

Abstract
Despite the fact that they are orphan diseases, congenital myasthenic syndromes (CMS) challenge those who suffer from it by causing fatigable muscle weakness, in the most benign cases, to a progressive wasting of muscles that may sentence patients to a wheelchair or even death. Compared to other more common neurological diseases, CMS are rare. Nevertheless, extensive research in CMS is performed in laboratories such as ours. Among the diverse neuromuscular disorders of CMS, we are focusing in the slow-channel congenital myasthenic syndrome (SCS), which is caused by mutations in genes encoding acetylcholine receptor subunits. The study of SCS has evolved from clinical electrophysiological studies to in vitro expression systems and transgenic mice models. The present review evaluates the methodological approaches that are most commonly employed to assess synaptic impairment in SCS and also provides perspectives for new approaches. Electrophysiological methodologies typically employed by physicians to diagnose patients include electromyography, whereas patient muscle samples are used for intracellular recordings, single-channel recordings and toxin binding experiments. In vitro expression systems allow the study of a particular mutation without the need of patient intervention. Indeed, in vitro expression systems have usually been implicated in the development of therapeutic strategies such as quinidine- and fluoxetine-based treatments and, more recently, RNA interference. A breakthrough in the study of SCS has been the development of transgenic mice bearing the mutations that cause SCS. These transgenic mice models have actually been key in the elucidation of the pathogenesis of the SCS mutations by linking IP-3 receptors to calcium overloading, as well as caspases and calpains to the hallmark of SCS, namely endplate myopathy. Finally, we summarize our experiences with suspected SCS patients from a local perspective and comment on one aspect of the contribution of our group in the study of SCS.
Keywords: Acetylcholine receptor, congenital myasthenic syndromes, slow-channel congenital myasthenic syndromes
Congenital myasthenic syndromes (CMS) are a group of orphan disorders of neuromuscular transmission characterized by muscle weakness and fatigability in the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The severity and course of the disease is highly variable, ranging from minor symptoms to progressive disabling weakness. CMS is due to a defect in an essential component of the transmission at the neuromuscular junction (NMJ) (1–3). Investigation of CMS has provided insights that have greatly enhanced our understanding of the development, maintenance, and function of the NMJ. The development of CMS animal models as a complementary approach to investigate the mechanisms for endplate degeneration in the aforementioned syndrome has confirmed the value of transgenic animal models in studying NMJ disorders. Understanding the molecular mechanisms leading to the pathogenesis of different forms of CMS is essential to develop new strategies for therapeutic intervention.
The study of CMS has been facilitated by the accessibility of the NMJ and the sophisticated techniques available for its study. Clinical electrophysiological studies have demonstrated the impairment of neuromuscular transmission in CMS in vivo. Pre- or post-synaptic defect localization have been determined by in vitro electrophysiological recordings of live biopsied muscle (4). An increasing number of known molecular targets in the NMJ have identified the genetic lesion and the type of CMS by ultrastructural and cytochemical studies, bungarotoxin-binding, molecular genetic test, and antibody binding assays (1, 3).
Many of the CMS cases reported are developmental disorders, resulting from absence or loss of function of a protein critical for NMJ development (2). These disorders are recessively inherited, usually non-progressive and result in fatigability and weakness, impaired skeletal muscle development and sometimes congenital joint and skeletal deformities. In most cases of CMS, the NMJ is frequently under-developed, providing a relatively forthright basis for the weakness. The exception to this is the slow-channel congenital myasthenic syndrome (SCS) because it increases the synaptic response to acetylcholine neurotransmitter. SCS is the focus of this review. SCS is usually an autosomal dominant inherited disorder caused by mutation in the genes encoding for the alpha1, beta1, delta, or epsilon subunit of the acetylcholine receptor (Table 1). The nature of the progressive weakness, impaired neuromuscular transmission, and degeneration of the NMJ in SCS has proven to be a complex interplay between developmental and degenerative processes acting at pre- and post-synaptic sites (5). The characterization of each of these processes may shed some light on the mechanisms governing both synaptic plasticity and synaptic disease. Several independent laboratories are working to elucidate the pathogenesis mechanisms and therapeutic strategies for the progressive synaptic degeneration seen in SCS.
Table 1.
Summary of the genes encoding human acetylcholine receptor at the neuromuscular junction.
| AchR subunit | GeneID/MIM number* | Gene symbol | Chromosome number | Gene map locus | Cloning reference |
|---|---|---|---|---|---|
| Alpha (α1) | 1134; 100960 | CHRNA1 | 2 | 2q24-q32 | (60–63) |
| Beta (β1) | 1140; 100710 | CHRNB1 | 17 | 17p13.1 | (62, 64) |
| Delta (δ) | 1144; 100720 | CHRND | 2 | 2q33-q34 | (62, 65–67) |
| Epsilon (ε) | 1145; 100725 | CHRNE | 17 | 17p13-p12 | (68) |
GeneID in Entrez Gene database and MIM number in Online Mendelian Inheritance in Man database.
Acetylcholine receptor structure and function
A principal component of the NMJ is the acetylcholine receptor (AChR), a ligand-gated ion channel consisting of homologous subunits in the stoichiometry of 2α1β1δε in the adult form or 2α1β1δγ in the fetal form. The AChR belongs to the superfamily of the ligand-gated ion channel receptors that are of fundamental importance in the chemical synaptic transmission throughout the nervous system (6–8). The AChR is composed of four homologous membrane-spanning subunits forming a cation-selective ion channel (9). Each subunit contains a large hydrophilic extracellular N-terminus, four putative transmembrane domains (M1–M4), a large cytoplasmic loop between the M3 and M4, and a short extracellular C-terminus (10). The M1 and M2 domains form the ion channel pore (11–12), whereas the M3 and M4 domains have the largest contact with the membrane lipids (13–14) and are distant from the ion channel pore and the ligand-binding sites (Figure 1). The AChR at the endplate (EP) of NMJ is activated by binding of the chemical neurotransmitter acetylcholine (ACh) released by the motor neuron. This produces an endplate potential (EPP) that spreads across the surface of the muscle fiber into the transverse tubules, eliciting the release of calcium from the sarcoplasmic reticulum, thus initiating muscle contraction (15).
Figure 1.

Slow-channel congenital myasthenic syndrome mutations and acetylcholine receptor structure. A) Top view of the AChR. The stereo-stoichiometry relationship of the five subunits, 2α1β1δγ, of the AChR are highlighted by colors. B) Side view of the AChR. The δ subunit was omitted for clarity. Some SCS mutations are shown and identified in the insert (the insert zooms in the transmembrane region). C) Top view of the transmembrane domains of the AChR The majority of the SCS mutations are clustered into the ion channel pore, M1 and M2 domains. The SCS mutations are colored according to legend.
Slow-Channel Congenital Myasthenic Syndrome
SCS, usually an autosomal dominant disorder of postsynaptic CMS, is caused by a kinetic abnormality of the AChR which comprises the healthy safety margin of neuromuscular transmission. SCS is characterized by the slow decay of endplate currents (Figure 2), destabilization of the closed channel state, prolonged activation episodes of AChR, and an endplate myopathy caused by cationic overloading of the muscle fibers. The precise diagnosis of SCS requires a multidisciplinary team comprising neurologists, geneticists, biochemists, and electrophysiologists to correlate the clinical phenotypes with in vitro electrophysiological, morphological, and molecular genetic studies.
Figure 2.

Classical electrophysiological features of slow-channel congenital myasthenic syndrome. A) Compound muscle action potentials (CMAPs) recorded over the hypothenar muscles evoked by single nerve stimulus. As is typical for SCS, there is a repetitive response to the single stimulus. B) CMAPs evoked by repetitive stimulation. The amplitude of each of four successive responses decreases, indicating failure of neuromuscular transmission. C) Superimposed miniature endplate currents recorded from anconeus muscle of control and the proband. The prolonged waveform of the patient miniature endplate current (MEPC) has a decay time constant of 31 msec, compared with control of 3.2 msec. The MEPC amplitude of the patient is about 50% of the control. Scale bars = A: 5 mV, 50 msec; B: 1 mV, 50 msec; C: 1 nA, 10 msec. (Figure 2 is reproduced by permission from Gómez et al. (2002) Annals of Neurology 51: 102–112).
Clinical features
Unlike other CMS, SCS tipically shows an autosomal dominant inheritance (Table 2). The clinical phenotype of SCS is variable as patients may express a severe early onset to moderate late onset (5, 16). In most cases, SCS patients present a severe weakness of the cervical, scapular, wrist, and finger extensor muscles (17). However, in selected cases ptosis, ophthalmoparesis, dysarthria, dysphagia, proximal limb weakness, and respiratory insufficiency may also occur.
Table 2.
Slow-channel congenital myasthenic syndrome kindred and acetylcholine receptor mutations.
| Mutation/Domain | DB1 | Onset/Activity2 | Medical Exam3 | Grade4 | MEPP amplitude5 | MEPC decay6 | EpM7 | αBT8 | References |
|---|---|---|---|---|---|---|---|---|---|
| αG153S N-terminal | 1 | Infant-53/sports | Ocular, hand, arm | 1–2 | 36–90% | 7.8/26.4 | Yes | Low | (20, 37) |
| αV156M N-terminal | - | 14 | No ocular; neck, jaw, limb | 1 | NA | NA | NA | NA | (37) |
| αN217K M1 | 9, 10 | 8–57 | Ocular, hand, arm, neck, gait | 2 | 43–69% | 3.6/22.1 | Yes | Low | (21, 69) |
| αV249F M2 | - | Infant/wc 8 | Severe generalized | 3 | 44% | 2.4/43.9 | Yes | 39% | (19) |
| αT254I M2 | - | 16 | No ocular; neck, arm | 1 | NA | NA | NA | NA | (37) |
| αS269I M2 | - | 23 | Arm, jaw, legs | 1 | Normal | 20/28* | NA | 44% | (37) |
| αC418W M4 | - | Infant-teens | Ocular, arm, legs | 2 | NA | NA | NA | NA | (42) |
| βV229F M1 | 7 | 18–50 | Hand, neck, arms | 1 | 20% 0.67 | 2.25/26.1 | Yes (ps) | NA | (41, 69) |
| βL262M M2 | 4 | inf/wc 13 | Severe generalized, atrophy | 3 | 19% | 38.6* | Yes | High | (36) |
| βV266M M2 | 13 | Infant | Ocular, face, limb | 2–4 | 43–69% | 3.5/35.6 | Yes | Low | (21, 69) |
| δS268F M2 | 6 | Infant/wc teens | No atrophy, severe generalized | 4 | 50% | 31* | Yes (ps) | NA | (39, 69) |
| εL78P N-terminal | 22–29 | Ocular, face, neck, shoulder, hand, hip | 1 | NA | NA | NA | NA | (40) | |
| εL221F M1 | - | 17–40s | Ocular, wrist, hand, neck | 1 | NA | NA | NA | NA | (40) |
| εT264P M2 | - | Infant/wc teens | Severe limb, neck | 3 | Reduced | 1.5/16.5 | Yes | 39% | (18) |
| εL269F M2 | 2, 5 | Infant | Severe focal, resp insuf. | 3 | 36–69% | 3.3/50 | Yes | NA | (21, 52, 55) |
Kindred/Proband number in the SCS database
Age or range of age of onset within kindred./Level of activity or disability. wc = wheelchair.
Most prominent features of weakness.
Score defined as 1: onset after infancy, limb, ocular weakness minimal limitation of activity; 2: onset in infancy, limitation of activity; 3: respiratory insufficiency, confinement to wheelchair; 4: death from respiratory failure.
Amplitude of miniature endplate currents or miniature endplate potentials. Shown is percent of control. NA=not available.
Time constant(s) of decay of miniature endplate current or potentials in milliseconds (ms). Normal = 2–3.3 ms.
MEPP.
noise analysis.
EpM=endplate myopathy confirmed. yes=present; n=absent. ps= presynaptic-predominant endplate myopathy.
α-Bungarotoxin binding sites on endplates. Shown is percent of control, reduced or increased.
Pathophysiology
SCS is etiologically produced by mutations in the genes encoding AChR subunits that cause a kinetic abnormality of the AChR (18–19). The SCS mutations produce an increase in the channel opening rate, a decrease in the channel closing rate, or an increase in the affinity of ACh for the AChR, resulting in the stabilization of the open state or the destabilization of the closed state of the AChR (18, 20–21) (Figure 3). The net effect of the mutation is a gain of function that abnormally prolongs channel opening events, slows the decay of endplate currents and potentials, and permits the cationic overload of the synaptic region of the muscle fiber. The prolonged opening episodes of the AChR channel lead to: 1) a slow decay of the synaptic potential, eventually causing a depolarization block, 2) evoke a repetitive compound muscle action potential (CMAP) caused by the EPP outlasting the absolute refractory period of the muscle fiber, 3) a cationic overload of the muscle endplate mainly contributed by the excessive accumulation of Ca2+, and 4) an endplate myopathy characterized by degeneration of the junctional folds and static weakness (5, 19, 21–22). The endplate myopathy is mainly attributed to the excessive accumulation of Ca2+ in muscle fiber and is characterized by the ultrastructure remodeling of the endplate, which consists in the widening of the synaptic space, calcium deposits, destruction of the junctional folds, vacuolizations, and tubular aggregates.
Figure 3.

Single-channel analysis of wild-type mouse and δS268F acetylcholine receptor. A) Current-voltage relationships for single-channel currents recorded from wild-type (WT) (open squares) or δS268F-acetylcholine receptor (AChR) (open circles) expressed in X. laevis oocytes. B) WT mouse and δS268F AChR single-channel currents recorded at low acetylcholine (ACh) concentration. AChR single-channel currents recorded from cell-attached patches on X. laevis oocytes expressing WT AChR (left) and AChRs expressing the δS268F mutation (right). All channels were recorded at a holding potential of 100 mV, 4 μM ACh, filtered at 5 kHz, sampling rate at 20 μsec per point and temperature of 22°C. Openings are shown as downward deflections, with close state denoted by the c, and the open state denoted by o. Representative histograms for current amplitude and open time distributions are shown for each of the AChR tested. Amplitude histograms were constructed from events list files created to include all event amplitudes that could be detected. The resulting distribution data was fitted with a Gaussian function with the appropriate number of components. Both the WT and mutant AChR show a single amplitude component of 7.874 ± 0.883 pA and 7.371 ± 0.465 pA, respectively. Open time histograms were fitted with exponential functions of the appropriate number of components using the maximum likelihood algorithm. The WT AChR shows two open time constants: τo1 = 0.411 msec (f = 0.229) and τo2 = 1.435 (f = 0.711), while the δS268F AChR shows two open time components of τo1 = 0.826 msec (f = 0.164) and τo2 = 10.582 msec (f = 0.896). Data were filtered at 5 kHz for each current sample display. Scale values are 5 pA for the vertical bar and 10 msec for the horizontal bar. C) Kinetics of activation of WT and δS268F AChR at high ACh concentrations. (Left) Single-channel currents elicited by the indicated concentration of ACh recorded from cell-attached patches from X. laevis oocytes expressing adult mouse AChRs containing the WT δ (upper) or mutated δS268F (lower) subunit. Recordings were done at holding potential of 100 mV, filtered at 5 kHz, sampling rate at 20 μsec per point and temperature of 22°C. Channel openings are shown as downward deflections. (Center and right) Open and closed time duration histograms for adult mouse AChRs containing the WT δ (upper) or mutated δS268F (lower) subunits generated and fitted using pSTAT6. Fitting was done using the simplex least-squares algorithm. Single-channel currents displayed were filtered at 4 kHz. (Figure 3 is reproduced by permission from Gómez et al. (2002) Annals of Neurology 51: 102–112).
Diagnosis
Diagnosis of SCS should be considered in any person that has been considered for myasthenia gravis (MG). Although there is a rare autoimmune form of the disease, SCS patients typically have no serum anti-AChR antibodies (23). The presence of a positive family history for autosomal dominant inheritance of a myasthenic syndrome should strongly suggest a SCS. In these patients and those with sporadic disease, the presence of repetitive CMAPs evoked by single nerve stimuli, particularly with decremental responses to repetitive stimulation in small muscles of the upper limb, strongly suggest the disease. Anti-cholinesterase toxicity and the condition of congenital endplate acetylcholinesterase must be excluded. In vitro electrophysiological studies, as described above, definitively establish the diagnosis. Genetic diagnosis, required to identify the responsible mutation, is available only on a research basis.
Diagnosis of SCS can be challenging for several reasons. The wide range in clinical presentation from severe congenital weakness to subtle fatigability noted in the fifth decade means that there is no classical presentation. Patients with SCS have been mistaken for having a variety of other disorders including MG, mitochondrial myopathy, and oculopharyngeal dystrophy. Difficulties relating predominantly to muscle sampling are sometimes encountered in eliciting repetitive CMAPs; the classic feature suggesting prolonged action of ACh. Both electrophysiological studies of biopsied muscle and molecular genetic testing are only available on a research basis.
Treatment
At present, SCS patients are treated with quinidine sulfate and fluoxetine hydrochloride (24–31). Both therapeutic medications are long-lived open-channel blockers of AChR that shorten the duration of channel opening events in a concentration dependent manner. Quinidine is orally administrated at a dose of 200 mg two to three times daily for one week in adults. After the first week, the dose is gradually increased to maintain a serum level of 1.0 – 2.5 μg/mL (3.0 – 7.5 μM/L) (27–28). In children, quinidine is administrated at a dose of 15 – 60 mg/kg/day in four to six divided doses. Fluoxetine, an alternative drug to treat SCS patients allergic to quinidine, is less prescribed and less effective than quinidine, but is eliminated more slowly, thus providing a more sustained serum levels (29). Fluoxetine is also orally administrated until a total daily dose of 80 to 120 mg (serum fluoxetine + norfluoxetine levels 8 to 11 μM/L) in adults (29). However, the maximal dose of fluoxetine for children has not been established hitherto. The therapy progress requires monitoring of routine laboratory analysis, physical, and electrophysiological examination (31). The physical evaluation includes measures of the arm elevation time, the number of times a patient can rise from squatting or from a low stool, the degree of eyelid ptosis, the number of steps they can climb or the distance they can walk before having to rest, the degree of weakness of selected muscles, maximal inspiratory and expiratory pressures, and maximal expiratory flow (27–28). The needle electromyographic (EMG) analysis includes measures of amplitude and area of the motor unit action potentials and CMAP (5).
In Vitro Expression Studies
The identification of a SCS is a clinical process. Physicians evaluate symptoms such as ocular and other muscle weakness, perform tests as EMGs, and check for anti-AChR antibodies. If anti-AChRs antibodies are not detected, congenital myasthenic syndromes are often suspected. On a research basis, a genetic analysis is then required to test this possibility. In addition, intracellular recordings as those required to probe miniature endplate potentials (MEPP), miniature endplate currents (MEPC), endplate potentials (EPP) and endplate currents (EPC) are performed directly on patient muscle biopsies. Patch clamp experiments to determine single-channel kinetics and conductance may also be performed on isolated muscle fibers from patients. Nevertheless, in vitro expression studies, more often than not, follow the clinical and ex vivo studies. These studies include the expression of recombinant AChR engineered to contain the mutation of interest in a suitable expression system such as Xenopus laevis oocytes and Human Embryonic Kidney 293 cells (HEK 293 cells). The characterization of AChR SCS mutations is facilitated by in vitro expression studies as the number of involved researchers is not limited by access to patients and because X. laevis oocytes and HEK 293 cells are amenable to patch clamping and toxin binding assays among other commonly employed techniques. An additional advantage of the in vitro studies is that they have facilitated the development of strategies to selectively silence the AChR mutations that cause SCS in an allele-specific manner through RNA interference. In the following paragraphs, a set of the methodological approaches most frequently followed to study SCS using in vitro expression systems and the more recent efforts to selectively silence pathogenic AChR mutations are briefly discussed in the context of the progress that has been achieved.
Patch clamp
AChR mutations that cause SCS are characterized by prolonged openings and as such, a direct approach for their identification is through the use of the patch clamp technique. Developed in 1976 by German scientists Erwin Neher and Bert Sakmann (32), the patch clamp technique enables the researcher to probe the activity of individual ion channels and to interpret the single-channel data in terms of mechanism. Indeed, not too long after the development of the technique, the necessary theory to interpret single-channel events in terms of functional mechanisms began to arise by David Colquhoun and Alan G. Hawkes (33–34). Soon after developing the patch clamp technique, Neher and Sakmann realized its full potential and in 1981 released a paper describing several patch clamp configuration, all of which are frequently used up to these days (35). Among these, the cell-attached configurations has been commonly used to study SCS as open times are readily measured (18–20, 36–43). Edwin Neher and Bert Sachmann were awarded the Nobel Prize in Physiology or Medicine in 1991 for “their discoveries concerning the function of single ion channels in cells”.
In the mid 1990’s, an increasing number of AChR mutations began to emerge as cause for SCS. Among these, the εT264P AChR mutation was identified by Andrew G. Engel’s group in a twenty-year-old woman suffering from myasthenia syndromes since birth (18). Patch clamp experiments on intercostals muscle fibers revealed markedly prolonged channel openings; an observation later confirmed during single-channel recordings in HEK 293 cells expressing the εT264P AChR mutation. Indeed, while both single-channel recordings in muscle fibers and HEK 293 cells revealed prolonged open times, the mean duration did result shorter in the patient than in the expression system. Furthermore, the amplitudes of the single-channel events in the patient were lower as well. However, as the εT264P was engineered in the mouse AChR rather than human, minor discrepancies in the data could be explained by differences in species (18). Interestingly, while recording single-channel activity in the absence of ACh, the authors also found that the εT264P expressed in HEK 293 cells displayed an unusually high rate of spontaneous openings (18).
Another AChR mutation identified in a patient displaying myasthenic symptoms and characterized through patch clamp experiments in transfected HEK 293 cells is the αG153S (20). Similar to the εT264P mutation, the αG153S was engineered into the mouse AChR and displayed prolonged openings as determined by single-channel recordings. Furthermore, single-channel events evoked by various ACh concentrations were fitted into a kinetic scheme to estimate rate constants thus revealing a remarkable decrease in the rate of agonist dissociation when compared to the wild-type (WT) AChR. Such a decrease in the rate of agonist dissociation would cause the ion channel to open repeatedly during ACh occupancy (20). The prolonged single-channel activations episodes displayed by the αN217K AChR mutation expressed in HEK 293 cells were also shown to result from a decrease in the rate of agonist dissociation (38). The αN217K had been previously shown to elicit prolonged openings through single-channel recordings along with two other AChR mutations identified in patients presenting myasthenic syndromes, namely εL269F and βV266M (21). The δQ267E AChR mutation was also studied by Engel et al., 1996; however, it did not evoke prolonged openings (21). Additional AChR mutations expressed in HEK 293 cells and characterized using the patch clamp technique include the αV249F, which showed prolonged openings and increased rate of spontaneous openings (19), and two mutations along the ε subunit, namely εL78P and εL221F, both of which revealed significant increases in the time constant of the longest component of the burst length distributions as compared to the WT (40).
Acetylcholine receptor mutations that cause SCS are located along transmembrane domains M1 and M2, the M2–M3 linker, and the N-terminal (Table 2). However, a mutation located in M4 was recently identified by Andrew Engel’s group (42). In this paper, the authors report a 24-year-old patient displaying bilateral lid ptosis, fatigable weakness of the lower extremities, high-arched palate, pectus carinatum, moderate limitation of eye movements, weak orbicularis oculi muscles, and selectively severe distal upper limb and extensor hallucis longus weakness. Furthermore, CMAP showed decremental response upon repetitive nerve stimulation. Repetitive action potentials were also elicited by a single stimulation. The patient did not present anti-AChR antibodies thus ruling out MG (42). Upon genetic testing, a cysteine to tryptophan substitution was identified in codon 418 of the α subunit (αC418W). The αC418W AChR mutation was engineered into the human α subunit cDNA, coexpressed with the remaining subunits in HEK 293 cells and patch-clamp experiments were performed, thus revealing an increase in the mean duration of the longest open component and bursts (42). An in-depth single-channel analysis of single-channel currents elicited by the weak-agonist choline also showed a fourfold increase the rate constant of channel opening and a sixfold decrease in the channel closing rate (42).
As indicated above, X. laevis oocytes have also been employed for in vitro expression studies and, as HEK 293 cells, oocytes are amenable for single-channel recordings provided the vitelline membrane is removed before attempting to form gigaseals. Indeed, patch clamp recordings of the βL263M mutation identified in a 32-year-old patient and expressed in X. laevis oocytes revealed an eightfold increase in channel open time (36). The αG153S mutation previously identified in a patient and characterized in vitro using HEK 293 cells (20), was also studied through single-channel recordings in the X. laevis oocyte expression system (37) thus confirming the previously observed prolonged openings. Three novel SCS mutations were also described in the aforementioned publication (37), namely αV156M, αT254I, and αS269I, all of which displayed prolonged channel activations, although a detailed kinetic analysis of the single-channel data was not carried out and thus rate constants were not estimated. Nevertheless, differences in the single-channel events within bursts evoked by the αG153S, αV156M, αT254I, and αS269I AChR mutations suggested different mechanisms responsible for the long duration of bursts (37). Besides, the δS268F and βV229F SCS mutations were also characterized using the X. laevis oocyte expression system by means of patch clamp recordings (39, 41). Single-channel currents evoked by the δS268F mutation revealed a more than sevenfold increase in open time while displaying a similar single-channel conductance as the WT (39) whereas an increased open time constant and burst durations where displayed by the βV229F and similar current amplitudes as the WT (41). Single-channel recordings performed at high ACh concentrations also revealed a decreased closing rate and increased effective opening rate in the SCS mutation βV229F (41).
Toxin binding assays
Toxin binding assays are used as means to estimate the cell-surface expression of the AChR in a given expression system. As α-bungarotoxin (α-BgTx) tightly binds AChR (44), exposing cells expressing the AChR to 125I-labeled α-BgTx ([125I]α-BgTx) and probing the remaining radioactivity upon extensive washing, constitutes an effective and sensitive methodological approach to quantify the cell-surface expression levels of the AChR. For instance, the surface expression of the εT264P AChR mutation engineered into the mouse AChR and expressed in HEK 293 cells was as estimated as ranging from 40% to 80% that of the WT receptor (18). Unlike the εT264P AChR mutation, the εL78P and εL221F mutations expressed in HEK 293 cells and the βV229F mutation expressed in X. laevis oocytes did not exhibit reduced surface expression levels as determined by [125I] α-BgTx binding assays, thus suggesting their normal assembly and oligomerization (40–41). These results also led the authors to conclude that the phenotype resulting from these AChR mutations probably results from altered AChR function rather than number (40). The αC418W AChR expressed in HEK 293 cells was also studied by [125I]α-BgTx binding assays (42). Indeed, these experiments showed that the surface expression of the mutant AChR was reduced to 70% of the WT. Furthermore, toxin binding assays, among other methodological approaches, were used to measure the downregulation of the AChR brought upon by short interference RNA (42).
RNA interference
As described above, Shen et al. characterized the αC418W AChR mutation expressed in HEK 293 cells through single-channel recordings and toxin binding assays. In addition, the authors employed a relatively new technique founded on the recently discovered post-transcriptional gene silencing by double-stranded RNA (dsRNA) known as RNA interference (RNAi) (45). RNAi is a gene-silencing mechanism initiated with the cleavage of dsRNA by the Dicer complex into 21–23 base pair fragments called short interfering RNA (siRNA) which upon being incorporated into RISC protein, are unwound and used as template to recognize the target mRNA. Depending on complementarity, this may result in the cleavage of the target mRNA. Cleaved mRNA is recognized as an intruder and degraded by a nuclease thus preventing its translation (46). Andrew Z. Fire and Craig C. Mello received the Nobel Prize in Physiology or Medicine, in 2006 for their discovery of RNAi.
The authors used this novel technology to selectively silence the αC418W AChR expressed in HEK 293 cells. To this aim, siRNAs were designed and transfected together with WT or mutant α cDNA in addition to β, ε, and δ cDNAs in a 2:1:1:1 ratio. The effect of siRNA on transcription and cell-surface expression was subsequently assessed by RT-PCR and toxin-binding experiments, respectively. Indeed, the authors were able to suppress the expression of the αC418W AChR to only 5% of its expression in HEK 293 cells not treated with siRNA, whereas the expression of the WT upon siRNA treatment remained at 84% of the non-treated control (42). Furthermore, the effect of siRNA on protein expression was corroborated using patch clamp experiments in the cell-attached configuration (35). For these experiments, a desensitizing concentration of ACh (10 μM) was used to observe clusters of single-channel events separated by relatively long periods of inactivity. The intracluster events are assumed to be evoked by a single channel, and the fraction of time spent in the open state with respect to the length of the cluster is known as the open probability (Popen) (47). The Popen determined using HEK 293 cells expressing the WT or mutant AChR differed markedly and thus, the activity of both WT and αC418W AChRs co-expressed in HEK 293 cells could be monitored independently to assess the selective downregulation of the mutant AChR. Using this approach, the authors were able to confirm the results gathered through toxin binding and RT-PCR experiments, demonstrating the RNAi-mediated effective and selective downregulation of the mutant AChR over the WT (42).
In fact, this methodological approach had been developed previously to selectively silence the SCS AChR mutation αS226F expressed in HEK 293 cells (48). The underlying rationale behind this study, as in (42), is that as little as only one mismatch between the designed siRNA and the target sequence is sufficient to significantly reduce the silencing effect (49). Thus, on the basis of this discrimination, the cell-surface expression of mutant AChR could be reduced over the WT. By utilizing siRNA bearing a mismatch in position 10 to the WT sequence, the authors were able to reduce the surface expression of the αS226F AChR to 28% of normalized surface levels, whereas the WT expression was reduced to 71% of normalized surface levels as determined by radioactively-labeled toxin binding assays. Moreover, the authors also probed the short hairpin RNAs (shRNA) to selectively silence mutant AChR (48). Sequences were designed and, utilizing the reporter gene DsRed2 as a model system initially, they discovered that, by placing a nucleotide mismatch at position 9, the ability of the shRNA to silence the expression of the reporter gene was almost obliterated. Using this knowledge, the authors set out to selectively silence the SCS AChR mutation αS226F. To this aim, HEK 293 cells were co-transfected with either the WT or mutant α subunit cDNA in addition to β, ε, and δ subunit cDNA and the shRNA molecules designed to selectively down-regulate the mutant AChR. Cell-surface AChR expression was quantified by radioactively-labeled toxin-binding assays and demonstrated that the expression of the αS226F was reduced to 38% of normalized surface levels, whereas in the WT the expression was only reduced to 83% of normalized surface levels. The selective down-regulation of the αS226F AChR by either siRNA or shRNA was confirmed qualitatively by immunofluorescence microscopy. These experiments demonstrated the feasibility of RNAi-based selective silencing of pathogenic mutant AChRs as a therapeutic approach to treat SCS.
Transgenic Mice Models
Not long after the first AChR mutations were identified in SCS patients, transgenic mice expressing AChR mutants were generated by microinjection of single-cell mouse embryos as models for SCS (50). The first of these transgenic mice expressed the δS262T AChR mutation (50). While this particular mutation is not known to be a spontaneously occurring mutation; it served, nevertheless, as model for SCS. Indeed, the success of this pioneering work paved the way for the generation of transgenic mice models expressing SCS mutations identified in patients (22, 51–54) with particular emphasis on the εL269F, which is among the first reported SCS mutations (21, 55). Methodological approaches employed on transgenic mice are not all that different to those employed on human muscle biopsies. In essence, intracellular recordings, single-channel recordings, toxin binding experiments, light microscopy, and electron microscopy, among others, are commonly employed. In addition, more recent articles have made use of additional methodological approaches to study the pathogenesis of the hallmark of SCS, namely endplate myopathy (52–54).
Electromyography
As stated herein, CMAPs typical of SCS show two distinctive features, namely repetitive muscle action potentials upon a single stimulus to nerve and a decremental response evoked by repetitive nerve stimulation (37, 39, 50). Thus far, the εL269F-transgenic has been characterized in terms of these two parameters through hindlimb and forelimb recordings (22, 54) while the δS262T-transgenic has been characterized in terms of repetitive muscle action potentials (50). For hindlimp recordings, animals were anesthetized and the sciatic nerve exposed to enable stimulation while responses from the gastrocnemius and intrinsic hindpaw muscles were recorded separately. Alternatively, forelimb recordings were achieved by placing the stimulus electrode into the brachial plexus (22). Repetitive CMAPs were observed in the εL269F-transgenic, being more prominent in the gastrocnemius and absent or diminished in the intrinsic hindpaw muscles. Indeed, up to two additional action potentials were observed in the gastrocnemius, an observation rationalized by the authors as the result of the prolongation of the endplate potential beyond the muscle fiber refractory period. Repetitive CMAPs were also observed in the δS262T-transgenic (50). In contrast, repetitive nerve stimulation in the εL269F-transgenic did not show decremental response in the gastrocnemius while intrinsic muscles of the hindpaws elicited decremental CMAPs, thus indicating a reduced safety margin of neuromuscular transmission (22). Repetitive nerve stimulation studies were also employed to study the effect of transgenic expression of calpastatin (CS), the natural inhibitor of calpain, in the decremental response observed in the εL269F-transgenic. Indeed, decremental responses in CS/εL269F mice were significantly lower when compared to the εL269F-transgenic (54). Further details on this subject will be presented in the following sections.
Intracellular recordings
MEPCs reflect the activation of postsynaptic AChRs by quantal release of ACh. Analogous to the exponential distribution of ion channel open times, miniature currents decay exponentially with rates defined by the time constant τ. MEPCs are analyzed in terms of their decay rate, amplitude, and frequency. While MEPCs are recorded in the absence of nerve stimulation, EPCs are typically recorded upon stimulation of the phrenic nerve. Several publications present results from transgenic mice MEPC and EPC recordings (50, 56), whereas others have focused on MEPC recordings (22, 51, 54). In essence, MEPC and EPC recordings of the δS262T-transgenic (38–39) and MEPC recordings of εL269F (22, 51, 54), αC418W (51), and αL251T (51) transgenics have been reported.
MEPC recordings from three different lines of δS262T-transgenics with different levels of the transgene mRNA in skeletal muscle as determined by RNA blot analysis, revealed that all three lines displayed prolonged decay phases and diminished amplitudes when compared to control mice (50). EPC recordings of the δS262T-transgenic evoked by phrenic nerve stimulation showed prolongation of the currents (50) and reduction in amplitude (56). Moreover, a greater proportion of the transgenic mice MEPCs displayed bi-exponential decays (25%) when compared to WT (6%). Indeed, this particular difference proved to be important, as reported on a paper published shortly after, to help explain the reduced MEPC amplitudes in terms of an activity-dependent reduction in sensitivity as neither a reduction in the number of endplate AChRs nor ion channel conductance was observed in the δS262T-transgenic (56). Other transgenic mice models studied employing intracellular recordings methods, namely εL269F (22, 51, 54), αC418W (51), and αL251T (51), also displayed the typical MEPC prolonged durations which increased successively in transgenic lines δS262T, αC418W, αL251T and εL269F. Furthermore, while differences in MEPC amplitude seldom reached statistical significance (statistical significance was only reached in the εL269F transgenic), the trend strongly reflected an inverse proportionality between mice strength and MEPC amplitude, thus suggesting the neuromuscular origin of the muscle weakness in these transgenic lines (51).
Single-channel recordings
The patch clamp electrophysiological technique (32) has been employed in the study of various transgenic lines. For example, flexor digitorum brevis muscles isolated by blunt dissection from δS262T-transgenic mice and dissociated through 0.2% collagenase two-hour incubations, followed by gentle trituration (56) were used for patch clamp experiments in the cell-attached configuration (35). Single-channel recordings were performed on single fibers bathed in Tyrode’s solution at different membrane potentials to determine the amplitude of single-channel events (current) as function of membrane potential and thus calculate single-channel conductance. The aim of these experiments was to assess the possibility of a decreased conductance as cause for the reduced MEPC amplitudes displayed by the δS262T-transgenic mice. The best linear fit of the current-voltage plots revealed a conductance of 61.8 pS for the transgenic and 61.4 pS for the control thus ruling out changes in single-channel conductance as the underlying cause for the reduced MEPC amplitudes (56). Open time histograms were also generated for the transgenic and the time constants were compared to those obtained while recording single-channel events from X. laevis oocytes expressing the δS262T AChR mutant (50). The similarity between the time constants determined in both expression systems suggested the predominance of the δS262T AChR in the δS262T-transgenic mice endplates (56).
An additional transgenic mouse model employed in patch experiments is the εL269F-transgenic. Open time histograms generated using single-channel data recorded from εL269F dissociated fibers revealed channels with prolonged open durations (22). In fact, single-channel recordings exhibited channel events of both short and long duration presumably evoked by single- and double-ligand occupancy AChR openings, and resembled the previously reported findings in a SCS patient bearing the εL269F mutation (21).
Electron microscopy
Electron micrographs of transgenic mice endplates enable researchers to study potential ultrastructural abnormalities (22, 51–52, 54, 56), as classical SCS are commonly associated with endplate myopathy, which consists of striking degenerative changes of the postsynaptic membrane (5). To this end, muscles are fixed in 2% glutaraldehyde, osmicated and embedded in epon following standard procedures in electron microscopy (56). An outstanding result was reported by Christopher M. Gomez’s group in which the δS262T-transgenic endplate displayed no recognizable pathological changes or ultrastructural abnormalities (22). Because MEPCs in this particular transgenic line have similar reductions in amplitude as other transgenic mice that display marked endplate myopathy (22), the δS262T-transgenic enabled the study of additional factors that contribute to MEPC reduction, which eventually led to insights into the role played by abnormal desensitization in determining the amplitude of endplate currents (56). The important role of desensitization was confirmed a few years later in SCS spontaneous mutations (43). Nevertheless, ultrastructural abnormalities are a common (5) feature of SCS cases as demonstrated by the degenerating neuromuscular synapse from εL269F mice displayed in Figure 4 (22).
Figure 4.
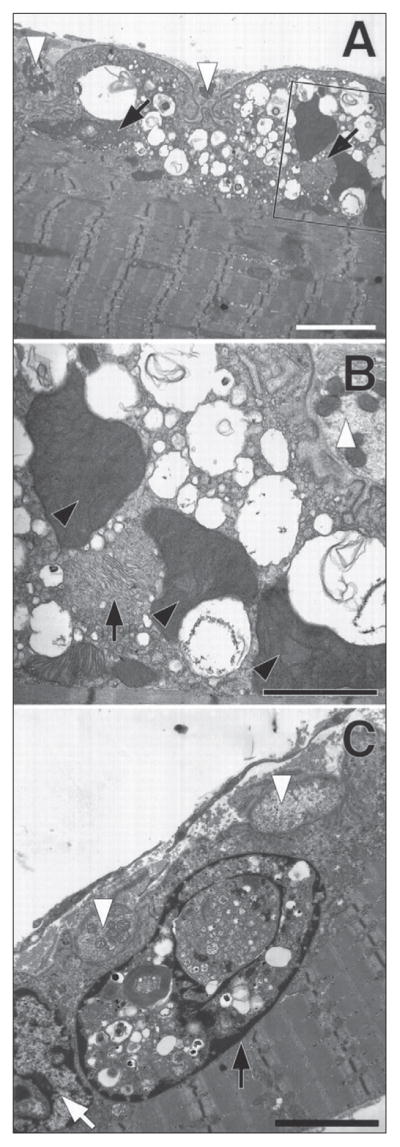
εL269F-mice have degeneration of the neuromuscular synapse. Neuromuscular junctions from forelimb flexor muscles of 4-month-old εL269F-transgenic mice. To facilitate orientation all nerve termini are indicated using white arrowheads. In all three views the postsynaptic folds are simplified, whereas the adjacent the nerve termini (white arrowheads) seem normal. A) Large vacuoles containing membranous or granular debris fill the junctional sarcoplasm. The composition of the vacuoles can be seen more clearly in the enlargement from the boxed section. B) The mitochondria in the muscle fiber in (A) and in (B) (inset) are greatly enlarged compared with those in the nerve termini. Black arrows indicate what seem to be two degenerating mitochondria. Three mitochondria are massively enlarged and have densely packed cristae (black arrowheads). C) A degenerating myonucleus (black arrow) filled with autophagic debris and cytoplasmic contents lies immediately beneath a neuromuscular junction and adjacent to a relatively normal-appearing nucleus (white arrow). Calibration bars: A, 20 nM; B, 10 nM; C, 15 nM. (Figure 4 is reproduced by permission from Gómez et al. (1997) Journal of Neuroscience 17: 4170–4179).
Additional SCS transgenic mice studied using electron microscopy include the αC418W- and αL251T-transgenics, being the εL269F the most extensively studied (22, 51, 52, 54). Similar to the δS262T-transgenic NMJ, the αC418W-transgenic displayed essentially normal NMJ with the exception of a few vacuoles observed in the endplates (51). However, both αL251T- and εL269F-transgenics had numerous endplates displaying marked ultrastructural changes such as collections of membrane-bound structures which in some cases filled the junctional sarcoplasm and that seemed as to arise from dilated sarcoplasmic reticulum. Other structural abnormalities present in both transgenic lines included degenerating nuclei, dilated or degenerating mitochondria, and membranous or granular debris (51). It is noteworthy that 66% of the εL269F endplates displayed abnormalities which may be observed even in two-day-old mice (52) compared to 56% in the αL251T mice (51). Also, endplate denervation in the εL269F-transgenic caused improvement of ultrastructural abnormalities as determined by electron microscopy (42).
Ca2+ overloading
The degeneration of motor endplates discussed above, and present in many SCS cases, has been attributed to Ca2+ overload in the sarcoplasm and the concomitant activation of Ca2+-sensitive degradative enzymes (5), thus sparking the interest of researchers in making use and developing metho-dological approaches to assess Ca2+ overloading in endplates (22, 51–53). For instance, histochemical methods relying on the Karnovsky and Roots cholinesterase method to localize endplates (57) and the glyoxal bis-(2-hydroxyanil) (GBHA) (58) or von Kossa (59) methods to localize Ca2+ were used to probe calcium deposits in εL269F-transgenic endplates (22). These experiments revealed Ca2+ staining over regions that co-localized with acetylcholinesterase, resembling the results obtained in the SCS patient bearing the εL269F AChR mutation (5, 55). Using the same approach, the proportion of endplates overloaded with Ca2+ was assessed on various transgenic mice lines revealing no GBHA staining in either control mice or the δS262T-transgenic, whereas the αC418W-, and αL251T-transgenic displayed some Ca2+ overloading, and the εL269F-transgenic showed a significantly greater proportion of Ca2+-overloaded endplates compared to the other transgenic lines (51). Indeed, endplates from three-day-old εL269F mice displayed intense GBHA staining (52). It was later revealed, using an epifluorescence microscopy-based approach, that the sarcoplasmic reticulum is the main source of the observed Ca2+ and implicating inositol-1,4,5-triphosphate receptors and L-type voltage gated Ca2+ channels as responsible for its efflux (53). Interestingly, it was also established that a 30-minute standardized exercise protocol results in a remarkable increase in the proportion of endplates overloaded with Ca2+ in the αC418W- αL251T- and εL269F-transgenic lines (51).
Personal Experience With Congenital Myasthenic Syndromes In Puerto Rico: A Local Perspective
CMS in Puerto Rico are rare, although the precise prevalence is not known. Over the last 10 years, one of the authors (CAL-R) has been able to identify five patients with features consistent with a CMS. These patients present ptosis and external ophthalmoplegia as the most prominent feature, although muscle weakness and fatigue are also prominent. In some instances, they have experienced episodes of respiratory insufficiency requiring temporary mechanical ventilation, although in our experience, this has been rare. With careful inquiry, it is possible to determine that the symptoms started during the neonatal period or during early childhood with delayed motor milestones. In most instances, the patients have been referred for the possibility of MG and the diagnosis of a CMS has been suspected on the basis of the early age at onset of symptoms and the lack or minimal response to immune suppressant therapies. The evaluation of these patients has included a thorough evaluation for possible Congenital Myopathies that may result in similar symptoms and the exclusion of autoimmune myasthenia with negative acetylcholine receptor or muscle specific kinase antibodies. The response to pyridostigmine has been variable indicating the possibility of different mutations within the group of patients. In some instances, we have been able to identify electrophysiologic features suggestive of a SCS mutation, such as the presence of a repetitive CMAP on the motor nerve conduction studies, with an abnormal decremental response on repetitive nerve stimulation. (Figure 5A and B). It is likely that many more of these patients are misdiagnosed and have not been identified. Although treatment for these syndromes is limited, an active effort to identify these patients is necessary to optimize their care and to avoid the needless exposure to immunotherapies with multiple and serious side effects.
Figure 5.

Electromyogram of a patient with suspected slow-channel congenital syndrome. A) Representative example of a repetitive compound action potential on a patient with a suspected slow channel syndrome. B) On repetitive nerve stimulation at low frequency (3 Hz) it is possible to observe an abnormal decrement (17%) in the amplitude between the first and fifth elicited compound motor action potential. (Amplitude 1st potential = 2.37 mV; amplitude 5th potential = 1.96 mV; decrement of 17%).
Future Perspectives
Electron microscopy micrographs such as those displayed in Figure 4 have had an important role in the study of endplate myopathies which are considered the hallmark of SCS. In spite of the inherent high resolution of electron microscopy, alternative imaging strategies must be thoroughly examined in order to more effectively monitor endplate changes associated to SCS. For instance, changes in the amount of endplates per muscle fiber or the extent of AChR expression within endplates among other valuable criteria are not easily examined through EM. To surpass these limitations, light microscopy-based experimental approaches have been developed in our laboratory to monitor endplates of transgenic mice bearing SCS mutations with respect to WT FVB mice. While the achievable resolution in light microscopy is much lower than that of electron microscopy, the optical sectioning capability of confocal microscopy and state-of-the-art three-dimensional analysis software packages such as Imaris x64 6.2.0 (www.bitplane.com) enable a detailed analysis of endplate morphology (based on the fluorescence of labeled-α-bungarotoxin) in the different transgenic lines bearing SCS mutations that we currently work with (Figure 6). Using this methodological approach, it is also possible to quantify the effects of any given variable on the number of transgenic mice endplates, the volume of endplates, and the number of AChRs. Furthermore, a common feature of degenerating NMJ, namely the widening of the synaptic space may also be studied using the optical sectioning capability of confocal microscopy. Certainly, by fluorescently labeling the pre-synaptic and post-synaptic membranes, endplate plasticity may be studied in SCS transgenics and compared to WT mice. The possibility that the expression of AChR displaying altered kinetics may concomitantly change endplate plasticity should provide additional insight into the pathogenesis of SCS and increase our understanding of the NMJ in general. Thus far, a significant number of novel observations have been enabled by this approach thus underscoring its importance in years to come.
Figure 6.
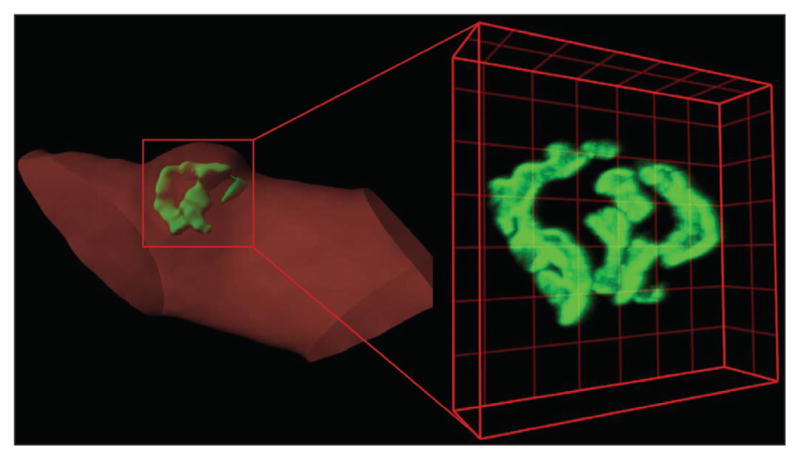
Imaging of a bungarotoxin-labeled mouse endplates. Three-dimensional confocal image and Imaris-generated surface from an FVB mouse endplate (green) on dissociated muscle fiber (red), highlight the capacity of an in-depth analysis of endplate structure using confocal microscopy. Images of bungarotoxin-labeled AChR were collected on a Zeiss LSM 510 Confocal Microscope as a series of optical slices (Z-stack). Three-dimensional reconstructions of Z-stacks were generated using Imaris x64 6.2.0 which enables accurate determination of mean fluorescence intensity of voxels (three-dimensional pixel) as well as endplate volume. Lines in grids are separated by 5 μm.
Final Remarks
The investigation of CMS has enhanced our understanding of the development, maintenance, and function of the NMJ. The development of CMS animal models to investigate the mechanisms for endplate degeneration has been a major breakthrough for the elucidation of molecular mechanisms leading to the CMS phenotype. All clinical, electrophysiological, pathologic, and biochemical aspects of the human disease process in CMS are fully replicated in the transgenic models. Moreover, the heterogeneous phenotypes of CMS are also replicated in the transgenic mice model. Several aspects of the CMS must be fully elucidated to define more efficient strategies for therapeutic intervention. The first aspect that deserves special attention is the alteration of endplate plasticity in the CMS: Is pre-synaptic feedback (i.e., decrease in release sites) playing a compensation role to attenuate the gain-of-function AChR mutation? What types of post-synaptic compensations are evident in the NMJ in response to a CMS (i.e. reduction of nAChR expression in endplate). Are there any environmental factors that can exacerbate or attenuate phenotypes within the same CMS? It is also important to understand the factors that regulate the expression of mutant AChR in the CMS endplate. Understanding the molecular mechanisms leading to synaptic remodeling of the NMJ in different forms of CMS is essential to develop new strategies for therapeutic intervention.
Summary
The study of SCS has led to a remarkably detailed explanation for the pathogenesis of the electrophysiological, pathological, and genetic basis of the disease. This understanding of SCS has led to design of therapeutic interventions that modulate the mutant AChR function,, as well as insights into further therapy development. These insights may have additional implications into understanding the pathological processes that pay a role in other disorders, such as epilepsy, stroke, Alzheimer’s disease, and myopathies. The diagnosis remains challenging due to failure to suspect the disease and the lack of clinically available genetic studies.
Acknowledgments
This work was partly supported by grants from the National Institutes of Health to J. A. Lasalde-Dominicci (grant no. 2RO1GM56371-12 and SNRP U54NS0430311), to C. M. Gómez (grant no. 2RO1-N33202), and to C. A. Luciano (grant no. P20RR011126). We especially thank Zayra E. Marrero-Burgos for proofreading the manuscript.
Abbreviations
- ACh
acetylcholine
- AChR
acetylcholine receptor
- α-BgTx
α-bungarotoxin
- [125I]-α-BgTx
125I-labeled α-bungarotoxin
- CA
calpastatin
- CMAP
compound muscle action potential
- CMS
congenital myasthenic syndrome
- dsRNA
double-stranded RNA
- EMG
electromyography
- EP
endplate
- EPC
endplate current
- EPP
endplate potentials
- GBHA
glyoxal bis-(2-hydroxyanil)
- HEK
human embryonic kidney
- MEPC
miniature endplate current
- MEPP
miniature endplate potential
- MG
myasthenia gravis
- NMJ
neuromuscular junction
- RNAi
RNA interference
- SCS
slow-channel congenital myasthenic syndrome
- shRNA
short hairpin RNA
- WT
wild-type
Footnotes
The authors have no conflict of interest to disclose.
References
- 1.Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes: recent advances. Arch Neurol. 1999;56:163–167. doi: 10.1001/archneur.56.2.163. [DOI] [PubMed] [Google Scholar]
- 2.Hantai D, Richard P, Koenig J, Eymard B. Congenital myasthenic syndromes. Curr Opin Neurol. 2004;17:539–551. doi: 10.1097/00019052-200410000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Harper CM. Congenital myasthenic syndromes. Semin Neurol. 2004;24:111–123. doi: 10.1055/s-2004-829592. [DOI] [PubMed] [Google Scholar]
- 4.Engel AG. Congenital myasthenic syndromes. Neurol Clin. 1994;12:401–437. [PubMed] [Google Scholar]
- 5.Engel AG, Lambert EH, Mulder DM, et al. A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Ann Neurol. 1982;11:553–569. doi: 10.1002/ana.410110603. [DOI] [PubMed] [Google Scholar]
- 6.Karlin A. Emerging structure of the nicotinic acetylcholine receptors. Nat Rev Neurosci. 2002;3:102–114. doi: 10.1038/nrn731. [DOI] [PubMed] [Google Scholar]
- 7.Corringer PJ, Le Novere N, Changeux JP. Nicotinic receptors at the amino acid level. Annu Rev Pharmacol Toxicol. 2000;40:431–458. doi: 10.1146/annurev.pharmtox.40.1.431. [DOI] [PubMed] [Google Scholar]
- 8.Le Novere N, Corringer PJ, Changeux JP. The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J Neurobiol. 2002;53:447–456. doi: 10.1002/neu.10153. [DOI] [PubMed] [Google Scholar]
- 9.Unwin N. Nicotinic acetylcholine receptor at 9 A resolution. J Mol Biol. 1993;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- 10.Devillers-Thiery A, Galzi JL, Eisele JL, Bertrand S, Bertrand D, Changeux JP. Functional architecture of the nicotinic acetylcholine receptor: a prototype of ligand-gated ion channels. J Membr Biol. 1993;136:97–112. doi: 10.1007/BF02505755. [DOI] [PubMed] [Google Scholar]
- 11.Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- 12.Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 13.Blanton MP, Cohen JB. Mapping the lipid-exposed regions in the Torpedo californica nicotinic acetylcholine receptor. Biochemistry. 1992;31:3738–3750. doi: 10.1021/bi00130a003. [DOI] [PubMed] [Google Scholar]
- 14.Blanton MP, Cohen JB. Identifying the lipid-protein interface of the Torpedo nicotinic acetylcholine receptor: secondary structure implications. Biochemistry. 1994;33:2859–2872. doi: 10.1021/bi00176a016. [DOI] [PubMed] [Google Scholar]
- 15.Hughes BW, Kusner LL, Kaminski HJ. Molecular architecture of the neuromuscular junction. Muscle Nerve. 2006;33:445–461. doi: 10.1002/mus.20440. [DOI] [PubMed] [Google Scholar]
- 16.Oosterhuis HJ, Newsom-Davis J, Wokke JH, et al. The slow channel syndrome. Two new cases. Brain. 1987;110:1061–1079. doi: 10.1093/brain/110.4.1061. [DOI] [PubMed] [Google Scholar]
- 17.Engel AG. Congenital myasthenic syndromes. Handb Clin Neurol. 2008;91:285–331. doi: 10.1016/S0072-9752(07)01510-2. [DOI] [PubMed] [Google Scholar]
- 18.Ohno K, Hutchinson DO, Milone M, et al. Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the epsilon subunit. Proc Natl Acad Sci U S A. 1995;92:758–762. doi: 10.1073/pnas.92.3.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milone M, Wang HL, Ohno K, et al. Slow-channel myasthenic syndrome caused by enhanced activation, desensitization, and agonist binding affinity attributable to mutation in the M2 domain of the acetylcholine receptor alpha subunit. J Neurosci. 1997;17:5651–5665. doi: 10.1523/JNEUROSCI.17-15-05651.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sine SM, Ohno K, Bouzat C, et al. Mutation of the acetylcholine receptor alpha subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron. 1995;15:229–239. doi: 10.1016/0896-6273(95)90080-2. [DOI] [PubMed] [Google Scholar]
- 21.Engel AG, Ohno K, Milone M, et al. New mutations in acetylcholine receptor subunit genes reveal heterogeneity in the slow-channel congenital myasthenic syndrome. Hum Mol Genet. 1996;5:1217–1227. doi: 10.1093/hmg/5.9.1217. [DOI] [PubMed] [Google Scholar]
- 22.Gómez CM, Maselli R, Gundeck JE, et al. Slow-channel transgenic mice: a model of postsynaptic organellar degeneration at the neuromuscular junction. J Neurosci. 1997;17:4170–4179. doi: 10.1523/JNEUROSCI.17-11-04170.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wintzen AR, Plomp JJ, Molenaar PC, et al. Acquired slow-channel syndrome: a form of myasthenia gravis with prolonged open time of the acetylcholine receptor channel. Ann Neurol. 1998;44:657–664. doi: 10.1002/ana.410440412. [DOI] [PubMed] [Google Scholar]
- 24.Sieb JP, Milone M, Engel AG. Effects of the quinoline derivatives quinine, quinidine, and chloroquine on neuromuscular transmission. Brain Res. 1996;712:179–189. doi: 10.1016/0006-8993(95)01349-0. [DOI] [PubMed] [Google Scholar]
- 25.Fukudome T, Ohno K, Brengman JM, Engel AG. AChR channel blockade by quinidine sulfate reduces channel open duration in the slow-channel congenital myasthenic syndrome. Ann N Y Acad Sci. 1998;841:199–202. doi: 10.1111/j.1749-6632.1998.tb10928.x. [DOI] [PubMed] [Google Scholar]
- 26.Fukudome T, Ohno K, Brengman JM, Engel AG. Quinidine normalizes the open duration of slow-channel mutants of the acetylcholine receptor. Neuroreport. 1998;9:1907–1911. doi: 10.1097/00001756-199806010-00044. [DOI] [PubMed] [Google Scholar]
- 27.Harper CM, Engel AG. Safety and efficacy of quinidine sulfate in slow-channel congenital myasthenic syndrome. Ann N Y Acad Sci. 1998;841:203–206. doi: 10.1111/j.1749-6632.1998.tb10929.x. [DOI] [PubMed] [Google Scholar]
- 28.Harper CM, Engel AG. Quinidine sulfate therapy for the slow-channel congenital myasthenic syndrome. Ann Neurol. 1998;43:480–484. doi: 10.1002/ana.410430411. [DOI] [PubMed] [Google Scholar]
- 29.Harper CM, Fukodome T, Engel AG. Treatment of slow-channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:1710–1713. doi: 10.1212/01.wnl.0000061483.11417.1b. [DOI] [PubMed] [Google Scholar]
- 30.Colomer J, Muller JS, Vernet A, et al. Long-term improvement of slow-channel congenital myasthenic syndrome with fluoxetine. Neuromuscul Disord. 2006;16:329–333. doi: 10.1016/j.nmd.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Engel AG. The therapy of congenital myasthenic syndromes. Neurotherapeutics. 2007;4:252–257. doi: 10.1016/j.nurt.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neher E, Sakmann B. Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature. 1976;260:799–802. doi: 10.1038/260799a0. [DOI] [PubMed] [Google Scholar]
- 33.Colquhoun D, Hawkes AG. On the stochastic properties of single ion channels. Proc R Soc Lond B Biol Sci. 1981;211:205–235. doi: 10.1098/rspb.1981.0003. [DOI] [PubMed] [Google Scholar]
- 34.Colquhoun D, Hawkes AG. On the stochastic properties of bursts of single ion channel openings and of clusters of bursts. Philos Trans R Soc Lond B Biol Sci. 1982;300:1–59. doi: 10.1098/rstb.1982.0156. [DOI] [PubMed] [Google Scholar]
- 35.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 36.Gómez CM, Maselli R, Gammack J, et al. A beta-subunit mutation in the acetylcholine receptor channel gate causes severe slow-channel syndrome. Ann Neurol. 1996;39:712–723. doi: 10.1002/ana.410390607. [DOI] [PubMed] [Google Scholar]
- 37.Croxen R, Newland C, Beeson D, et al. Mutations in different functional domains of the human muscle acetylcholine receptor alpha subunit in patients with the slow-channel congenital myasthenic syndrome. Hum Mol Genet. 1997;6:767–774. doi: 10.1093/hmg/6.5.767. [DOI] [PubMed] [Google Scholar]
- 38.Wang HL, Auerbach A, Bren N, Ohno K, Engel AG, Sine SM. Mutation in the M1 domain of the acetylcholine receptor alpha subunit decreases the rate of agonist dissociation. J Gen Physiol. 1997;109:757–766. doi: 10.1085/jgp.109.6.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gómez CM, Maselli RA, Vohra BP, et al. Novel delta subunit mutation in slow-channel syndrome causes severe weakness by novel mechanisms. Ann Neurol. 2002;51:102–112. doi: 10.1002/ana.10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Croxen R, Hatton C, Shelley C, et al. Recessive inheritance and variable penetrance of slow-channel congenital myasthenic syndromes. Neurology. 2002;59:162–168. doi: 10.1212/wnl.59.2.162. [DOI] [PubMed] [Google Scholar]
- 41.Navedo MF, Lasalde-Dominicci JA, Baez-Pagán CA, et al. Novel beta subunit mutation causes a slow-channel syndrome by enhancing activation and decreasing the rate of agonist dissociation. Mol Cell Neurosci. 2006;32:82–90. doi: 10.1016/j.mcn.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen XM, Deymeer F, Sine SM, Engel AG. Slow-channel mutation in acetylcholine receptor alphaM4 domain and its efficient knockdown. Ann Neurol. 2006;60:128–136. doi: 10.1002/ana.20861. [DOI] [PubMed] [Google Scholar]
- 43.Zayas R, Lasalde-Dominicci J, Gomez CM. Macroscopic properties of spontaneous mutations in slow-channel syndrome: correlation by domain and disease severity. Synapse. 2006;60:441–449. doi: 10.1002/syn.20317. [DOI] [PubMed] [Google Scholar]
- 44.Nirthanan S, Gwee MC. Three-finger alpha-neurotoxins and the nicotinic acetylcholine receptor, forty years on. J Pharmacol Sci. 2004;94:1–17. doi: 10.1254/jphs.94.1. [DOI] [PubMed] [Google Scholar]
- 45.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 46.Leung RK, Whittaker PA. RNA interference: from gene silencing to gene-specific therapeutics. Pharmacol Ther. 2005;107:222–239. doi: 10.1016/j.pharmthera.2005.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colquhoun D, Ogden DC. Activation of ion channels in the frog end-plate by high concentrations of acetylcholine. J Physiol. 1988;395:131–159. doi: 10.1113/jphysiol.1988.sp016912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abdelgany A, Wood M, Beeson D. Allele-specific silencing of a pathogenic mutant acetylcholine receptor subunit by RNA interference. Hum Mol Genet. 2003;12:2637–2644. doi: 10.1093/hmg/ddg280. [DOI] [PubMed] [Google Scholar]
- 49.Elbashir SM, Martínez J, Patkaniowska A, Lendeckel W, Tuschl T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. Embo J. 2001;20:6877–6888. doi: 10.1093/emboj/20.23.6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gómez CM, Bhattacharyya BB, Charnet P, et al. A transgenic mouse model of the slow-channel syndrome. Muscle Nerve. 1996;19:79–87. doi: 10.1002/(SICI)1097-4598(199601)19:1<79::AID-MUS11>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 51.Gómez CM, Maselli RA, Groshong J, et al. Active calcium accumulation underlies severe weakness in a panel of mice with slow-channel syndrome. J Neurosci. 2002;22:6447–6457. doi: 10.1523/JNEUROSCI.22-15-06447.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vohra BP, Groshong JS, Zayas R, Wollmann RL, Gómez CM. Activation of apoptotic pathways at muscle fiber synapses is circumscribed and reversible in a slow-channel syndrome model. Neurobiol Dis. 2006;23:462–470. doi: 10.1016/j.nbd.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 53.Zayas R, Groshong JS, Gómez CM. Inositol-1,4,5-triphosphate receptors mediate activity-induced synaptic Ca2+ signals in muscle fibers and Ca2+ overload in slow-channel syndrome. Cell Calcium. 2007;41:343–352. doi: 10.1016/j.ceca.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 54.Groshong JS, Spencer MJ, Bhattacharyya BJ, et al. Calpain activation impairs neuromuscular transmission in a mouse model of the slow-channel myasthenic syndrome. J Clin Invest. 2007;117:2903–2912. doi: 10.1172/JCI30383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gómez CM, Gammack JT. A leucine-to-phenylalanine substitution in the acetylcholine receptor ion channel in a family with the slow-channel syndrome. Neurology. 1995;45:982–985. doi: 10.1212/wnl.45.5.982. [DOI] [PubMed] [Google Scholar]
- 56.Bhattacharyya BJ, Day JW, Gundeck JE, Leonard S, Wollmann RL, Gómez CM. Desensitization of mutant acetylcholine receptors in transgenic mice reduces the amplitude of neuromuscular synaptic currents. Synapse. 1997;27:367–377. doi: 10.1002/(SICI)1098-2396(199712)27:4<367::AID-SYN10>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 57.Namba T, Nakamura T, Grob D. Staining for nerve fiber and cholinesterase activity in fresh frozen sections. Am J Clin Pathol. 1967;47:74–77. doi: 10.1093/ajcp/47.1.74. [DOI] [PubMed] [Google Scholar]
- 58.Kashiwa HK. Calcium phosphate in osteogenic cells. A critique of the glyoxal bis(2-hydroxyanil) and the dilute silver acetate methods. Clin Orthop Relat Res. 1970;70:200–211. [PubMed] [Google Scholar]
- 59.Rungby J, Kassem M, Eriksen EF, Danscher G. The von Kossa reaction for calcium deposits: silver lactate staining increases sensitivity and reduces background. Histochem J. 1993;25:446–451. doi: 10.1007/BF00157809. [DOI] [PubMed] [Google Scholar]
- 60.Noda M, Furutani Y, Takahashi H, et al. Cloning and sequence analysis of calf cDNA and human genomic DNA encoding alpha-subunit precursor of muscle acetylcholine receptor. Nature. 1983;305:818–823. doi: 10.1038/305818a0. [DOI] [PubMed] [Google Scholar]
- 61.Schoepfer R, Luther M, Lindstrom J. The human medulloblastoma cell line TE671 expresses a muscle-like acetylcholine receptor. Cloning of the alpha-subunit cDNA. FEBS Lett. 1988;226:235–240. doi: 10.1016/0014-5793(88)81430-3. [DOI] [PubMed] [Google Scholar]
- 62.Beeson D, Morris A, Vincent A, Newsom-Davis J. The human muscle nicotinic acetylcholine receptor alpha-subunit exist as two isoforms: a novel exon. Embo J. 1990;9:2101–2106. doi: 10.1002/j.1460-2075.1990.tb07378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Keiger CJ, Prevette D, Conroy WG, Oppenheim RW. Developmental expression of nicotinic receptors in the chick and human spinal cord. J Comp Neurol. 2003;455:86–99. doi: 10.1002/cne.10468. [DOI] [PubMed] [Google Scholar]
- 64.Beeson D, Brydson M, Newsom-Davis J. Nucleotide sequence of human muscle acetylcholine receptor beta-subunit. Nucleic Acids Res. 1989;17:4391. doi: 10.1093/nar/17.11.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heidmann O, Buonanno A, Geoffroy B, et al. Chromosomal localization of muscle nicotinic acetylcholine receptor genes in the mouse. Science. 1986;234:866–868. doi: 10.1126/science.3022377. [DOI] [PubMed] [Google Scholar]
- 66.Lobos EA, Rudnick CH, Watson MS, Isenberg KE. Linkage disequilibrium study of RFLPs detected at the human muscle nicotinic acetylcholine receptor subunit genes. Am J Hum Genet. 1989;44:522–533. [PMC free article] [PubMed] [Google Scholar]
- 67.Pasteris NG, Trask BJ, Sheldon S, Gorski JL. Discordant phenotype of two overlapping deletions involving the PAX3 gene in chromosome 2q35. Hum Mol Genet. 1993;2:953–959. doi: 10.1093/hmg/2.7.953. [DOI] [PubMed] [Google Scholar]
- 68.Beeson D, Brydson M, Betty M, et al. Primary structure of the human muscle acetylcholine receptor. cDNA cloning of the gamma and epsilon subunits. Eur J Biochem. 1993;215:229–238. doi: 10.1111/j.1432-1033.1993.tb18027.x. [DOI] [PubMed] [Google Scholar]
- 69.Vohra BP, Groshong JS, Maselli RA, Verity MA, Wollmann RL, Gómez CM. Focal caspase activation underlies the endplate myopathy in slow-channel syndrome. Ann Neurol. 2004;55:347–352. doi: 10.1002/ana.10823. [DOI] [PubMed] [Google Scholar]