

Abstract
Acetaminophen protein adducts (APAP adducts) were quantified in 157 adolescents and children presenting at eight pediatric hospitals with the chief complaint of APAP overdose. Two of the patients required liver transplantation, whereas all the others recovered spontaneously. Peak APAP adducts correlated with peak hepatic transaminase values, time-to-treatment with N-acetylcysteine (NAC), and risk determination per the Rumack–Matthews nomogram. A population pharmacokinetic analysis (NONMEM) was performed with post hoc empiric Bayesian estimates determined for the elimination rate constants (ke), elimination half-lives (t½), and maximum concentration of adducts (Cmax) of the subjects. The mean (±SD) ke and half-life were 0.486 ± 0.084 days−1 and 1.47 ± 0.30 days, respectively, and the Cmax was 1.2 (±2.92) nmol/ml serum. The model-derived, predicted adduct value at 48 h (Adduct 48) correlated with adduct Cmax, adduct Tmax, Rumack–Matthews risk determination, peak aspartate aminotransferase (AST), and peak alanine aminotransferase (ALT). The pharmacokinetics and clinical correlates of APAP adducts in pediatric and adolescent patients with APAP overdose support the need for a further examination of the role of APAP adducts as clinically relevant and specific biomarkers of APAP toxicity.
Acetaminophen (APAP) overdose is a common cause of pediatric ingestions,1 and in severe cases it may result in acute liver failure (ALF).2 Recent data have shown that APAP overdose is a major cause of ALF in adults in the United States.3 In adults, APAP accounts for ~50% of cases of ALF,3,4 and in children it accounts for ~13% of cases.2 It is generally recognized that morbidity in children following an APAP overdose is lower than that in adults.5,6 However, the etiology of ALF is unknown in ~50% of pediatric cases, and it is possible that APAP overdose may be a contributory factor in some of these cases. The development of new biomarkers of drug toxicity may help to facilitate diagnosis and improve the existing knowledge base of the epidemiology of pediatric ALF.
The Rumack–Matthews nomogram is widely used in hospital emergency departments to make determinations on the need for treatment with the antidote, N-acetylcysteine (NAC), following an APAP overdose. The nomogram was derived from time-dependent APAP concentration data generated from adults with single, acute ingestions of APAP who presented to medical centers within 24 h of the APAP overdose.7 However, assessing the risk of developing toxicity is difficult in patients who do not meet these criteria. Examples of patient subgroups for which the Rumack–Matthews nomogram is not applicable include patients presenting in the late stages of toxicity (>24 h after the overdose), patients with chronic ingestion of APAP, acute ingestion in alcoholics,8 patients who concurrently ingest other drugs that may alter gastric emptying and therefore alter the known kinetic profile of APAP (e.g., opiods, anticholinergic drugs),9 and patients ingesting sustained-release APAP.10 The development of new biomarkers of APAP toxicity may prove helpful in the diagnosis and/or management of these patient subgroups.
The toxicity of an overdose of APAP has been recognized since the 1960s,11,12 and the mechanisms responsible for the toxicity have been studied and reviewed extensively.13–15 After absorption from the gastrointestinal tract, APAP enters the enterohepatic circulation, where most of the parent drug is removed by conjugation.16 A small fraction is oxidized by cytochrome P450 to form the reactive metabolite (N-acetyl-p-benzoquinone imine, NAPQI). The most significant P450 isoforms in the oxidation of APAP are CYP2E1, CYP1A2, and CYP3A4.17 Following low doses of APAP, NAPQI is efficiently detoxified by glutathione, forming the conjugate 3-(glutathione-S-yl)APAP, which is converted to APAP mercapturate and excreted by the kidney.18 In a situation of APAP overdose or in circumstances that lead to the depletion of glutathione, the reactive metabolite covalently binds to cellular proteins. The available evidence indicates that the covalent binding of APAP to protein is the result of the reaction between NAPQI and cysteinyl sulfhydryl groups on proteins to produce the corresponding 3-(cystein-S-yl) APAP protein adduct.19,20 However, despite extensive research, the molecular and cellular events associated with APAP overdose are not adequately understood, and APAP toxicity remains a significant clinical problem.
In previous studies, our laboratory showed that measurement of APAP protein adducts (hereafter referred to as “ APAP adducts” or just “adducts”) can be used to identify patients with occult APAP toxicity.4,21 Most recently, we characterized the pharmacokinetics of APAP adducts in adults who were having ALF secondary to APAP overdose. Adducts were detected up to 12 days after APAP overdose,22 and the elimination half-life of adducts far exceeded that reported for the parent compound, APAP, in overdose.23 The t½ of APAP in adults with APAP-related ALF has been reported to be 6.4 h in patients without encephalopathy and 18.4 h in patients with encephalopathy.23
This study was conducted to examine the pharmacokinetics and clinical correlations of APAP adducts in children and adolescents presenting to pediatric medical centers with APAP overdose. A knowledge of the toxicokinetics of adducts in patients across the pediatric age spectrum is necessary to explore the potential applications of this biomarker to the clinical diagnosis and management of children and adolescents with APAP overdose, particularly those in whom the Rumack–Matthews nomogram cannot be utilized.
RESULTS
Patient data
The gender composition of the study population was 122 female subjects (77.7%) and 35 male subjects (22.3%). The majority of the subjects were >12 years of age (130 subjects, 82.8%). Of the rest, 15 (9.6%) subjects were <2 years old; 4 (2.5%) subjects were 2–6 years old; and 8 (5.1%) subjects were 7–12 years old. The majority of the subjects were Caucasian (129 subjects, 82%). The rest were African-American (16; 10%); Hispanic (6; 4%), and other (6; 4%). Spontaneous recovery occurred in 155 (98.7%) of the study subjects. Two subjects required liver transplantation, and there were no deaths in the study population. The vast majority of the overdoses were acute (94.3%; n = 148), and nine were chronic overdoses. Dose information was available for 78 and 74% of the acute and chronic overdoses, respectively. The mean reported (±SD) dose of APAP ingested in the acute overdose patients was 633 (±1,647) mg/kg, and in the chronic overdose patients it was 139 (±30) mg/kg/day. NAC was administered to 141 (90%) of the subjects; gastric decontamination agents used in the study population were activated charcoal (65, 42.2%), syrup of ipecac (1, 0.6%), and gastric lavage (18, 11.9%).
APAP adduct values were compared with clinical outcomes and laboratory parameters. Given that multiple measures were available from each patient, the peak value for APAP adducts was used for initial correlations with demographic, laboratory, and treatment data. The levels of adducts did not significantly vary as functions of gender, race, ethnicity, age, or reported dose of APAP ingested.
Significant correlations were noted between peak APAP adducts and peak aspartate aminotransferase (AST) (Figure 1, R = 0.843) and also between peak APAP adducts and peak alanine aminotransferase (ALT) (R = 0.828). These correlations did not vary significantly according to whether patients were victims of acute ingestions (R = 0.837; n = 148) or chronic ingestions (R = 0.830; n = 9). The peak APAP adduct level also varied as a function of the severity of hepatotoxicity, stratified by three toxicity severity groups by peak AST (Figure 2). Peak APAP adduct levels did not correlate with peak prothrombin time, peak international normalized ratio, or peak creatinine (data not shown). These data indicate that APAP adducts correlate with common clinical markers of hepatotoxicity in children and adolescents with APAP overdose (Figures 1–2).
Figure 1.
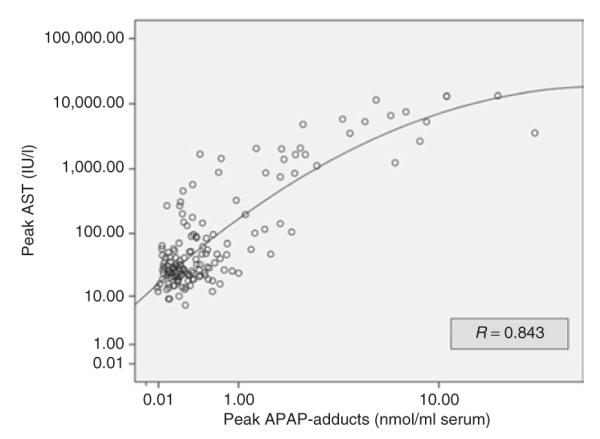
Correlation of peak acetaminophen (APAP) adducts with peak aspartate aminotransferase (AST) in children and adolescents with APAP overdose.
Figure 2.
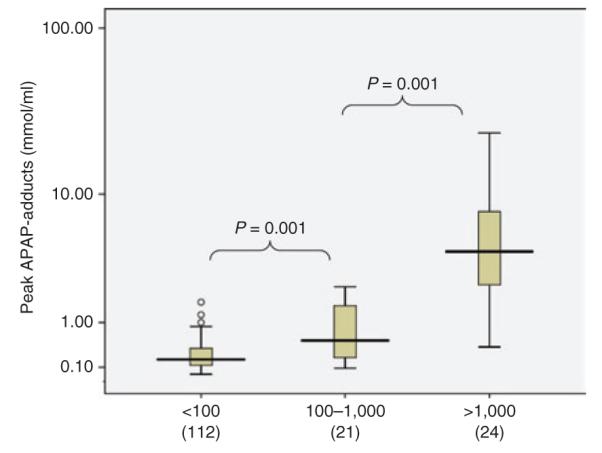
Box plots depicting the relationship of adducts to toxicity as a function of severity subgroups determined by aspartate aminotransferase level. The box plots show the median (line) and 25–75% interquartile range for adducts by toxicity severity subgroups (circles represent outliers).
As mentioned earlier, the Rumack–Matthews nomogram is the risk-assessment tool used in hospital emergency departments to determine the risk of serious liver toxicity and therefore the need for treatment with antidotal NAC. Adduct values were analyzed relative to the nomogram-predicted risk. As shown in Figure 3, peak APAP adducts varied as a function of risk. A significant percentage of the study cohort did not meet the criteria for use of the Rumack–Matthews nomogram. In these patients, either the time of the ingestion was unknown (n = 20) or they presented for medical management >24 h after the APAP overdose (n = 2). Adduct values were higher in this group than in the other two groups, suggesting that many of the patients for whom use of the Rumack–Matthews nomogram was not appropriate did in fact have serious toxicity.
Figure 3.
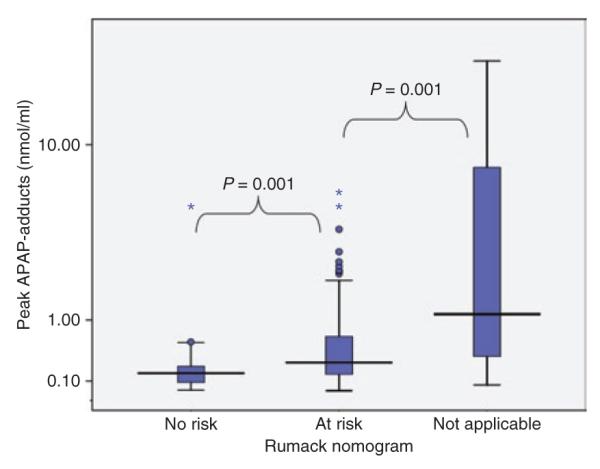
Relationship of adducts to Rumack–Matthews nomogram-based risk stratification. The box plots show the median (line) and 25–75% interquartile range for adducts by toxicity severity subgroups (circles represent outliers and asterisks represent extreme values).
Treatment with NAC prevents the development of toxicity when administered within 10 h of an APAP overdose.7,24 In this study, 90% (141) of the study subjects received treatment with NAC and 54% of those received NAC within 10 h of the overdose. Adduct values were higher in patients for whom NAC treatment was delayed relative to the APAP overdose, as indicated in Figure 4.
Figure 4.
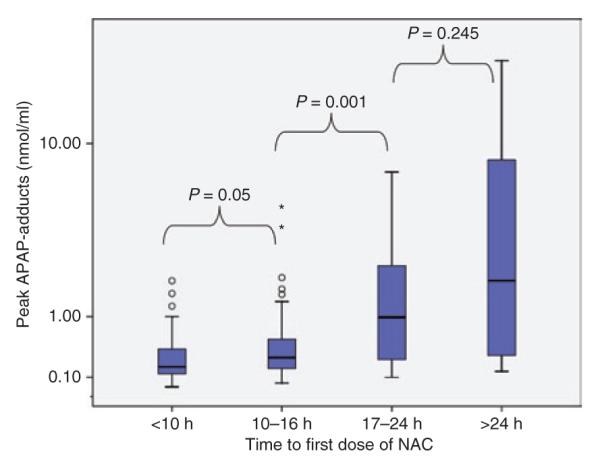
Relationship of APAP adducts to time-to-first-dose of N-acetylcysteine (NAC). The box plots show the median (line) and 25–75% interquartile range for adducts stratified by time-to-first-dose of NAC (circles represent outliers and asterisks represent extreme values).
Overall, the APAP adduct concentrations were well described by a mono-exponential decay (Figure 5). The population estimate (and SE) for ke was 0.0197 ± 0.00128 h−1, and subjects sampled within 2 days of ingestion exhibited a population lag time of 1.4 (±1.5 SE) h with an absorption rate constant, ka, of 0.086 (±0.010 SE) days−1. The intersubject variability for ke was 23% (±11.6% SE), and the residual error was 22% (±9.0% SE). Adding age (<7 vs. ≥7 years) or Cmax (used as a surrogate for degree of overdose) as potential covariates on ke, did not improve the model. In the post hoc analysis, subjects with fewer than two samples had lower Cmax values but ke similar to that in subjects with more than two samples.
Figure 5.
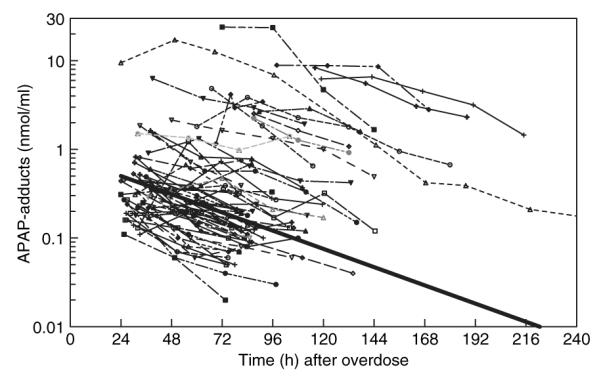
Adduct concentration vs. time plot for individual subjects with >2 samples available for analysis. The thick solid line represents the population elimination.
A summary of the data relating to the pharmacokinetic analysis of APAP adducts is presented in Table 1. The data reflect the pharmacokinetics in subjects with two or more samples available for analysis. Because of the convenience sampling design of the study, a varied number of samples and sample collection points existed in the study database. On the basis of the model, the parameter “Adduct 48” was used as an estimate of the projected APAP adduct value at 48 h after ingestion. (This time point was selected because the greater proportion of samples in the data set were from time points >24 h after ingestion rather than <24 h after ingestion and because the peak of APAP toxicity is typically thought to occur between 48 and 72 h after ingestion.24) Adduct 48 included data from all subjects, including those with only one sample. Adduct 48 was compared with toxicity end points. As demonstrated in Figure 6, Adduct 48 strongly correlated with peak AST (R = 0.625; P < 0.001), and no age association was present. In addition, Adduct 48 correlated with peak ALT (R = 0.649, P < 0.0001), the model-predicted Cmax (R = 0.91; P < 0.0001), and the Rumack–Matthews risk prediction (R = 0.62; P < 0.0001).
Table 1.
S ummary of pharmacokinetic data for APAP adducts in 125 subjects
| ke (h−1) | Half-life (h) | ke (days−1) | Half-life (days) |
Cmax (nmol/ ml serum) |
|
|---|---|---|---|---|---|
| Mean | 0.020 | 35.32 | 0.486 | 1.47 | 1.20 |
| SD | 0.004 | 7.10 | 0.084 | 0.30 | 2.92 |
| Median | 0.020 | 34.95 | 0.476 | 1.46 | 0.36 |
| 25th Percentile | 0.119 | 31.40 | 0.448 | 1.31 | 0.20 |
| 75th Percentile | 0.022 | 37.11 | 0.530 | 1.55 | 0.81 |
Figure 6.
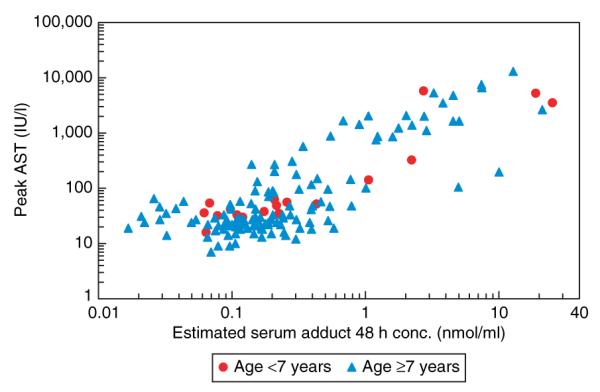
Raw data points showing correlation of Adduct 48 with peak aspartate aminotransferase (AST). No association was noted for age.
DISCUSSION
Covalent binding of the APAP reactive metabolite NAPQI to cysteinyl sulfhydryl groups on protein to produce the corresponding 3-(cystein-S-yl) APAP protein adduct was recognized as a correlate of APAP toxicity in animal models in the 1980s.25,26 Antisera with specificity for the 3-(cystein-S-yl) APAP epitope were used to elucidate dose–response and temporal relationships between the appearance of APAP protein adducts in the liver and in serum following the administration of hepatotoxic doses of APAP to laboratory mice.25,27 As hepatocytes lyse, hepatic transaminases and adducts are concurrently released into the serum. The initial translation of these findings from the experimental setting to the clinical arena occurred in 1990 when Hinson et al. reported the detection of adducts in adults with severe APAP overdoses.28 This initial study in humans utilized an ELISA assay and demonstrated high levels of adducts in patients with treatment delays and severe hepatotoxicity following APAP overdose. Further studies used western blot assays to examine blood samples obtained from adolescents and children with APAP overdose and revealed that this approach was labor-intensive and not adequately sensitive for diagnostic work with human subjects.29 In recent years, a more precise and sensitive analytical method based on high-performance liquid chromatography with electrochemical detection30 has been used to quantify APAP-cysteine from proteolytically digested serum proteins as a measure of the total levels of the APAP protein adduct biomarker in human peripheral blood. An examination of blood samples from adults and children with ALF showed that high levels of adducts were invariably present in well-characterized cases of APAP overdose resulting in ALF.4,21 In addition, no adducts were found in patients with other known causes of ALF.4 Further, relatively lower levels of adducts were found in acute overdose patients who received early antidotal treatment with NAC to protect against toxicity. In subsequent studies, post hoc analysis of samples from patients with ALF of unknown etiology was performed, and adducts were detected in 19% of patient samples, strongly suggesting that APAP was the etiology of the liver injury.4 The conclusion of likely APAP etiology was further supported by extensive testing for other causes of ALF, including viral hepatitis, autoimmune liver disease, and metabolic etiologies. Adducts were also detected in the samples from 15% of children with ALF of unknown etiology.21
In our study of children and adolescents with APAP overdose, significant relationships were noted between APAP adducts and common, but nonspecific, clinical indicators of liver injury (Figures 1,2). APAP adducts had a strong correlation with both AST (Figure 1) and ALT. In addition, the model-derived estimate, Adduct 48, also correlated with peak AST (Figure 6) and peak ALT. As reported previously for adults with ALF,22 this study also showed the correlation between adducts and AST (Figure 1) to be slightly stronger than the correlation between adducts and ALT. Both AST and ALT may be of extrahepatic (in addition to hepatic) origin. In this regard, the relative abundance of AST in extrahepatic tissues (e.g., heart, skeletal muscle, blood cells) is greater than that of ALT.31,32 In addition, the primary cytochrome for APAP metabolism, CYP2E1, is present in extrahepatic tissues (e.g., nasal mucosa, olfactory epithelium, lung, and kidney)33 and adducts occur in these tissues.34 Therefore it is possible that a small proportion of adduct formation may be extrahepatic, and necrotic lysis of these extrahepatic cells contributes, in addition to serum adducts, to a greater proportion of AST than of ALT, and this may account for the slightly higher correlation of AST with APAP adducts.
As expected, APAP adduct values were higher in patients for whom there had been delays in treatment with NAC and who had consequently experienced greater toxicity. This finding is consistent with previously published data showing the effect of NAC treatment on the risk of developing acute liver injury.5,24 APAP adduct values were also higher in patients at risk for toxicity per the Rumack–Matthews nomogram (Figure 4). This finding was further supported by the correlation seen between the model-derived Adduct 48 and the Rumack–Matthews risk prediction. As mentioned earlier, the Rumack–Matthews nomogram7 is widely used for assessing the risk of developing toxicity within the first 24 h of acute APAP overdose. The assessment of risk of hepatotoxicity and determination of the need for treatment with NAC is difficult in other patient groups, including patients who present in the late stages of toxicity (>24 h after the overdose), patients with chronic ingestions of APAP, acute ingestions in alcoholics,8 patients who concurrently ingest other drugs that may alter gastric emptying and therefore alter the known kinetic profile of APAP (e.g., opiods, anticholinergic drugs),9 and patients with ingestions of sustained-release APAP.10 The development of biomarkers of APAP toxicity would have potential clinical applications for these groups of patients with APAP toxicity. An important difference between assessment based on the Rumack–Matthews nomogram and one based on determination of adducts is that the former relies on levels of the parent drug regardless of metabolism, whereas the latter (adduct level) is a measure of the NAPQI remaining after detoxification by glutathione and also reflects the relative contributions of phase I and phase II metabolism.
We recently reported that APAP adducts persist in human sera for 12 days in adults with APAP-related ALF.22 In the adult population with liver failure, the elimination half-life of adducts was 1.72 (±0.34) days.22 In the less severely ill population in our study, the elimination half-life was remarkably similar at 1.47 ± 0.30 days. Moreover, the variability for the elimination half-life for adducts was low (Table 1), and the t1/2 did not significantly vary as a function of Cmax or of the number of samples obtained (data not shown). The similarity in the values of elimination half-life in the two populations likely reflects similar rates of proteolytic digestion for APAP adducts among them. As APAP adducts become small peptide adducts, they are removed either through renal clearance or by the sample preparation methodology, which removes small molecules. Despite the similarity of this parameter to the corresponding data in adults, the maximum value of adduct observed (Cmax, Table 1) in this study was significantly lower (1.2 ± 2.9 nmol/ml serum) than the one previously reported for adults with APAP-related ALF (10.9 ± 9.3 nmol/ml serum). A number of factors are likely to have contributed to the lower value for Cmax of APAP adducts in this study, including a lower degree of disease severity. The mortality rate in the adult ALF population was 16.9%, whereas no deaths occurred in the pediatric and adolescent population. Furthermore, a large percentage of the patients in our cohort (71.3%) did not develop significant liver injury (peak AST > 1,000 IU/l), either because of subtoxic ingestions (Figure 2) or because of early NAC intervention (Figure 4).
Several limitations of our study should be noted. As previously mentioned, a significant percentage of the patients were those with acute overdoses who received timely treatment with NAC and therefore did not develop significant liver injury. Consequently, the data do not represent the development of liver toxicity that would be observed in an untreated population. However, the demographics of our study population are consistent with previous literature describing the demographic profile of APAP overdose in adolescents and children presenting to pediatric medical centers. For example, Alander et al. characterized the demographics of APAP overdose in adolescents and children and noted that 78% of the patients who had intentionally overdosed were female, 62% were Caucasian, and the mean age of this subgroup was 14.3 ± 1.3 years.35 In addition, several reports have documented that adolescents fail to recognize the toxicity potential of APAP taken as an overdose.36,37 Although adduct values did not significantly vary with gender, race, ethnicity, or age in this study, the fact that there were only a small number of subjects in the lower age groups may have limited the conclusions regarding the effect of age on APAP adducts. Another limitation was that the study did not have the ability to systematically verify exposure to co-ingestants. It is possible that the kinetics of APAP adducts may be affected by drugs that alter gastric emptying, such as opiods and anticholinergic drugs. The potential effects of these co-ingestants on the pharmacokinetics of this biomarker should be examined in future studies.
A troubling aspect in the pediatric population is the occurrence of inadvertent APAP overdose. A number of possible causes have been cited for the increasing occurrence of this phenomenon, including the administration of adult formulations to children, the widespread availability of APAP in over- the-counter cough and cold remedies, and the misconception by the general public that APAP is nontoxic.36,38,39 Previous literature suggests that patients who present with unintentional APAP overdose are typically young persons. The diagnosis may be challenging because of vague histories of APAP dosing and nonspecific symptoms of overdose. In this setting, the Rumack–Matthews nomogram is of limited use, and the development of new diagnostic biomarkers would greatly facilitate diagnosis. A recent development in adolescent drug abuse is the shift toward greater use of prescription drugs,40 as opposed to street drugs. Opioid/APAP combination products represent a likely target of drug abuse in adolescents, and the Rumack–Matthews nomogram would not be applicable in this setting of chronic exposure. Because the appearance of the APAP adduct biomarker factors in the influences of absorption, phase I and phase II metabolism, and glutathione reserve, this biomarker has relevance in a number of clinical scenarios involving APAP use and exposure in susceptible children and adolescents.
In summary, this study found that APAP adducts correlate with measures of toxicity in children and adolescents with APAP overdose who received timely treatment with NAC. In addition, elimination characteristics of APAP adducts in this population were similar to those reported for adults with ALF following APAP overdose. Adducts were also shown to correlate with time to treatment with NAC and with Rumack–Matthews risk assessment. Further studies are needed to examine the effects of co-ingestants on APAP adduct formation and elimination. With further refinement and testing, this biomarker may ultimately prove to be useful in the diagnostic evaluation of children with ALF of unknown etiology, given that the elimination half-life of adducts appears to be greater than that of the parent compound.23 In addition, the biomarker could potentially be used in patients who present >24 h after APAP ingestion but before the development of severe liver injury. This study, and recent data in adults with APAP-related ALF,22 extends our knowledge regarding the relationship of APAP adduct formation to nonspecific clinical measures of toxicity in humans and further confirms the position of APAP protein adducts as a clinically relevant and specific biomarker of APAP toxicity.
METHODS
Study population
Serum samples were obtained from 157 adolescents and children from eight participating sites within the Network of Pediatric Pharmacology Research Units funded by the National Institutes of Child Health and Human Development. Patients eligible for enrollment presented with a chief complaint of APAP ingestion. The study was reviewed by the institutional review boards of all participating sites, informed consent was obtained from the parents of the children, and assent was obtained from children >7 years of age.
After the subjects were enrolled, serum samples were collected from the study subjects by convenience sampling at the same time that samples for the routine monitoring of APAP overdose were obtained. Sampling continued until the time of hospital discharge. Clinical chemistry samples for the routine monitoring of APAP overdose (hepatic transaminases, coagulation studies) were obtained at the discretion of the treating physician. The duration of treatment with NAC and the route of administration of NAC were determined by the treating physician.
Data collection forms that included historical, demographic, clinical, and laboratory data were completed by the staff at the enrolling site and sent to the lead site, where they were entered into an Access data base (Microsoft Office, Redmond, WA).
Analytical method
Serum samples were assayed for APAP adducts using a modification of the previously reported high-performance liquid chromatography with electrochemical detection assay that has greater sensitivity than the one we used for our initial reports.4,21,29,30 Assay modifications, including gel filtration and enhanced proteolytic digestion, resulted in improved sensitivity and efficiency of the assay.22 The lower limit of quantitation for the assay was 0.03 μmol/l. Final assay results were expressed as nmol/ml serum.
Clinical end points
Patient data include reported dose (mg/kg) and time of APAP ingestion; history of concomitantly ingested drugs; administration, duration, and route (intravenous, oral, or both) of NAC; Rumack–Matthews risk classification (no risk; at risk; or not applicable, defined as time of overdose unknown, or patient presented at the medical facility >24 h after the overdose) and outcome (spontaneous recovery, liver transplantation, or death). The overdoses were categorized as acute (one single ingestion) or chronic (multiple ingestions). The laboratory tests included ALT, AST, total bilirubin, prothrombin time, international normalized ratio, and creatinine. Individual subject peak values for each laboratory parameter were analyzed relative to the peak levels of APAP adducts for the corresponding subject, indicating the peak or maximum measurement of APAP adduct for the subject. In addition, clinical end points and APAP adduct values were analyzed relative to the time of reported overdose and expressed in 24-h increments relative to the time of the overdose. The day of the overdose was defined as day 0 for these analyses.
Statistical analysis
Nonparametric tests were used for examining differences between subgroups (Kruskal–Wallis, Mann–Whitney tests). Associations among variables were determined using Spearman’s correlations. Statistical analysis was performed using SPSS (version 15, SPSS, Chicago, IL).
Pharmacokinetic analysis
The elimination of APAP adducts was analyzed with a population pharmacokinetic approach using the NONMEM program (version VI, first-order conditional estimation subroutine with interaction).41 A one-compartment model was used. Other than testing for possible influence of age and Cmax on elimination, more complex structural models were not evaluated because of the limited range and number of samples available for the analysis. Given that >90% of the subjects received treatment with NAC, adduct formation was assumed to be complete for subjects sampled 3 or more days after ingestion. A first-order input model that included a lag time was used for subjects sampled within 2 days of ingestion to account for ongoing APAP adduct production. The amount of APAP adduct was not estimated from APAP dose or other patient characteristics because the model focused solely on the rate of APAP adduct elimination. The “apparent doses” of adduct were scaled on the basis of the initially observed APAP adduct concentrations. The first-order conditional estimation–plus-interaction estimation method was used, and the model incorporated a combined proportional plus an additive residual error equal to half of the limit of quantitation. Individual empiric Bayesian estimates for the elimination rate ke, t½, and Cmax were determined using the post hoc subroutine. The mean (±SD) number of samples available per patient was 2.48 (±1.29) and sample availability for the entire population was as follows: 1 sample (32 subjects); 2 samples (61 subjects); 3 samples (38 subjects); 4 samples (16 subjects); 5 samples (7 subjects); 6 samples (0 subjects); 7 samples (1 subject); 8 samples (2 subjects).
AcknowleDgMents
This work was supported by grants DK06799 and HD031324 to L.P.J. We acknowledge the invaluable contributions of the study coordinators and research nurses of the Network of Pediatric Pharmacology Research Units (PPRU). The work of the following participating PPRU centers and personnel is acknowledged: Lisa Bomgaars, Baylor College of Medicine PPRU; Mary Lei Lai, Wayne State PPRU; Kosair Children’s Hospital PPRU; Arkansas Children’s Hospital PPRU; Mercy Children’s Hospital PPRU; Mike Reed, Rainbow Babies Children’s Hospital PPRU; Mike Christenson, PharmD, University of Tennessee PPRU (1994–1998); and John van den Anker, (Ohio State PPRU 2001–2002, currently at National Children’s Medical Center PPRU). This research was also supported, in part, by the Arkansas Children’s Hospital Research Institute and the Arkansas Biosciences Institute, the major research component of the Tobacco Settlement Proceeds Act of 2000.
Footnotes
CONFLICT OF INTEREST L.P.J., D.R., and J.A.H. have filed a patent for technology relating to the measurement of acetaminophen protein adducts. The other authors declared no conflict of interest.
References
- 1.Litovitz TL, et al. 2001. Annual report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am. J. Emerg. Med. 2002;20:391–452. doi: 10.1053/ajem.2002.34955. [DOI] [PubMed] [Google Scholar]
- 2.Squires RH, Jr., et al. Acute liver failure in children: the first 348 patients in the pediatric acute liver failure study group. J. Pediatr. 2006;148:652–658. doi: 10.1016/j.jpeds.2005.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larson AM, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–1372. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- 4.Davern TJ, 2nd, et al. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology. 2006;130:687–694. doi: 10.1053/j.gastro.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 5.Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics. 1975;55:871–876. [PubMed] [Google Scholar]
- 6.Rumack BH. Acetaminophen overdose in children and adolescents. Pediatr. Clin. North Am. 1986;33:691–701. doi: 10.1016/s0031-3955(16)36050-3. [DOI] [PubMed] [Google Scholar]
- 7.Rumack BH, Peterson RC, Koch GG, Amara IA. Acetaminophen overdose. 662 cases with evaluation of oral acetylcysteine treatment. Arch. Intern. Med. 1981;141:380–385. doi: 10.1001/archinte.141.3.380. [DOI] [PubMed] [Google Scholar]
- 8.Ali FM, Boyer EW, Bird SB. Estimated risk of hepatotoxicity after an acute acetaminophen overdose in alcoholics. Alcohol. 2008;42:213–218. doi: 10.1016/j.alcohol.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Halcomb SE, Sivilotti ML, Goklaney A, Mullins ME. Pharmacokinetic effects of diphenhydramine or oxycodone in simulated acetaminophen overdose. Acad. Emerg. Med. 2005;12:169–172. doi: 10.1197/j.aem.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 10.Bizovi KE, Aks SE, Paloucek F, Gross R, Keys N, Rivas J. Late increase in acetaminophen concentration after overdose of Tylenol Extended Relief. Ann. Emerg. Med. 1996;28:549–551. doi: 10.1016/s0196-0644(96)70119-1. [DOI] [PubMed] [Google Scholar]
- 11.Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J. Pharmacol. Exp. Ther. 1973;187:185–194. [PubMed] [Google Scholar]
- 12.Davidson DG, Eastham WN. Acute liver necrosis following overdose of paracetamol. Br. Med. J. 1966;2:497–499. doi: 10.1136/bmj.2.5512.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos. 2003;31:1499–1506. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- 14.Hinson JA, Pohl LR, Monks TJ, Gillette JR. Acetaminophen-induced hepatotoxicity. Life Sci. 1981;29:107–116. doi: 10.1016/0024-3205(81)90278-2. [DOI] [PubMed] [Google Scholar]
- 15.Jaeschke H. Role of inflammation in the mechanism of acetaminophen-induced hepatotoxicity. Expert Opin. Drug Metab. Toxicol. 2005;1:389–397. doi: 10.1517/17425255.1.3.389. [DOI] [PubMed] [Google Scholar]
- 16.Nelson SD, Tirmenstein MA, Rashed MS, Myers TG. Acetaminophen and protein thiol modification. Adv. Exp. Med. Biol. 1991;283:579–588. doi: 10.1007/978-1-4684-5877-0_73. [DOI] [PubMed] [Google Scholar]
- 17.Hinson JA, Pumford NR, Roberts DW. Mechanisms of acetaminophen toxicity: immunochemical detection of drug-protein adducts. Drug Metab. Rev. 1995;27:73–92. doi: 10.3109/03602539509029816. [DOI] [PubMed] [Google Scholar]
- 18.Gillette JR, et al. Formation of chemically reactive metabolites of phenacetin and acetaminophen. Adv. Exp. Med. Biol. 1981;136(Pt B):931–950. [PubMed] [Google Scholar]
- 19.Streeter AJ, Harvison PJ, Nelson SD, Baillie TA. Cross-linking of protein molecules by the reactive metabolite of acetaminophen, N-acetyl-p-benzoquinone imine, and related quinoid compounds. Adv. Exp. Med. Biol. 1986;197:727–737. doi: 10.1007/978-1-4684-5134-4_67. [DOI] [PubMed] [Google Scholar]
- 20.Hoffmann KJ, Streeter AJ, Axworthy DB, Baillie TA. Identification of the major covalent adduct formed in vitro and in vivo between acetaminophen and mouse liver proteins. Mol. Pharmacol. 1985;27:566–573. [PubMed] [Google Scholar]
- 21.James LP, et al. Detection of acetaminophen protein adducts in children with acute liver failure of indeterminate cause. Pediatrics. 2006;118:e676–e681. doi: 10.1542/peds.2006-0069. [DOI] [PubMed] [Google Scholar]
- 22.James LP, et al. Pharmacokinetic analysis of acetaminophen protein adducts in adults with acute liver failure. Clin. Pharmacol. Ther. 2007;81 (Abstract) [Google Scholar]
- 23.Schiødt FV, Ott P, Christensen E, Bondesen S. The value of plasma acetaminophen half-life in antidote-treated acetaminophen overdosage. Clin. Pharmacol. Ther. 2002;71:221–225. doi: 10.1067/mcp.2002.121857. [DOI] [PubMed] [Google Scholar]
- 24.Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985) N. Engl. J. Med. 1988;319:1557–1562. doi: 10.1056/NEJM198812153192401. [DOI] [PubMed] [Google Scholar]
- 25.Pumford NR, Hinson JA, Potter DW, Rowland KL, Benson RW, Roberts DW. Immunochemical quantitation of 3-(cystein-S-yl) acetaminophen adducts in serum and liver proteins of acetaminophen-treated mice. J. Pharmacol. Exp. Ther. 1989;248:190–196. [PubMed] [Google Scholar]
- 26.Roberts DW, Pumford NR, Potter DW, Benson RW, Hinson JA. A sensitive immunochemical assay for acetaminophen-protein adducts. J. Pharmacol. Exp. Ther. 1987;241:527–533. [PubMed] [Google Scholar]
- 27.Roberts DW, et al. Immunohistochemical localization and quantification of the 3-(cystein-S-yl)-acetaminophen protein adduct in acetaminophen hepatotoxicity. Am. J. Pathol. 1991;138:359–371. [PMC free article] [PubMed] [Google Scholar]
- 28.Hinson JA, Roberts DW, Benson RW, Dalhoff K, Loft S, Poulsen HE. Mechanism of paracetamol toxicity. Lancet. 1990;335:732. doi: 10.1016/0140-6736(90)90851-u. [DOI] [PubMed] [Google Scholar]
- 29.James LP, et al. Measurement of acetaminophen-protein adducts in children and adolescents with acetaminophen overdoses. Pediatric Pharmacology Research Unit Network, NICHD. J. Clin. Pharmacol. 2001;41:846–851. doi: 10.1177/00912700122010744. [DOI] [PubMed] [Google Scholar]
- 30.Muldrew KL, et al. Determination of acetaminophen-protein adducts in mouse liver and serum and human serum after hepatotoxic doses of acetaminophen using high-performance liquid chromatography with electrochemical detection. Drug Metab. Dispos. 2002;30:446–451. doi: 10.1124/dmd.30.4.446. [DOI] [PubMed] [Google Scholar]
- 31.Green RM, Flamm S. AGA technical review on the evaluation of liver chemistry tests. Gastroenterology. 2002;123:1367–1384. doi: 10.1053/gast.2002.36061. [DOI] [PubMed] [Google Scholar]
- 32.Wroblewski F. The clinical significance of transaminase activities of serum. Am. J. Med. 1959;27:911–923. doi: 10.1016/0002-9343(59)90175-5. [DOI] [PubMed] [Google Scholar]
- 33.Gu J, et al. In vivo mechanisms of tissue-selective drug toxicity: effects of liver-specific knockout of the NADPH-cytochrome P450 reductase gene on acetaminophen toxicity in kidney, lung, and nasal mucosa. Mol. Pharmacol. 2005;67:623–630. doi: 10.1124/mol.104.007898. [DOI] [PubMed] [Google Scholar]
- 34.Hart SG, Cartun RW, Wyand DS, Khairallah EA, Cohen SD. Immunohistochemical localization of acetaminophen in target tissues of the CD-1 mouse: correspondence of covalent binding with toxicity. Fundam. Appl. Toxicol. 1995;24:260–274. doi: 10.1006/faat.1995.1029. [DOI] [PubMed] [Google Scholar]
- 35.Alander SW, Dowd MD, Bratton SL, Kearns GL. Pediatric acetaminophen overdose: risk factors associated with hepatocellular injury. Arch. Pediatr. Adolesc. Med. 2000;154:346–350. doi: 10.1001/archpedi.154.4.346. [DOI] [PubMed] [Google Scholar]
- 36.Harris HE, Myers WC. Adolescents’ misperceptions of the dangerousness of acetaminophen in overdose. Suicide Life Threat Behav. 1997;27:274–277. [PubMed] [Google Scholar]
- 37.Myers WC, Otto TA, Harris E, Diaco D, Moreno A. Acetaminophen overdose as a suicidal gesture: a survey of adolescents’ knowledge of its potential for toxicity. J. Am. Acad. Child Adolesc. Psychiatry. 1992;31:686–690. doi: 10.1097/00004583-199207000-00016. [DOI] [PubMed] [Google Scholar]
- 38.Heubi JE, Barbacci MB, Zimmerman HJ. Therapeutic misadventures with acetaminophen: hepatoxicity after multiple doses in children. J. Pediatr. 1998;132:22–27. doi: 10.1016/s0022-3476(98)70479-2. [DOI] [PubMed] [Google Scholar]
- 39.Kearns GL, Leeder JS, Wasserman GS. Acetaminophen overdose with therapeutic intent. J. Pediatr. 1998;132:5–8. doi: 10.1016/s0022-3476(98)70476-7. [DOI] [PubMed] [Google Scholar]
- 40.Friedman RA. The changing face of teenage drug abuse—the trend toward prescription drugs. N. Engl. J. Med. 2006;354:1448–1450. doi: 10.1056/NEJMp068010. [DOI] [PubMed] [Google Scholar]
- 41.Beal SL, Sheiner LB. NONMEM Users Guides Parts I-VIII. NONMEM Project Group. University of California; San Francisco, CA: [Google Scholar]