

Abstract
A flexible molecular scaffold bearing varying numbers of terminal alkyne groups was synthesized in five steps from solanesol. R(CO)-MSH(4)-NH2 ligands, which have a relatively low affinity for binding at the human melanocortin 4 receptor (hMC4R), were prepared by solid phase synthesis and were N-terminally acylated using 6-azidohexanoic acid. Multiple copies of the azide N3(CH2)5(CO)-MSH(4)-NH2 were attached to the alkyne-bearing, solanesol-derived molecular scaffold via the copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reaction. Control studies showed that the binding affinity of the triazole-containing ligand, CH3(CH2)3(C2N3)(CH2)5(CO)-MSH(4)-NH2, was not significantly diminished relative to the corresponding parental ligand, CH3(CO)-MSH(4)-NH2. In a competitive binding assay using a Eu-labeled probe based on the superpotent ligand NDP-α-MSH, the monovalent and multivalent constructs appear to bind to hMC4R as monovalent species. In a similar assay using a Eu-labeled probe based on MSH(4), modest increases in binding potency with increased MSH(4) content per scaffold were observed.
Introduction
Early detection and diagnosis of many human cancers would be facilitated by the availability of reagents that could seek out and selectively bind to cancer cells and report their existence and location by non-invasive molecular imaging.1 One strategy for development of such reagents involves linking imaging agents to molecules that contain multiple copies of individual binding units, or ligands, targeted to cell surface receptors that are displayed by cancer cells.2 Such multivalent constructs should display enhanced affinity and selectivity for cancer cells based on cooperative binding.1–3
The foundation for ligand-guided multivalent attachment of reporter groups to cell surface receptors was laid in part by studies that employed a poly(vinyl alcohol) scaffold (PVA) decorated with fluorescein and R(CO)-NDP-α-MSH-NH2 ligands.4 Such constructs bound specifically and irreversibly to mouse and human melanoma cells that expressed melanocortin receptors. The PVA-based multimer system was not extended to other peptide hormone/receptor systems at that time due to problems with the attachment chemistry and solubility of the multimeric constructs. Recent advances in polymer-supported multivalent binding5 prompted a reexamination of this earlier approach to ligand multimerization with the intent of using more efficient attachment chemistry and developing more soluble scaffolds.
With regard to ligand attachment, the copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) reaction, which generates 1,4-disubstituted triazole products,6 is a reasonable choice to replace the maleimide electrophile/thiol nucleophile and thiol/disulfide exchange attachment chemistries used previously with PVA.4c,4d
With regard to scaffold, PVA derived by incomplete hydrolysis of poly(vinyl acetate) is often more water-soluble at room temperature than is more completely hydrolyzed poly(vinyl acetate).7 This is presumably due to interruption of hydrogen bonded microcrystalline domains, and suggested that a polymer bearing fewer, stereorandom hydroxyl groups might be less crystalline and more water-soluble. Such a polymer can be prepared from polyisoprene.8 Since analysis of polymer products is complicated by high molecular weight and polydispersity, we elected to employ more tractable model systems for the establishment of the synthetic methodology necessary to this approach.
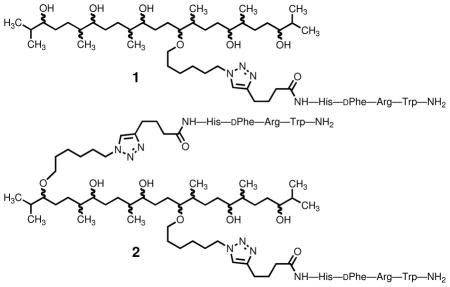
Previously we described the preparation from squalene of regiochemically and stereochemically mixed hexol derivatives 1 and 2 which carry one and two copies, respectively, of the linear tetrapeptide amide R(CO)-MSH(4)-NH2.2d In contrast with CH3(CO)-NDP-α-MSH-NH2,9 CH3(CO)-MSH(4)-NH2 ligands have relatively low affinity for binding at the human melanocortin 4 receptor (hMC4R).10 R(CO)-MSH(4)-NH2 was selected for this work because synergistic effects are more easily detected for multivalent constructs of low-affinity ligands.3c Mixture 2 was found to be about 18 times more potent than mixture 1 in a competitive binding assay against a europium-labeled derivative of MSH(4).11 The synthetic and biological results with 1 and 2 were encouraging, and work toward a polyisoprene-derived polyol scaffold was begun. However, when solubility problems were encountered during hydroboration and oxidation of polyisoprene, a study of the chemistry of a second model system intermediate in size was deemed appropriate. It was also recognized that study of a larger model system would permit testing of the biological effects of higher levels of ligand multimerization. The synthesis of a scaffold derived from the polyterpene solanesol (3), decoration of the scaffold with copies of MSH(4) via CuAAC, and results of competitive binding studies using cells expressing hMC4R are presented herein.
Results
Synthesis
The ready availability of solanesol (3) and solanesyl bromide (4)12 made these compounds appealing starting materials for construction of an intermediate-sized polyisoprene scaffold (Scheme 1). Double alkylation of dimethyl malonate with 4 gave 5 in 72% yield. Reduction of 5 using LAH produced diol 6 in 82% yield. Hydroboration and oxidation of 6 under standard conditions proved to be unsatisfactory since the use of BH3/THF left an unacceptably high percentage of alkene groups in the product polyol.13 The use of excess disiamylborane in THF for extended periods gave more satisfactory results.14 For example, hydroboration of 6 using 1.7 equivalents of disiamyborane per alkene for a period of 72 hours left 4% of the alkene groups intact in the product polyol as determined by mass spectrometry (Figure 1) and titration with bromine (see Experimental Procedures for details). The principal polyols present in the product mixture are formulated as the stereochemically mixed eicosols 7 based on mass spectral and NMR data (Figure 1). While hydroboration of 6 with BH3 should yield products containing both secondary and tertiary alcohol groups,8d disiamylborane was expected to be more regioselective and lead mostly to secondary alcohol groups. The 13C NMR spectrum of 7 (Figure 1) suggests that this expectation was met. Compound 7 was obtained in about 90% yield based on diol 6. This result was shown to be repeatable in three separate runs. Reaction of 7 with 40 equivalents of sodium hydride and 12 equivalents of 1-bromo-5-hexyne (8)15 in DMF afforded a mixture of polyol/polyalkynes 9. The product distribution was determined by mass spectrometry (Figure 2). The average number of alkynes was approximately six per molecular scaffold. The product yield based on this level of alkyne incorporation was 70%. This result was shown to be repeatable in three separate runs.
Scheme 1. Synthesis of Polyol/Polyalkyne Scaffold 9a–c.

aReagents: (a) PBr3, ether (Ref. 12). (b) NaCH(CO2Me)2, EtOH. (c) LiAlH4, ether. (d) disiamyborane, THF; H2O2, NaOH. (e) NaH, Br(CH2)4C≡CH (8). bProduct 7 resulting from anti-Markovnikov hydration is shown. It is assumed that mixtures of all possible stereoisomers of 7 are produced. cStructure 9 is a representative hexaalkyne scaffold. The sites of attachment of the 5-hexyn-1-yl groups shown in 9 are arbitrary.
Figure 1.

(a) A portion of the DEPT 135 13C NMR spectrum of 7 in CD3OD (see Ref 8d). The lines from 76–79 ppm are due to methine carbons bearing an OH group. The line at 62.8 ppm is due to the two methylene carbons that bear OH groups. The relatively sharp line at 73.3 ppm is due to residual byproduct 3-methyl-2-butanol in this sample. (b) MS showing the distribution of products in the mixture of polyols 7. Peaks near 1572, 1590, 1608, and 1626 represent solanesol-derived heptadecaols, octadecadols, nonadecaols, and eicosaols, respectively.
Figure 2.

MS showing the distribution of products in the mixture 9 resulting from alkylation of polyols 7. Peaks near 1888, 1968, 2048, 2128, 2208, 2288, and 2368 represent incorporation of 3, 4, 5, 6, 7, 8, and 9 alkyne units, respectively, onto the solanesol-derived scaffold.
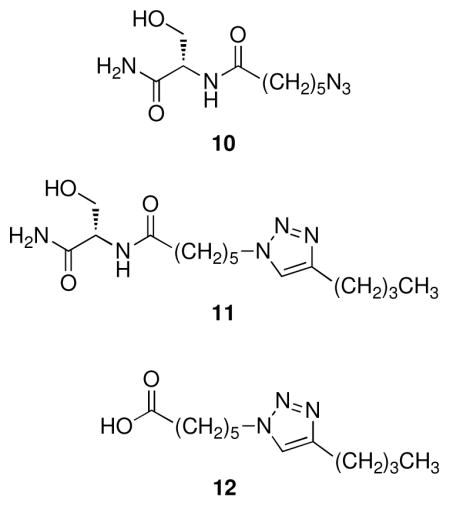
Azide 10 was prepared in 84% yield from serine amide hydrochloride16 and 6-azidohexanoic acid.17 Triazole 11 was prepared in 83% yield by reaction of azide 10 with 1-hexyne at room temperature in methanol in the presence of tetrakis(acetonitrile)copper(I) hexafluorophosphate and TBTA.18 6-(4-Butyl-1H-1,2,3-triazol-1-yl)hexanoic acid (12) was prepared in 60% yield by reaction of 6-azidohexanoic acid with 1-hexyne at room temperature in a mixture of water and t-butanol in the presence of copper sulfate and sodium ascorbate.
Solid Phase Synthesis
The low affinity ligand MSH(4)10 was constructed on Rink amide Tentagel S resin (initial loading 0.62 mmol/g) as depicted in Scheme 2.19 The product resin retained all side chain protecting groups. 6-Azidohexanoic acid17 was coupled to the N-terminus of the resin-bound tetrapeptide. Simultaneous side chain deprotection and cleavage of the tetrapeptide from the resin was effected using a mixture of trifluoroacetic acid, 1,2-ethanedithiol, thioanisole, and water (91:3:3:3), producing the desired azide-terminated ligand, N3(CH2)5(CO)-MSH(4)-NH2 (13). Triazole-containing ligand CH3(CH2)3(C2N3)(CH2)5(CO)-MSH(4)-NH2 (14) was similarly prepared by N-terminal acylation of the resin-bound tetrapeptide with acid 12 in place of 6-azidohexanoic acid. Compounds 13 and 14 were purified by reverse-phase C18 preparative HPLC and were characterized by ESI-MS and MALDI-TOF. Details appear in Table 1.
Scheme 2. Solid Phase Synthesisa.

aReagents: (a) piperidine. (b) Fmoc/tBu solid phase synthesis. (c) N3(CH2)5(CO)OH, Cl-HOBt, DIC. (d) TFA/1,2-ethanedithiol/thioanisole/water (91/3/3/3).
Table 1.
Mass spectral and HPLC characterization of compounds 13 and 14.
| Compound | Formula [M] | Calc Mass [Ion] | Mass Found (error) | tRa |
|---|---|---|---|---|
| 13 | C38H50N14O5 | 783.4161 [M+1]+ | 783.4158 (0.4 ppm) | 15.10 |
| 14 | C44H60N14O5 | 865.4949 [M+1]+ | 865.4936 (0.9 ppm) | 13.77 |
Linear gradient of from 10→90% acetonitrile in water containing 0.1% TFA over 50 min.
Multimer Assembly
Reaction of the polyol/polyalkyne mixture 9 with an excess of 10 in the presence of copper(I) and TBTA in methanol under microwave irradiation (temperature held constant at 100 °C) gave a mixture of serine amide multivalent constructs 15 (Scheme 3 and Table 2).20 After dilution with water, copper ions were removed by complexation with dithizone and extraction of the complex with CHCl3.21 Other organic-soluble molecules (TBTA, excess 10) were also removed by this extraction. The serine amide-containing product mixture 15 was recovered from the aqueous solution by lyophilization as a white powder and was characterized by MALDI-TOF mass spectrometry (see Supporting Information). The average number of serine amide residues per molecule of 15 was determined to be six.
Scheme 3. Synthesis of mixtures of multivalent constructs 15 and 16a–16e from solanesol-derived molecular scaffold 9 via CuAAC a.

aOnly one set of hexameric products from each mixture is depicted here for illustrative purposes. The sites of sidechain attachment shown are arbitrary.
Table 2.
Synthesis and compositions of 15 and 16a–e.
| Product | mg 9 | mg 13 (equiv) | mg 10 (equiv) | Yield, mg (%) | MALDI-TOF |
|---|---|---|---|---|---|
| 15 | 10 | 0 | 15 (15) | 16 (88) | Figure S12 |
| 16a | 20 | 8 (1) | 0 | 22 (76) | Figure S13 |
| 16b | 20 | 16 (2) | 0 | 29 (78) | Figure S14 |
| 16c | 20 | 8 (1) | 15 (7) | 31 (76) | Figure S15 |
| 16d | 20 | 16 (2) | 12 (6) | 19 (41) | Figure S16 |
| 16e | 10 | 16 (4) | 4 (4) | 22 (76) | Figure S17 |
Reaction of 9 with one or two equivalents of 13 in the presence of copper(I) and TBTA in methanol under microwave irradiation20 gave mixtures of MSH(4)-bearing multivalent constructs 16a and 16b, respectively (Scheme 3 and Table 2). Reaction of 9 with from one, two, or four equivalents of 13, followed by reaction of the intermediate MSH(4)-containing multivalent constructs with an excess of 10 under these same conditions, gave mixtures of MSH(4)/serine amide multivalent constructs 16c–e. After dilution with water, copper ions were removed by complexation with dithizone and extraction of the complex with CHCl3.21 Other organic-soluble molecules (TBTA, excess 10) were also removed by this extraction. The resulting aqueous solutions of multivalent constructs were subjected to lyophilization, producing 16a–e as white powders in good to very good yields. These mixtures were characterized by MALDI-TOF and by UV analysis (see Supporting Information).
Binding Studies
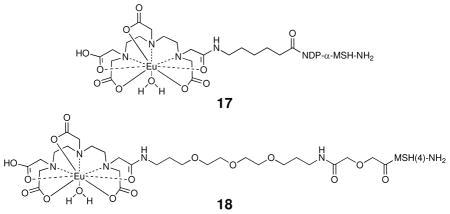
Hek293 cells overexpressing hMC4R were used to assess ligand binding22 using two previously described europium-based competitive binding assays.11,23 Assay A23a employed Eu-DTPA-NDP-α-MSH-NH2 (17) as the labeled probe, and Assay B11 employed Eu-DTPA-PEGO-MSH(4)-NH2 (18) as the labeled probe. The Ki values for the serine amide-containing control compounds 11 and 15, for the parental ligand MSH(4), for the triazole-containing MSH(4) analog 14, and for MSH(4)-containing multivalent constructs 16a–e are given in Table 3.
Table 3.
Assays of competitive binding of MSH(4), 11, 14, 15, and 16a–e to hMC4R.
| Assay Aa | Assay Bb | |||
|---|---|---|---|---|
| Compound | Kic | n | Kic | n |
| 11 | nbd | 3 | nbe | 3 |
| 15 | nbd | 3 | nbe | 3 |
| MSH(4) | 1.9 ± 0.55 μM | 5 | 0.76 ± 0.04 μM | 4 |
| 14 | 2.7 ± 0.21 μM | 5 | 0.82 ± 0.26 μM | 3 |
| 16a | ndf | 1.7 ± 0.39 μM | 4 | |
| 16b | ndf | 1.2 ± 0.38 μM | 6 | |
| 16c | ndf | 0.84 ± 0.10 μM | 4 | |
| 16d | ndf | 0.52 ± 0.02 μM | 6 | |
| 16e | 2.2 ± 0.17 μM | 4 | 0.22 ± 0.06 μM | 4 |
This assay employed probe 17.
This assay employed probe 18.
Ki values were calculated using the equation Ki = EC50/(1 + ([ligand]/KD)) where [ligand] refers to the concentration of probe used as the labeled competed ligand. For probe 17, [ligand] = 10 nM and KD = 18.8 nM. For probe 18, [ligand] = 0.5 μM and KD = 9.1 μM. The value given represents the average of n independent competition binding experiments, each done in quadruplicate.
This compound was unable to prevent probe 17 from binding in the concentration range tested (10−5–10−12 M in serine amide).
This compound was unable to prevent probe 18 from binding in the concentration range tested (10−5–10−12 M in serine amide).
Not determined.
Discussion
The four-step synthesis of the mixture of polyols 7 starting from solanesol was efficient and highly regioselective. The water-soluble product 7, which is available in gram quantities, was statistically alkylated using a 12-fold molar excess of 1-bromo-5-hexyne to produce a mixture of polyol/polyalkynes 9 that bore an average of six alkyne residues per scaffold. Presumably, the identity of the alkylating agent and the extent of alkylation might be varied. Microwave-driven CuAAC was used to attach copies of serine amide and MSH(4) derivatives bearing N-terminal azide groups. In principle, this method might be used to attach other ligands, imaging agents, and/or therapeutic agents to scaffold 9. The constructs so produced were purified from copper and from small molecules, characterized by MALDI-TOF and by UV spectroscopy, and subjected to testing using two previously described competitive binding assays.
In Assay A,23a serine amide derivative 11 and the mixture of serine amide multivalent constructs 15 were both ineffective at blocking probe 17 from binding to hMC4R over the range of concentrations tested. The Ki for the monovalent control compound 14 was 1.4 times the value for the parental ligand, indicating that attachment of the triazole-containing “spacer” to the N-terminus of MSH(4) has a modest detrimental effect on ligand binding to hMC4R. In keeping with prior results obtained with the squalene-derived monovalent and divalent MSH(4) constructs 1 and 2,11 multimer 16e was shown to be nearly as potent as MSH(4), but not more so, in Assay A. This result suggests monovalent binding of 16e at available hMC4R. Monovalent binding of 16e would be expected whenever the off-rate of monovalently bound 16e is faster than the binding of a second “anchoring” ligand arm to hMC4R on the cell surface. The NDP-α-MSH ligand,9 upon which 17 is based,23a is a much stronger binder than MSH(4) and is known to have a slow off-rate (~8 h).24 Thus, monopolization of neighboring receptors by the superpotent probe 17 would disfavor the binding of a second ligand arm of 16e. Depletion of receptors at the cell surface by cycling may also have contributed, since internalization results in fewer neighboring surface receptors to which a second ligand arm of 16e might bind.
Assay B11 was developed to better balance the off-rates of competed and competing ligands, probe 18 and 16a–16e, respectively. In this assay, serine amide derivative 11 and serine amide multivalent constructs 15 were ineffective at blocking probe 18 from binding to hMC4R over the range of concentrations tested. The Ki for compound 14 was 1.1 times higher than the value for the parental ligand, indicating that attachment of the triazole-containing “spacer” to the N-terminus of MSH(4) had a very modest detrimental effect on ligand binding to hMC4R. The Ki values measured in Assay B differed among the various solanesol-derived constructs 16a–16e (Table 3). Construct 16a contained, on average, one copy of MSH(4) and five unreacted alkyne groups per scaffold and was 2.2 times less potent than the parental ligand, MSH(4). Construct 16b contained, on average, two copies of MSH(4) and four unreacted alkyne groups per scaffold and was 1.6 times less potent than MSH(4). Construct 16c contained, on average, one copy of MSH(4) and five serinamide residues per scaffold and was 1.1 times less potent than MSH(4). Construct 16d contained, on average, two copies of MSH(4) and four serinamide residues per scaffold and was 1.5 times more potent than MSH(4). Construct 16e contained, on average, four copies of MSH(4) and two serinamide residues per scaffold and was 3.4 times more potent than MSH(4). As demonstrated by comparison of the Ki values for 16a and 16b with those for 16c and 16d, conversion of the remaining hydrophobic terminal alkyne spacers to the longer, bulkier, but more hydrophilic triazole-serinamide sidechains enhances binding. That this enhancement is not due to specific binding by the serinamide residues is supported by the inactivity of compounds 11 and 15 in Assay B. Enhanced binding for solanesol-derived constructs with higher levels of MSH(4) incorporation was observed, but the small magnitude of the changes in the Ki values was unexpected and inconsistent with prior studies with multivalent MSH(4) constructs.25 The observed binding enhancements for the series 16c–16e can be entirely attributed to statistics and also suggest monovalent binding of 16a–e at available hMC4R. It may be that mixtures 16a–e are competent binders as monovalent species, but 16b and 16d–e are incompetent binders as multivalent species due to improper ligand spacing or presentation.26 Incompetence could also be due to release of the monovalently-bound multivalent construct from the cell surface before binding of a second ligand arm can occur (vide supra). At present, the on rates and off-rates of MSH(4) and related constructs, such as 18 and 16a–e, are unknown. As before, depletion of receptors at the cell surface by cycling should reduce the number of neighboring receptors to which a second ligand arm might bind and limit opportunities for multivalent binding. In support of this point, internalization of probes 17 and 18 contributes significantly to the fluorescence measured in these assays, which may more properly be termed “binding and uptake” assays. In a preliminary study with probe 17 that compared measured fluorescence at 37 °C and 4 °C, as much as 90% of the fluorescence at 37 °C was attributable to internalized probe.27 Work is presently underway to delineate the details of binding and uptake in the further characterization of the interactions of solanesol-derived multivalent constructs with hMC4R.
Experimental Procedures28
Chemical Synthesis (Scheme 1)
Dimethyl 2,2-Bis-[(2E,6E,10E,14E,18E,22E,26E,30E)-3,7,11,15,19,23,27,31,35-nonamethylhexatriaconta-2,6,10,14,18,22,26,30,34-nonaenyl]malonate (5)
To a suspension of NaH (60 mg, 2.5 mmol) in dry THF (10 mL) at 0 °C was added dimethyl malonate (80 μL, 92 mg, 0.7 mmol). The mixture was allowed to warm to RT and was stirred for 3 h. To the mixture was added a solution of solanesyl bromide12 (4, 1.00 g, 1.45 mmol) in dry THF (10 mL) dropwise over 20 min. After 1 h, additional NaH (60 mg, 2.5 mmol) and 4 (200 mg, 0.28 mmol) were added. After 5 h, the reaction was quenched with saturated aqueous NH4Cl solution and the mixture was extracted with ether (200 mL). The organic extract was washed with water, brine, dried over anhydrous MgSO4, and filtered. Approximately 3 g of NaHCO3 was added to the filtrate to neutralize any HBr formed during evaporation of volatiles using a rotary evaporator. Column chromatography on silica gel 60 eluted with 2% ether/hexanes gave 684 mg (0.50 mmol, 72%) of 5 (Rf 0.45, 5% EtOAc/hexanes) as a viscous pale yellow oil; 1H NMR (500 MHz, CDCl3) δ 1.58 (s, 54H), 1.66 (s, 6H), 1.96–2.09 (m, 64H), 2.59 (d, J = 7.2 Hz, 2H), 3.67 (s, 6H), 4.96 (t, J = 7.2 Hz, 2H), 5.10 (t, J = 6.6 Hz, 16H); 13C NMR (125 MHz, CDCl3) δ 16.0, 17.6, 25.7, 26.7, 30.8, 39.7, 52.2, 57.8, 117.7, 123.9, 124.2, 131.1, 134.8, 135.2, 139.1, 171.8; HRMS calculated for C95H152O4Na (M+Na)+ 1380.1588, observed 1380.1505.
2,2-Bis-[(2E,6E,10E,14E,18E,22E,26E,30E)-3,7,11,15,19,23,27,31,35-nonamethylhexatriaconta-2,6,10,14,18,22,26,30,34-nonaenyl]propane-1,3-diol (6)
To a suspension of LiAlH4 (12 mg, 0.29 mmol) in dry ether (6 mL) was added diester 5 (200 mg, 0.147 mmol) dropwise at room temperature. The suspension was stirred at RT for 8 h. After completion of the reaction (TLC), the reaction mixture was quenched with saturated aqueous Na2SO4 solution and filtered. The filtrate was extracted with diethyl ether and the combined organic extracts were washed with saturated NaHCO3 (15 mL) and brine (15 mL). The ether layer was dried over MgSO4, filtered, and concentrated. Column chromatography on silica gel 60 eluted with 30% ether/hexanes gave 157 mg (0.12 mmol, 82%) of 6 (Rf 0.60, 10% EtOAc/hexanes) as a colorless, viscous oil; 1H NMR (500 MHz, CDCl3) δ 1.59 (s, 54H), 1.68 (s, 6H), 1.97–2.07 (m, 68H), 3.56 (s, 4H), 5.10 (t, J = 6 Hz, 18H); 13C NMR (125 MHz, CDCl3) δ 15.9, 17.6, 25.6, 26.6, 29.9, 40.1, 43.4, 68.7, 74.7, 119.4, 124.2, 131.2, 134.8, 137.8. Elemental Analysis. Calculated for C93H152O2 C 85.78, H 11.77; Found C 85.70; H 11.87.
37,37-Bis(hydroxymethyl)-2,6,10,14,18,22,26,30,34,40,44,48,52,56,60,64,68,72-octadecamethyltriheptacontan-3,7,11,15,19,23,27,31,35,39,43,47,51,55,59,63,67,71-octadecaol (7)
[N.B. All reagents and solutions were deoxygenated with argon before use.] In a three-necked flask (500 mL) a solution of borane in THF (1M, 84 mL, 84 mmol) was deoxygenated with argon gas. The flask was immersed in an ice bath, and a solution of 2-methyl-2-butene in THF (2M, 63 mL, 126 mmol), also deoxygenated with argon gas, was added to the borane solution dropwise with stirring at 0 °C. The resulting mixture was maintained at 0 °C for 3 h prior to use. Diol 6 (1.80 g, 1.38 mmol) in THF was then added dropwise at 0 °C. The mixture was stirred at 0 °C for 5 h, kept at 4 °C for 3 days, and then 3N NaOH (26 mL) and 30% H2O2 (26 mL) were added. After 36 h at rt, the reaction mixture was diluted with water and extracted with ether. The ether extracts were combined, and the volatile materials, including most of the byproduct 3-methyl-2-butanol, were removed under reduced pressure. The crude product was then washed twice with hexane (20 mL). Alternatively, after dilution with water, the reaction mixture and the aqueous layer were placed in a liquid-liquid extraction apparatus, the aqueous phase saturated with NaCl, and the mixture continuously extracted with ether. After 72 h the ether extracts were separated, volatile components removed under reduced pressure, and the residue washed with hexane. Yield: 1.58 g (0.97 mmol, 70%). The product was characterized by 13C NMR and by mass spectral analysis (see Figure 1). A 10 mM solution of 7 in methanol was titrated with a 10 mM solution of bromine in methanol. It was found that 3.8% of the double bonds remained in the sample of polyol 7 produced in the experiment described above as determined by the amount of bromine decolorized by 7.29
Synthesis of Polyol/Polyalkynes 9
To a suspension of NaH (60% in oil, 183 mg, 4.59 mmol) in dry DMF (10 mL) at room temperature was added polyol 7 (250 mg, 0.153 mmol). After stirring for 20 min, a solution of 1-bromo-5-hexyne15 (8, 298 mg, 1.84 mmol) in dry DMF (2 mL) was added. The mixture was stirred under argon for 3 days, then quenched with aqueous NH4Cl (20 mL) and extracted with EtOAc (3 × 30 mL). The EtOAc extracts were washed with water (3 × 20 mL) and brine (3 × 15 mL). Volatiles were evaporated and the residue washed with hexanes (2 × 10 mL) to afford a mixture of polyol/polyalkynes 9 (218 mg). Based on mass spectral analysis (see Figure 2) it was found that an average of six alkylations per molecule had occurred. Based on this average, the calculated yield is 69%. This reaction was repeated twice under similar conditions with similar results.
N-(1-Amino-3-hydroxy-1-oxopropan-2-yl)-6-azidohexanamide (10)
To a solution of serine amide hydrochloride16 (1.10 gm, 7.9 mmol) in DMF (20 mL) at room temperature was added triethylamine (0.80 g, 7.9 mmol). To the resulting white suspension was added 2,5-dioxopyrrolidin-1-yl 6-azidohexanoate17 (1.88 g, 7.1 mmol) and the reaction mixture was stirred at room temperature. After completion of the reaction (disappearance of 2,5-dioxopyrrolidin-1-yl 6-azidohexanoate as monitored by TLC), volatiles were evaporated under reduced pressure. Column chromatography on silica gel 60 (50 g) eluted with 10% EtOAc/methanol gave 1.46 g (6.0 mmol, 84%) of 10 (Rf 0.5, 15% methanol/CHCl3) as a white solid, mp 94–96 °C. 1H NMR (500 MHz, CDCl3, CD3OD) δ 4.36 (t, J = 5 Hz, 1H), 3.79 (dd, 1H), 3.58 (dd, 1H), 3.19 (t, J = 7 Hz, 2H), 2.19 (t, J = 7 Hz, 2H), 1.56 (m, 4H), 1.32 (m, 2H); 13C NMR (125 MHz, CDCl3, CD3OD) δ 24.8, 26.1, 28.4, 35.7, 51.0, 54.2, 62.2, 173.9, 174.0; HRMS (ESI) calculated for C9H17N5O3 (MH)+ 244.1411, observed 244.1412.
N-(1-Amino-3-hydroxy-1-oxopropan-2-yl)-6-(4-butyl-1H-1,2,3-triazol-1-yl)hexanamide (11)
To a solution of azide 10 (100 mg, 0.41 mmole) in methanol (5 mL) were added 1-hexyne (337 mg, 4.11 mmole), TBTA (42 mg, 0.08 mmol), and tetrakis(acetonitrile)copper(I) hexafluorophosphate (30 mg, 0.08 mmol). The reaction mixture was stirred for 15 h at room temperature. Volatiles were removed under reduced pressure and residue chromatographed on silica gel 60 (10 g) eluted with 10% methanol/CHCl3 to afford 11 (110 mg, 0.34 mmol, 83%) as a solid, mp 124–126 °C. 1H NMR (500 MHz, CD3OD) δ 0.94 (t, J = 7.5 Hz, 3H), 1.35 (m, 4H), 1.65 (m, 4H), 1.90 (m, 2H), 2.28 (t, J = 7.5 Hz, 2H), 2.67 (t, J = 7.5 Hz, 2H), 3.76 (m, 2H), 4.35 (t, J = 7 Hz, 2H), 4.41 (t, J = 5 Hz, 1H), 7.73(s, 1H); 13C NMR (125 MHz, CD3OD) δ 14.1, 23.2, 26.0, 27.0, 30.9, 32.7, 36.5, 51.0, 56.5, 63.1, 123.1, 175.0, 175.8; HRMS (ESI) calculated for C15H28N5O3 (MH)+ 326.2187, observed 326.2189.
6-(4-Butyl-1H-1,2,3-triazol-1-yl)hexanoic acid (12)
1-Hexyne (5.00 g, 63 mmol), CuSO4 (1.5 g, 6.3 mmol), and sodium ascorbate (2.5 g, 12.6 mmol) were added to a round bottom flask containing a 1:1 mixture of t-BuOH and water (20 mL). 6-Azidohexanoic acid17 (1.00 gm, 6.32 mmol) was added to the solution with stirring. The flask was purged with argon and sealed with a glass stopper to avoid evaporation of 1-hexyne. The reaction mixture was stirred overnight, and then additional 1-hexyne (1.03 g, 12.6 mmol) was added. After 24 h the reaction mixture was diluted with EtOAc (100 mL), washed with 1N HCl (2 × 20 mL) and brine (2 × 20 mL), the organic phase separated, dried over Na2SO4, filtered, and concentrated in vacuo. Column chromatography on silica gel 60 (40 g) eluted with 2% methanol/CHCl3 gave the product 12 (910 mg, 3.79 mmol, 60%) as a white solid, mp 41–43 °C. 1H NMR (500 MHz, CDCl3) δ 0.87 (t, J = 6.9 Hz, 3H), 1.33 (m, 4H), 1.58 (m, 4H), 1.84 (m, 2H), 2.28 (t, J = 7.2 Hz, 2H) 2.66 (t, J = 7.5 Hz, 2H), 4.27 (t, J = 7.2 Hz, 2H), 7.24 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 13.7, 22.2, 23.9, 25.0, 25.8, 29.9, 31.4, 33.7, 49.9, 120.6, 148.2, 178.0; HRMS (ESI) calculated for C12H22N3O2 (MH)+ 240.1707, observed 240.1706.
Solid Phase Synthesis (Scheme 2)
In a syringe (polypropylene reaction tube equipped with a polypropylene frit) Rink amide resin (1 gm, 0.68 mmol) was allowed to swell in THF for 1 hr. THF was removed and addition of 20% piperadine in DMF (15 mL) for 2 min led to the deprotection of the Fmoc functionality. DMF was removed, 20% piperadine solution in DMF (15 mL) was again added and the mixture shaken for 18 min. DMF was removed and the resin washed with DMF (3 × 15 mL), CH2Cl2 (3 × 15 mL), DMF (3 × 15 mL), 0.5 M HOBt in DMF (1 × 15 mL), 0.5 M HOBt in DMF (1×15 mL) plus a drop of bromophenol blue, DMF (2 × 15 mL), and CH2Cl2 (1 × 15 mL), in that order. A solution of the next Fmoc-amino acid (1.05 gm, 2.04 mmol), Cl-HOBt (345 mg, 2.04 mmol), and DIC (512 mg, 4.08 mmol) in DMF (15 mL) was allowed to react for 2 min, then added to the resin and the mixture shaken for 1hr. The resin was then washed with DMF (3 × 15 mL), CH2Cl2 (3 × 15 mL), and DMF (3 × 15 mL). Free NH2 groups were then capped by addition of a 1:1 mixture of acetic anhydride and pyridine (6 mL). After the mixture was shaken for 20 min, the resin was washed with DMF (3 × 15 mL), CH2Cl2 (3 × 15 mL), and DMF (3 × 15 mL). The absence of free amine groups was confirmed by the Kaiser test. The same cycle of procedures was repeated for coupling of the other amino acids in the sequence, and finally for attachment of the N-terminal 6-azidohexanoic acid residue or the N-terminal 6-(4-butyl-1H-1,2,3-triazol-1-yl)hexanoic acid residue, thus producing the resin-bound tetrapeptide derivatives related to compounds 13 and 14, respectively. Cleavage and deprotection were achieved using a 91:3:3:3 mixture of trifluoroacetic acid, triisopropylsilane, thioanisole, and water (10 mL). The mixture of cleavage cocktail and resin was shaken for overnight, the solution was separated from the resin, volatiles were evaporated, the residue triturated with ether, and the crude product separated by centrifugation. Purification of the tetrapeptide amides 13 and 14 was accomplished by reverse phase chromatography using a 19 × 256 mm X-Bridge Preparative C18 column. The mobile phase used was 10–90% acetonitrile and water containing 0.1% TFA within 50 min; the flow rate was 15 mL/min and the UV detector system operated at 230 nm. The purity of compounds 13 and 14 was checked by reverse phase HPLC using a 4.6 × 75 mm Symmetry Analytical C18 column. The mobile phase was 10–90% acetonitrile and water containing 0.1% TFA within 50 min; the flow rate was 1 mL/min and the UV detector system operated at 230 nm. Compounds 13 and 14 were characterized by MALDI-TOF mass spectrometry. Both the reflectron and linear techniques were used for positive ion detection. The matrix, sinapic acid, and the analyte were dissolved in water:acetonitrile 1:1 containing 0.1% of formic acid and the solutions mixed in a ratio of 100:1. ESI was also used to ionize some of the samples. The samples were dissolved in methanol:water (1:1) at a concentration of ca 50 μM. Standard ESI conditions were applied to detect positively charged ions.
Multimer Assembly (Scheme 3)
Serine Amide Multivalent Constructs 15
A mixture of polyol/polyalkynes 9 (10 mg, 5 mmol), azide 10 (10 mg, 50 mmol), TBTA (3 mg, 5 mmol), and tetrakis(acetonitrile)copper(I) hexafluorophosphate (2 mg, 5 mmol) in dry methanol (1 mL) was irradiated for 4 hr in a Biotage microwave reactor (100 °C). Additional azide 10 (5 mg, 25 mmol) was then added to the reaction mixture and irradiation was resumed for another 4 h. After the reaction was complete, water (20 mL) was added and the mixture was extracted with CHCl3 containing dithizone (20 mg/L, 3 × 30 mL) to remove copper.21 The water layer was then washed with CHCl3 (2 × 20 mL) to remove any remaining 10 and TBTA. After lyophilization, 16 mg (88% yield) of 15 was obtained as a white powder. Product 15 was analyzed by MALDI-TOF (see Figure S12 in the Supporting Information).
General Procedure for Reaction of Azide-functionalized MSH(4) Derivative 13 and Azide-functionalized Serine Amide Derivative 10 with Polyol/Polyalkynes 9 to Produce Compounds 16a–e
Mixtures of polyol/polyalkynes 9 (10 or 20 mg, 5 or 10 mmol), MSH(4) azide 13 (variable amount, see Table 2), TBTA (3 mg, 5 mmol), and tetrakis(acetonitrile)copper(I) hexafluorophosphate (2 mg, 5 mmol) in dry methanol (1 mL/5 mmol of 9) were irradiated for 6 hr in a Biotage microwave reactor (100 °C). Azide 10 (variable amount, see Table 2) was then added to the reaction mixtures and irradiation was resumed for another 4 h. After the reactions were complete, water (20 mL/5 mmol of 9) was added and the mixtures were extracted with CHCl3 containing dithizone (20 mg/L, 3 × 30 mL) to remove copper.21 The water layers were then washed with CHCl3 (2 × 20 mL) to remove any remaining 10 and TBTA. After lyophilization, white powders were obtained (yields are given in Table 2). Products were analyzed by MALDI-TOF and by UV spectroscopy (see Figures S13–17 in the Supporting Information).
Formulation of Solutions
Solutions of multivalent constructs for binding assays were prepared in DMSO (HYBRI-MAX) based on the average incorporation of MSH(4) and/or serinamide as determined by mass spectrometric analysis. Concentrations of MSH(4) ligand in solution were confirmed by measurement of the UV absorbance at 280 nm using a standard calibration plot (see Supporting Information). In all cases the concentrations calculated from the mass spectral analysis and the weight of the compound used agreed within 5% with the measured UV absorption.
Binding Assays
Quantitative receptor-binding assays were carried out following a previously described method.11 Hek293 cells overexpressing hMC4R were used to assess ligand binding. Cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS. Cells were seeded in a 96-well plate at a density of 15,000 cells per well and were allowed to reach 80–90% confluence. On the day of the experiment, media was aspirated from all wells, and 50 μL of the compounds to be tested (dilutions ranging from 2 × 10−4 to 1 × 10−11 M) and 50 μL of Eu-labeled ligand (Assay A, 10 nM probe 17, Assay B, 0.5 μM probe 18) were added to each well. Ligands were diluted in binding media (DMEM, 1 mM 1,10-phenanthroline, 200 mg/L bacitracin, 0.5 mg/L leupeptin, 0.3% BSA) and each concentration was tested in quadruplicate. Cells were incubated in the presence of unlabeled and labeled ligands at 37 °C and 5% CO2 for 1 hour. The cells were washed with wash buffer (50 mM Tris-HCl, 0.2% BSA, 30 mM NaCl), enhancement solution (PerkinElmer 1244-105) was added (100 μl/well), and fluorescence was measured on a Wallac VICTOR3 instrument using standard Eu TRF measurement conditions (340 nm excitation, 400 μs delay, and emission collection for 400 μs at 615 nm). Competitive binding data were analyzed with GraphPad Prism software using nonlinear regression analysis and fitted to a classic one site binding competition equation. Each EC50 value was generated from individual competitive binding assays and converted to a Ki value using the equation Ki = EC50/(1 + ([ligand]/KD)) where [ligand] refers to the concentration of the probe used as the labeled competed ligand. For probe 17, [ligand] = 10 nM and KD = 18.8 nM. For probe 18, [ligand] = 0.5 μM and KD = 9.1 μM. Results are given in Table 3. The value given represents the average of n independent competition binding experiments, with the error bars indicating standard error of the mean.
Supplementary Material
Acknowledgments
This work was supported by grants R33 CA 95944, RO1 CA 97360, RO1 CA 123547, and P30 CA 23074 from the National Cancer Institute.
Footnotes
Supporting Information Available: A general experimental section, a list of nonstandard abbreviations, copies of the 1H and 13C NMR spectra of compounds 5, 6, 10, 11, and 12, the APT of 7, MALDI-TOF spectra of multivalent construct mixtures 15 and 16a–e, and a description of UV studies. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Footnotes
- 1.(a) Weissleder R, Mahmood U. Radiology. 2001;219:316–333. doi: 10.1148/radiology.219.2.r01ma19316. [DOI] [PubMed] [Google Scholar]; (b) Gillies RJ, Hruby VJ. Expert Opin Ther Targets. 2003;7:137–139. doi: 10.1517/14728222.7.2.137. [DOI] [PubMed] [Google Scholar]; (c) Gillies RJ, Hoffman JM, Lam KS, Menkens AE, Piwnica-Worms DR, Sullivan DC, Weissleder R. Molecular Imaging. 2005;4:98–103. doi: 10.1162/15353500200505115. [DOI] [PubMed] [Google Scholar]; (d) Cassidy PJ, Radda GK. J R Soc Interface. 2005;2:133–144. doi: 10.1098/rsif.2005.0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Handl HL, Vagner J, Han H, Mash E, Hruby VJ, Gillies RJ. Expert Opin Ther Targets. 2004;8:565–586. doi: 10.1517/14728222.8.6.565. [DOI] [PubMed] [Google Scholar]; (b) Vagner J, Handl HL, Monguchi Y, Jana U, Begay LJ, Mash EA, Hruby VJ, Gillies RJ. Bioconjugate Chem. 2006;17:1545–1550. doi: 10.1021/bc060154p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bowen ME, Monguchi Y, Sankaranarayanan R, Vagner J, Begay LJ, Xu L, Jagadish B, Hruby VJ, Gillies RJ, Mash EA. J Org Chem. 2007;72:1675–1680. 3608. doi: 10.1021/jo062276g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jagadish B, Sankaranarayanan R, Xu L, Richards R, Vagner J, Hruby VJ, Gillies RJ, Mash EA. Bioorg Med Chem Lett. 2007;17:3310–3313. doi: 10.1016/j.bmcl.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Handl HL, Sankaranarayanan R, Josan JS, Vagner J, Mash EA, Gillies RJ, Hruby VJ. Bioconjugate Chem. 2007;18:1101–1109. doi: 10.1021/bc0603642. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Vagner J, Xu L, Handl HL, Josan JS, Morse DL, Mash EA, Gillies RJ, Hruby VJ. Angew Chem Int Ed. 2008;47:1685–1688. doi: 10.1002/anie.200702770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Mammen M, Choi SK, Whitesides GM. Angew Chem Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; (b) Kiessling LL, Gestwicki JE, Strong LE. Angew Chem Int Ed. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Carlson CB, Mowery P, Owen RM, Dykhuizen EC, Kiessling LL. ACS Chemical Biology. 2007;2:119–127. doi: 10.1021/cb6003788. [DOI] [PubMed] [Google Scholar]; (d) Trouche N, Wieckowski S, Sun W, Chaloin O, Hoebeke J, Fournel S, Guichard G. J Am Chem Soc. 2007;129:13480–13492. doi: 10.1021/ja073169m. [DOI] [PubMed] [Google Scholar]; (e) Diestler DJ, Knapp EW. Phys Rev Lett. 2008;100:178101/1–178101/4. doi: 10.1103/PhysRevLett.100.178101. [DOI] [PubMed] [Google Scholar]; (f) Choi S-K. Synthetic Multivalent Molecules. Wiley-Interscience; Hoboken, NJ: 2004. [Google Scholar]
- 4.(a) Schwyzer R, Kriwaczek VM. Biopolymers. 1981;20:2011–2020. doi: 10.1002/bip.1981.360200923. [DOI] [PubMed] [Google Scholar]; (b) Wunderlin R, Sharma SD, Minakakis P, Schwyzer R. Helv Chim Acta. 1985;68:12–22. [Google Scholar]; (c) Sharma SD, Granberry ME, Jiang J, Leong SPL, Hadley ME, Hruby VJ. Bioconjugate Chem. 1994;5:591–601. doi: 10.1021/bc00030a015. [DOI] [PubMed] [Google Scholar]; (d) Sharma SD, Jiang J, Hadley ME, Bentley DL, Hruby VJ. Proc Natl Acad Sci USA. 1996;93:13715–13720. doi: 10.1073/pnas.93.24.13715. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Carrithers MD, Lerner MR. Chem & Biol. 1996;3:537–542. doi: 10.1016/s1074-5521(96)90144-1. [DOI] [PubMed] [Google Scholar]
- 5.(a) Cairo CW, Gestwicki JE, Kanai M, Kiessling LL. J Am Chem Soc. 2002;124:1615–1619. doi: 10.1021/ja016727k. [DOI] [PubMed] [Google Scholar]; (b) Griffith BR, Allen BL, Rapraeger AC, Kiessling LL. J Am Chem Soc. 2004;126:1608–1609. doi: 10.1021/ja037646m. [DOI] [PubMed] [Google Scholar]; (c) Line BR, Mitra A, Nan A, Ghandehari H. J Nucl Med. 2005;46:1552–1560. [PubMed] [Google Scholar]; (d) Li RC, Broyer RM, Maynard HD. J Polym Sci, Part A: Polym Chem. 2006;44:5004–5013. [Google Scholar]; (e) Cilliers R, Song Y, Kohlmeir EK, Larson AC, Omary RA, Meade TJ. Mag Res Med. 2008;59:898–902. doi: 10.1002/mrm.21518. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; (b) Gil MV, Arévalo MJ, López Ó. Synthesis. 2007:1589–1620. [Google Scholar]; (c) Angell YL, Burgess K. Chem Soc Rev. 2007;36:1674–1689. doi: 10.1039/b701444a. [DOI] [PubMed] [Google Scholar]; (d) Holub JM, Garabedian MJ, Kirshenbaum K. QSAR Comb Sci. 2007;26:1175–1180. [Google Scholar]
- 7.Moore WRAD, O’Dowd M. In: Properties and Applications of Polyvinyl Alcohol. Finch CA, editor. Chapter 4. Staples Printers Ltd; Kent, England: 1968. pp. 77–87. [Google Scholar]
- 8.(a) Levesque G, Pinazzi C. Bull Soc Chim Fr. 1971:1008–1010. [Google Scholar]; (b) Yamaguchi H, Azuma K, Minoura Y. Polym J. 1972;3:12–20. [Google Scholar]; (c) Pinazzi CP, Guillaume P, Reyx D. Eur Polym J. 1976;12:9–15. [Google Scholar]; (d) Perera MCS, Elix JA, Bradbury JH. J Appl Polym Sci. 1987;33:2731–2742. [Google Scholar]
- 9.The “high-affinity” ligand, NDP-α-MSH, is Ac-Ser-Tyr-Ser-Nle-Glu-His-DPhe-Arg-Trp-Gly-Lys-Pro-Val-NH2; see Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. Proc Natl Acad Sci USA. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754.Hadley ME, Anderson B, Heward CB, Sawyer TK, Hruby VJ. Science. 1981;213:1025–1027. doi: 10.1126/science.6973820.
- 10.The “low-affinity” ligand MSH(4), Ac-His-DPhe-Arg-Trp-NH2, was based on the minimal active sequence for full agonist activity of α-MSH; see Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, de Vaux AE, Dym O, de L, Castrucci AM, Hintz MF, Riehm JP, Rao KR. J Med Chem. 1987;30:2126–2130. doi: 10.1021/jm00394a033.Castrucci AML, Hadley ME, Sawyer TK, Wilkes BC, Al-Obiedi F, Staples DJ, de Vaux AE, Dym O, Hintz MF, Riehm JP, Rao KR, Hruby VJ. Gen Comp Endocrinol. 1989;73:157–163. doi: 10.1016/0016-6480(89)90066-x.Haskell-Luevano C, Hendrata S, North C, Sawyer TK, Hadley ME, Hruby VJ, Dickinson C, Gantz I. J Med Chem. 1997;40:2133–2139. doi: 10.1021/jm960840h.
- 11.Xu L, Vagner J, Alleti R, Rao V, Jagadish B, Morse DL, Hruby VJ, Gillies RJ, Mash EA. Bioorg Med Chem Lett. 2010;20:2489–2492. doi: 10.1016/j.bmcl.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamamura K, Yamatsu I, Minami N, Yamagishi Y, Inai Y, Kijima S, Nakamura T. J Label Compd Radiopharm. 2002;45:823–829. Solanesol (3) and solanesyl bromide (4) are commercially available from NetChem. [Google Scholar]
- 13.This presumably was due to temporary intermolecular and intramolecular crosslinking during hydroboration, leading to sterically inaccessible, unreactive double bonds. Similar behavior was noted in previous work on the hydroboration of polyisoprene; see Ref. 8a–d.
- 14.Use of triisopinocampheyldiborane as described in reference 8b gave mixtures of polyols and isopinocampheol that were difficult to separate. The byproduct from use of disiamylborane, 3-methyl-2-butanol, was more easily removed from the polyol product under vacuum.
- 15.Sharma S, Oehlschlager AC. J Org Chem. 1989;54:5064–5073. [Google Scholar]
- 16.Hidy PH, Hodge EB, Young VV, Harned RL, Brewer GA, Phillips WF, Runge WF, Stavely HE, Pohland A, Boaz H, Sullivan HR. J Am Chem Soc. 1955;77:2345–2346. Serine amide is commercially available from Senn Chemicals USA. [Google Scholar]
- 17.Grandjean C, Boutonnier A, Guerreiro C, Fournier JM, Mulard LA. J Org Chem. 2005;70:7123–7132. doi: 10.1021/jo0505472. [DOI] [PubMed] [Google Scholar]
- 18.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 19.(a) Merrifield RB. J Am Chem Soc. 1963;85:2149–2154. [Google Scholar]; (b) Hruby VJ, Meyer J-P. In: Bioorganic Chemistry: Peptides and Proteins. Hecht SM, editor. Oxford Univ. Press; New York: 1998. pp. 27–64. [Google Scholar]
- 20.Kappe CO, Dallinger D, Murphree SS. Practical Microwave Synthesis for Organic Chemists. Wiley-VCH; Weinheim: 2009. [Google Scholar]
- 21.Caplin S. Tissue Culture Association Manual. 1976;2:439–440. Copper ion could not be removed by dialysis, presumably due to the strong chelatory properties of MSH(4) [Google Scholar]
- 22.The hMC4R vector was originally received from Dr. Ira Gantz; see Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. J Biol Chem. 1993;268:15174–15179.
- 23.(a) Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Anal Biochem. 2004;330:242–250. doi: 10.1016/j.ab.2004.04.012. [DOI] [PubMed] [Google Scholar]; (b) De Silva CR, Vagner J, Lynch R, Gillies RJ, Hruby VJ. Anal Biochem. 2010;398:15–23. doi: 10.1016/j.ab.2009.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haskell-Luevano C, Miwa H, Dickinson C, Hadley ME, Hruby VJ, Yamada T, Gantz I. J Med Chem. 1996;39:432–435. doi: 10.1021/jm950407s. [DOI] [PubMed] [Google Scholar]
- 25.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Bioorg Med Chem Lett. 2004;14:211–215. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 26.Shewmake TA, Solis FJ, Gillies RJ, Caplan MR. Biomacromol. 2008;9:3057–3064. doi: 10.1021/bm800529b. [DOI] [PubMed] [Google Scholar]
- 27.Xu, L. Unpublished work.
- 28.The General Experimental section appears in the Supporting Information.
-
29.This method was validated by titration of 0.10 M solutions of citronellal and geraniol in methanol with a 0.10 M solution of bromine in methanol. A negative control experiment using hexol A (see Ref. 2d for the preparation of A) gave a persistent bromine color upon addition of the first drop of bromine solution.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.