, dichloromethane; ERK, extracellular signal-regulated kinase; ES, electrospray; FDA, Food and Drug Administration; GSK3β, glycogen synthase kinase 3 β isoform; HRP, horseradish peroxidase; MAPK, mitogen-activated protein kinase; MEK, dual-specificity mitogen-activated protein kinase kinase; PDB, Protein Data Bank; PDGFR, platelet-derived growth factor receptor; PI3K, phosphatidylinositol 3-kinase; RAF, RAF proto-oncogene serine/threonine-protein kinase (subtypes A, B, and C); RAS, small G protein Ras; RTK, receptor tyrosine kinase; TBAF, tetrabutylammonium fluoride; THF, tetrahydrofuran; TMSEtOH, trimethylsilylethanol; VEGFR, vascular endothelial growth factor receptor.
References
- 1.Rapp UR, Goldsborough MD, Mark GE, Bonner TI, Groffen J, Reynolds FH, Jr, Stephenson JR. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc Natl Acad Sci USA. 1983;80:4218–4222. doi: 10.1073/pnas.80.14.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jansen HW, Lurz R, Bister K, Bonner TI, Mark GE, Rapp UR. Homologous cell-derived oncogenes in avian carcinoma virus MH2 and murine sarcoma virus 3611. Nature. 1984;307:281–284. doi: 10.1038/307281a0. [DOI] [PubMed] [Google Scholar]
- 3.Sutrave P, Bonner TI, Rapp UR, Jansen HW, Patschinsky T, Bister K. Nucleotide sequence of avian retroviral oncogene v-mil: Homologue of murine retroviral oncogene v-raf. Nature. 1984;309:85–88. doi: 10.1038/309085a0. [DOI] [PubMed] [Google Scholar]
- 4.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 5.Jaiswal RK, Moodie SA, Wolfman A, Landreth GE. The mitogen-activated protein kinase cascade is activated by B-Raf in response to nerve growth factor through interaction with p21ras. Mol Cell Biol. 1994;14:6944–6953. doi: 10.1128/mcb.14.10.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catling AD, Reuter CW, Cox ME, Parsons SJ, Weber MJ. Partial purification of a mitogen-activated protein kinase kinase activator from bovine brain. Identification as B-Raf or a B-Raf-associated activity. J Biol Chem. 1994;269:30014–30021. [PubMed] [Google Scholar]
- 7.Moodie SA, Paris MJ, Kolch W, Wolfman A. Association of MEK1 with p21ras.GMPPNP is dependent on B-Raf. Mol Cell Biol. 1994;14:7153–7162. doi: 10.1128/mcb.14.11.7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pritchard CA, Samuels ML, Bosch E, McMahon M. Mol Cell Biol. Vol. 15. 1995. Conditionally oncogenic forms of the A-Raf and B-Raf protein kinases display different biological and biochemical properties in NIH 3T3 cells; pp. 6430–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wojnowski L, Stancato LF, Larner AC, Rapp UR, Zimmer A. Overlapping and specific functions of Braf and Craf-1 proto-oncogenes during mouseembryogenesis. Mech Dev. 2000;91:97–104. doi: 10.1016/s0925-4773(99)00276-2. [DOI] [PubMed] [Google Scholar]
- 10.Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 1999;18:2137–2148. doi: 10.1093/emboj/18.8.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 12.Ikenoue T, Hikiba Y, Kanai F, Tanaka Y, Imamura J, Imamura T, Ohta M, Ijichi H, Tateishi K, Kawakami T, Aragaki J, Matsumura M, Kawabe T, Omata M. Functional analysis of mutations within the kinase activation segment of B-Raf in human colorectal tumors. Cancer Res. 2003;63:8132–8137. [PubMed] [Google Scholar]
- 13.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 14.Satyamoorthy K, Li G, Gerrero MR, Brose MS, Volpe P, Weber BL, Van Belle P, Elder DE, Herlyn M. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res. 2003;63:756–759. [PubMed] [Google Scholar]
- 15.Wellbrock C, Ogilvie L, Hedley D, Karasarides M, Martin J, Niculescu-Duvaz D, Springer CJ, Marais R. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004;64:2338–2342. doi: 10.1158/0008-5472.can-03-3433. [DOI] [PubMed] [Google Scholar]
- 16.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/ MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 17.Fecher LA, Amaravadi RK, Flaherty KT. The MAPK pathway in melanoma. Curr Opin Oncol. 2008;20:183–189. doi: 10.1097/CCO.0b013e3282f5271c. [DOI] [PubMed] [Google Scholar]
- 18.Venetsanakos E, Stuart D, Tan N, Ye H, Salangsang F, Aardalen K, Faure M, Heise C, Mendel D, Jallal B. CHIR-265, a novel inhibitor that targets B-Raf and VEGFR, shows efficacy in a broad range of preclinical models. 97th AACR Annual Meeting; Washington, DC. 2006. Abstract 4854. [Google Scholar]
- 19.Tsai J, Zhang J, Bremer R, Artis R, Hirth P, Bollag G. Development of a novel inhibitor of oncogenic B-Raf. 97th AACR Annual Meeting; Washington, DC. 2006. Abstract 2412. [Google Scholar]
- 20.Stuart DS, Aardalen KM, Lorenzana EG, Salangsang FD, Venetsanakos E, Tan N, Zhang W, Garrett E, Jallal B, Mendel DB. Characterization of a novel Raf kinase inhibitor that causes target dependent tumor regression in human melanoma xenografts expressing mutant B-Raf. 97th AACR Annual Meeting; Washington, DC. 2006. Abstract 4856. [Google Scholar]
- 21.Amiri P, Aikawa ME, Dove J, Stuart DD, Poon D, Pick T, Ramurthy S, Subramanian S, Levine B, Costales A, Harris A, Paul R. CHIR-265 is a potent selective inhibitor of c-Raf/ B-Raf/mutB-Raf that effectively inhibits proliferation and survival of cancer cell lines with Ras/Raf pathway mutations. 97th AACR Annual Meeting; Washington, DC. 2006. Abstract 4855. [Google Scholar]
- 22.Niculescu-Duvaz I, Roman E, Whittaker SR, Friedlos F, Kirk R, Scanlon IJ, Davies LC, Niculescu-Duvaz D, Marais R, Springer CJ. J Med Chem. Vol. 49. 2006. Novel inhibitors of B-RAF based on a disubstituted pyrazine scaffold. Generation of a nanomolar lead; pp. 407–416. [DOI] [PubMed] [Google Scholar]
- 23.Newbatt Y, Burns S, Hayward R, Whittaker S, Kirk R, Marshall C, Springer C, McDonald E, Marais R, Workman P, Aherne W. Identification of inhibitors of the kinase activity of oncogenic V600E BRAF in an enzyme cascade high-throughput screen. J Biomol Screening. 2006;11:145–154. doi: 10.1177/1087057105283584. [DOI] [PubMed] [Google Scholar]
- 24.Takle AK, Brown MJ, Davies S, Dean DK, Francis G, Gaiba A, Hird AW, King FD, Lovell PJ, Naylor A, Reith AD, Steadman JG, Wilson DM. The identification of potent and selective imidazole-based inhibitors of B-Raf kinase. Bioorg Med Chem Lett. 2006;16:378–381. doi: 10.1016/j.bmcl.2005.09.072. [DOI] [PubMed] [Google Scholar]
- 25.Ouyang B, Knauf JA, Smith EP, Zhang L, Ramsey T, Yusuff N, Batt D, Fagin JA. Inhibitors of Raf kinase activity block growth of thyroid cancer cells with RET/PTC or BRAF mutations in vitro and in vivo. Clin Cancer Res. 2006;12:1785–1793. doi: 10.1158/1078-0432.CCR-05-1729. [DOI] [PubMed] [Google Scholar]
- 26.Khire UR, Bankston D, Barbosa J, Brittelli DR, Caringal Y, Carlson R, Dumas J, Gane T, Heald SL, Hibner B, Johnson JS, Katz ME, Kennure N, Kingery-Wood J, Lee W, Liu XG, Lowinger TB, McAlexander I, Monahan MK, Natero R, Renick J, Riedl B, Rong H, Sibley RN, Smith RA, Wolanin D. Omega-carboxypyridyl substituted ureas as Raf kinase inhibitors: SAR of the amide substituent. Bioorg Med Chem Lett. 2004;14:783–786. doi: 10.1016/j.bmcl.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 27.Meggers E, Atilla-Gokcumenm GE, Bregman H, Maksimoska J, Mulcahy SP, Pagano N, Williams DS. Exploring chemical space with organometallics: Ruthenium complexes as protein kinase inhibitors. Synlett. 2007;8:1177–1189. [Google Scholar]
- 28.Williams DS, Carroll PJ, Meggers E. Platinum complex as a nanomolar protein kinase inhibitor. Inorg Chem. 2007;46:2944–2946. doi: 10.1021/ic062055t. [DOI] [PubMed] [Google Scholar]
- 29.Debreczeni JE, Bullock AN, Atilla GE, Williams DS, Bregman H, Knapp S, Meggers E. Ruthenium half-sandwich complexes bound to protein kinase Pim-1. Angew Chem, Int Ed. 2006;45:1580–1585. doi: 10.1002/anie.200503468. [DOI] [PubMed] [Google Scholar]
- 30.Bregman H, Carroll PJ, Meggers E. Rapid access to unexplored chemical space by ligand scanning around a ruthenium center: Discovery of potent and selective protein kinase inhibitors. J Am Chem Soc. 2006;128:877–884. doi: 10.1021/ja055523r. [DOI] [PubMed] [Google Scholar]
- 31.Smalley KS, Contractor R, Haass NK, Kulp AN, Atilla-Gokcumen GE, Williams DS, Bregman H, Flaherty KT, Soengas MS, Meggers E, Herlyn M. An organometallic protein kinase inhibitor pharmacologically activates p53 and induces apoptosis in human melanoma cells. Cancer Res. 2007;67:209–217. doi: 10.1158/0008-5472.CAN-06-1538. [DOI] [PubMed] [Google Scholar]
- 32.Atilla-Gokcumen GE, Williams DS, Bregman H, Pagano N, Meggers E. Organometallic compounds with biological activity: A very selective and highly potent cellular inhibitor for glycogen synthase kinase 3. ChemBioChem. 2006;7:1443–1450. doi: 10.1002/cbic.200600117. [DOI] [PubMed] [Google Scholar]
- 33.Williams DS, Atilla GE, Bregman H, Arzoumanian A, Klein PS, Meggers E. Switching on a signaling pathway with an organoruthenium complex. Angew Chem, Int Ed. 2005;44:1984–1987. doi: 10.1002/anie.200462501. [DOI] [PubMed] [Google Scholar]
- 34.Xie P, Williams DS, Atilla-Gokcumen GE, Milk L, Xiao M, Smalley KS, Herlyn M, Meggers E, Marmorstein R. Structure-based design of an organoruthenium phosphatidyl-inositol-3-kinase inhibitor reveals a switch governing lipid kinase potency and selectivity. ACS Chem Biol. 2008;3:305–316. doi: 10.1021/cb800039y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knighton DR, Zheng JH, Ten Eyck LF, Xuong NH, Taylor SS, Sowadski JM. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- 36.Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 37.King AJ, Patrick DR, Batorsky RS, Ho ML, Do HT, Zhang SY, Kumar R, Rusnak DW, Takle AK, Wilson DM, Hugger E, Wang L, Karreth F, Lougheed JC, Lee J, Chau D, Stout TJ, May EW, Rominger CM, Schaber MD, Luo L, Lakdawala AS, Adams JL, Contractor RG, Smalley KS, Herlyn M, Morrissey MM, Tuveson DA, Huang PS. Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885. Cancer Res. 2006;66:11100–11105. doi: 10.1158/0008-5472.CAN-06-2554. [DOI] [PubMed] [Google Scholar]
- 38.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, Sproesser K, Li L, Smalley KS, Fong D, Zhu YL, Marimuthu A, Nguyen H, Lam B, Liu J, Cheung I, Rice J, Suzuki Y, Luu C, Settachatgul C, Shellooe R, Cantwell J, Kim SH, Schlessinger J, Zhang KY, West BL, Powell B, Habets G, Zhang C, Ibrahim PN, Hirth P, Artis DR, Herlyn M, Bollag G. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimela-noma activity. Proc Natl Acad Sci USA. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collaborative Computational Project Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 40.Vagin A, Teplyakov A. MOLREP: An automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 41.Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 43.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. 2004;D60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 44.Bregman H, Williams DS, Atilla GE, Carroll PJ, Meggers E. An Organometallic Inhibitor for Glycogen Synthase Kinase 3. J Am Chem Soc. 2004;126:13594–13595. doi: 10.1021/ja046049c. [DOI] [PubMed] [Google Scholar]
- 45.Pagano N, Maksimoska J, Bregman H, Williams DS, Webster RD, Xue F, Meggers E. Ruthenium half-sandwich complexes as protein kinase inhibitors: derivation of the pyridocarbazole pharmacophore ligand. Org Biomol Chem. 2007;5:1218–1227. doi: 10.1039/b700433h. [DOI] [PubMed] [Google Scholar]
- 46.Zelonka RA, Baird MC. Benzene complexes of ruthenium (II) Can J Chem. 1972;503063 [Google Scholar]
- 47.Bregman H, Carroll PJ, Meggers E. Rapid Access to Unexplored Chemical Space by Ligand Scanning around a Ruthenium Center: Discovery of Potent and Selective Protein Kinase Inhibitors. J Am Chem Soc. 2006;128:877–884. doi: 10.1021/ja055523r. [DOI] [PubMed] [Google Scholar]
- 48.Kuehne ME, Lambert BF. The Reduction of Aromatic Acids and Amides by Sodium in Liquid Ammonia. J Am Chem Soc. 1959;81:4278–4287. [Google Scholar]