

Abstract

Addition of nitrosobenzene to pinacol allylboronates leads to oxidation of the organoboron with concomitant rearrangement of the substrate alkene. This reaction appears to proceed by allylboration of the nitroso group in analogy to carbonyl and imine allylation reactions. Remarkably, the N-O bond is cleaved during the reaction such that simple alcohols are the final reaction product.
Due to the utility of allylboron reagents in organic synthesis, their preparation has been studied intensely.1 Along these lines, recent efforts from our laboratory have focused on the development of the catalytic hydroboration of dienes2 and the catalytic diboration of both allenes3 and dienes.4 These reactions convert simple hydrocarbon building blocks into substituted pinacolato allylboronates. These types of allylmetal reagents participate in a wide range of allylations with carbonyl and imine derivatives.5 However, the only other reactions that have been developed for these species are a narrow range of oxidation,6 cross-coupling,7 conjugate addition,8 and homologation reactions.9 To expand the utility of allyl boronates in organic synthesis we have begun to study other reactions that might apply to these reagents. Considering the isoelectronic relationship between the nitroso group and carbonyl groups, we were prompted to study the reaction between nitrosobenzene and allyl boronates. While a single example by Bubnov describes the reaction between nitrosobenzene and highly reactive triallyborane, a number of critical issues remain unaddressed.10 First, it is not clear whether the diminished electrophilicty of boronic esters relative to boranes will impede the reaction. Second, with substituted allylic boronates, it is not readily apparent whether the reaction with nitrosobenzene will occur with allylic transposition as occurs with carbonyls, or whether it will occur by coordination and subsequent 1,2 alkyl migration as occurs in the reaction between organoboranes and many reagents.11 Lastly, while Bubnov has demonstrated that triallylborane and nitrosobenzene react with relatively non-selective formation of both N-O and C-O bonds, the issue of N-versus O-allylation with allylic boronates was uncertain.12
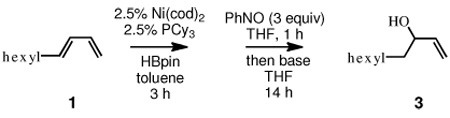
Two preliminary experiments revealed much about the reaction between allylboronates and nitrosobenzene. In the first (eq 1, Scheme 1), 1,3-decadiene (1) was subjected to Ni-catalyzed 1,4-hydroboration, a reaction that delivers cis allyl boronate 2.2a After diluting the reaction mixture with THF it was treated with 1.05 equivalents of nitrosobenzene. Subsequent oxidative treatment furnished allylic alcohols 3 and 4 in 44% and 23% yield, respectively. In the second experiment (eq 2, Scheme 1), intermediate allyl boronate 2 was treated with nitrosobenzene for 13 hours at −78 °C prior to work-up with brine and passage through a short silica gel plug. This reaction did not provide any of the terminal alcohol 4, but did produce internal allylic alcohol 3 and alkoxyamine 5. The observation that allylic alcohol 4 is only produced when oxidative work-up is employed suggests that 4 arises from unreacted 2. The observation that regioisomers 3 and 5, but not 4, are produced in the absence of hydrogen peroxide suggests that the predominant reaction pathway for allylic boronate 2 and nitrosobenzene occurs by addition of the allylboron to the oxygen atom and with allylic rearrangement.13
Scheme 1.

Reaction of Nitrosobenzene with Allylboronate 2
Considering the robustness of the N-O bond, it is somewhat surprising that N-O bond-cleaved compound 3 is produced in equation 2 (Scheme 1). A clue to its formation can be found in the fact that compound 5 is not isolated from equation 1 and that mass spectral analysis of the unpurified reaction mixture from equation 2 revealed the presence of a compound with an m/z ratio corresponding to compound 6 (Scheme 1). 1H NMR analysis also reveals the presence of 6. In line with these observations, it was surmised that a second molecule of nitrosobenzene and the Brønsted base might conspire to cleave the N-O bond of the initial allylation product in a fashion such as that depicted in Scheme 2. This type of reaction has been observed by Barbas and appears consistent with the reaction outcome.14,15
Scheme 2.

Proposed Mechanism for N-O Bond Cleavage
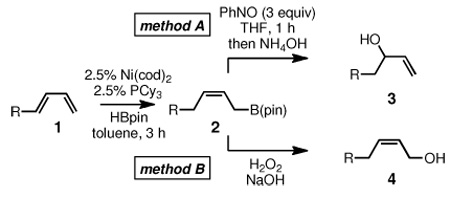
In view of the mechanism depicted in Scheme 2, it might be anticipated that additional nitrosobenzene and alternate bases would provide an improved yield of allylation product 3. As depicted in Table 1 (entry 1), addition of three equivalents of nitrosobenzene, as opposed to one equivalent (Scheme 1), results in a significantly enhanced yield of the secondary allylic alcohol 3 and, with oxidative work-up, none of the terminal allylic alcohol 4 could be detected. According to the mechanism in Scheme 2, hydrogen peroxide is not required for the reaction and the experiment in entry 2 (Table 1) bears this out. In the absence of base, however, the reaction yield is significantly diminished (entry 3). With the requirement for addition of H2O2 apparently obviated, we examined work-up under basic, non-oxidative conditions. As depicted in Table 1, a number of bases suffice for the nitrosobenzene-mediated oxidation of allylboronates. In all cases reasonable yields of secondary allylic alcohol 3 were isolated.
Table 1.
Tandem Diene Hydroboration/Nitrosobenzene Oxidation.
 | ||
|---|---|---|
| Entry | base | % yielda |
| 1 | NaOH/H2O2 | 69 |
| 2 | NaOH | 61 |
| 3 | none | 37 |
| 4 | CsOH | 55 |
| 5 | LiOH | 62 |
| 6 | KOH | 59 |
| 7 | NH4OH | 67 |
Isolated yield of purified product.
To study the generality of the nitrosobenzene-mediated allylboronate oxidation, a number of substrates were explored in the tandem hydroboration/oxidation sequence and both nitrosobenzene/NH4OH (method A) and NaOH/H2O2 (method B) oxidation procedures were examined. As depicted in Table 2, a number of different terminally-substituted butadienes participate in the reaction and deliver moderate yields of the secondary allylic alcohol from method A. While protected oxygen functional groups (entries 3, 5–7) and steric encumbrance (entries 2 and 5) at the diene terminus are tolerated, substitution at the 2 position of the diene leads to substantially diminished reactivity (entry 8). As noted in Table 2, oxidation with H2O2/NaOH consistently delivers the terminal allylic alcohol 4. The fact that method B is efficient with all substrates in Table 2 suggests that the diminished reactivity observed with method A in entry 8 arises due to inefficient reaction of the intermediate allylic boronate with nitrosobenzene. Surprisingly, substitution at the 3 position is tolerated in the nitrosobenzene reaction and the derived tertiary alcohol was isolated in good yield (entry 9).
Table 2.
Substrate Scope for Tandem Diene Hydroboration/Oxidation.
 | |||||
|---|---|---|---|---|---|
| entry | diene |
method A product/yield (%)a |
method B product/yield (%)a |
||
| 1 | 66 | 85 | |||
| 2 | 62 | 81 | |||
| 3 | 57 |  |
56 | ||
| 4 |  |
64 | 91 | ||
| 5 | 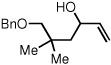 |
44 | 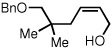 |
89 | |
| 6 | 58 | 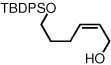 |
95 | ||
| 7 | 63 | 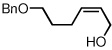 |
93 | ||
| 8 | 33 | 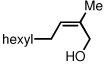 |
93 | ||
| 9 | 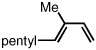 |
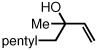 |
58 | 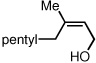 |
81 |
Isolated yield of purified product. Value is an average of two experiments.
With achiral allylboronates such as 2 the nitrosobenzene oxidation products 3 are racemic. However, stereogenicity in the allylic boronate may impact the oxidation if the reacting alkene is prochiral. To study the capacity for diastereoinduction in the nitrosobenzene-mediated oxidation reaction, the chiral 1,4-diboryl-2-alkene 7 (Scheme 3) was generated by Ni-catalyzed diene diboration and subjected to the nitrosobenzene allylation reaction. Whereas reaction with nitrosobenzene at room temperature delivered diol 8 in 2.6:1 diastereoselection (data not shown), when nitrosobenzene was added to diboronate 7 at −78 °C and followed by oxidative work-up, diol 8 was isolated in 10:1 anti:syn stereoselection and in moderate yield (eq 3, Scheme 3). For comparison, the direct oxidation with hydrogen peroxide furnishes regioisomeric 1,4-diol 9 in 85% yield (eq 4).
Scheme 3.

Diastereoinduction in the Oxidation of Chiral Allylboronate 6
The fact that the allylation of nitrosobenzene proceeds with allylic rearrangement suggests that a cyclic transition structure operates. This hypothesis was further supported by subjecting octylB(pin) to nitrosobenzene and NH4OH; less than 5% of 1-octanol could be detected. With these observations in mind, the stereochemical outcome of the reaction depicted in equation 3 may be rationalized by considering transition structure 10 as the predominating reaction pathway. Presumably, the enhanced basicity of nitrogen relative to oxygen results in N-B bonded complex 10 wherein nitrosobenzene has coordinated to the least hindered boronate. The stereogenic carbon atom in transition structure 10 is oriented in a manner where the electron rich C-B bond is aligned with the π-system of the reacting alkene in a manner that should enhance π nucleophilicity. If the small hydrogen is placed inside with respect to the alkene to minimize A[1,3] strain, the model correctly predicts the stereochemical outcome of the reaction. Similar features account for the stereochemistry of hydroboration of allylic silanes and boranes.16
In conclusion, treatment of simple allylboronates with nitrosobenzene and base can be considered a reliable strategy for oxidation with allylic rearrangement. Future studies will expand upon this reactivity pattern and will be reported in due course.
Supplementary Material
Acknowledgment
This work was supported by the NIH (GM 59417). We thank BASF for a generous donation of pinacolborane.
Footnotes
Supporting Information Available. Complete experimental procedures, characterization data (1H and 13C NMR, IR, and mass spectrometry). This material is free of charge via the internet at http://pubs.acs.org.
References
- 1.For a recent review see: Lachance H, Hall DG. In: Organic Reactions. Denmark SE, editor. Vol. 73. New York: Wiley; 2009.
- 2. Ely RJ, Morken JP. J. Am. Chem. Soc. 2010;132:2534. doi: 10.1021/ja910750b. See also: Zaidlewicz M, Meller J. Tetrahedron Lett. 1997;38:7279. Satoh M, Nomoto Y, Miyaura N, Suzuki A. Tetrahedron Lett. 1989;30:3789. Matsumoto Y, Hayashi T. Tetrahedron Lett. 1991;32:3387. Wu JY, Moreau B, Ritter T. J. Am. Chem. Soc. 2009;131:12915. doi: 10.1021/ja9048493.
- 3. Pelz NF, Woodward AR, Burks HE, Sieber JD, Morken JP. J. Am. Chem. Soc. 2004;126:16328. doi: 10.1021/ja044167u. Burks HE, Liu S, Morken JP. J. Am. Chem. Soc. 2007;129:8766. doi: 10.1021/ja070572k. See also: Ishiyama T, Kitano T, Miyaura N. Tetrahedon Lett. 1998;39:2357. Yang FY, Cheng CH. J. Am. Chem. Soc. 2001;123:761. doi: 10.1021/ja005589g.
- 4. Morgan JB, Morken JP. Org. Lett. 2003;5:2573. doi: 10.1021/ol034936z. Burks HE, Kliman LT, Morken JP. J. Am. Chem. Soc. 2009;131:9134. doi: 10.1021/ja809610h. See also: Ishiyama T, Yamamoto M, Miyaura N. Chem. Commun. 1996:2073. Ishiyama T, Yamamoto M, Miyaura N. Chem. Commun. 1997:689. Clegg W, Thorsten J, Marder TB, Norman NC, Orpen AG, Peakman TM, Quayle MJ, Rice CR, Scott AJ. J. Chem. Soc., Dalton Trans. 1998:1431.
- 5.For a review of allylboration of carbonyl compounds see: Hall DG. Pure Appl. Chem. 2008;80:913. See also: Zhang P, Morken JP. J. Am. Chem. Soc. 2009;131:12550. doi: 10.1021/ja9058537. For allylboration of imines see: Sugiura M, Hirano K, Kobayashi S. Org. Synth. 2006;83:170. Sieber JD, Morken JP. J. Am. Chem. Soc. 2006;128:74. doi: 10.1021/ja057020r. Elford TG, Hall DG. Tetrahedron Lett. 2008;49:6995.
- 6.Review of oxidation of organoboron compounds: Brown HC, Snyder C, Rao BCS, Zweifel G. Tetrahedron. 1986;42:5505.
- 7.Selected examples: Nilsson K, Hallberg A. Acta Chem. Scand. B. 1987;41:569. Kalinin VN, Denisov FS, Bubnov YN. Mendeleev Commun. 1996:206. Kotha S, Behera M, Shah VR. Synlett. 2005;12:1877. Yamamoto Y, Takada S, Miyaura N. Chem. Lett. 2006;35:704. Kotha S, Shah VR, Mandal K. Adv. Synth. Catal. 2007;349:1159. Kotha S, Shah VR. Eur. J. Org. Chem. 2008;6:1054. Gerbino DC, Mandolesi SD, Schmalz H, Podestá JC. Eur. J. Org. Chem. 2009;23:3964. Flegeau EF, Schneider U, Kobayashi S. Chem. Eur. J. 2009;15:12247. doi: 10.1002/chem.200902221.
- 8.(a) Sieber JD, Liu S, Morken JP. J. Am. Chem. Soc. 2007;129:2214. doi: 10.1021/ja067878w. [DOI] [PubMed] [Google Scholar]; (b) Sieber JD, Morken JP. J. Am. Chem. Soc. 2008;130:4978. doi: 10.1021/ja710922h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Homologation of allyl boronates: Hoffmann RW, Stiasny HC. Tetrahedron Lett. 1995;36:4595. Matteson DS, Majumdar D. J. Am. Chem. Soc. 1980;102:7588. Matteson DS, Majumdar DJ. Orgmet. Chem. 1980;184:C41. Stymiest JL, Bagutski V, French RM, Aggarwal VK. Nature. 2008;456:778. doi: 10.1038/nature07592. Bagutski V, Ros A, Aggarwal VK. Tetrahedron. 2009;65:9956.
- 10.Bubnov YN, Pershin DG, Karionova AL, Gurskii ME. Mendeleev Commun. 2002;12:202. [Google Scholar]
- 11.(a) Brown HC, Singaram B. Pure Appl. Chem. 1987;59:879. [Google Scholar]; (b) Crudden CM, Edwards D. Eur. J. Org. Chem. 2003;24:4695. [Google Scholar]
- 12.For recent examples that use PhNO as an electrophile see: Momiyama N, Yamamoto H. J. Am. Chem. Soc. 2003;125:6038. doi: 10.1021/ja0298702. Brown SP, Brochu MP, Sinz CJ, MacMillan DWC. J. Am. Chem. Soc. 2003;125:10808. doi: 10.1021/ja037096s. Yamamoto Y, Momiyama N, Yamamoto H. J. Am. Chem. Soc. 2004;126:5962. doi: 10.1021/ja049741g. Momiyama N, Yamamoto H. J. Am. Chem. Soc. 2005;127:1080. doi: 10.1021/ja0444637. Simmons B, Walji AM, MacMillan DWC. Angew. Chem. Int. Ed. 2009;48:4349. doi: 10.1002/anie.200900220.
- 13.The N-allylation product was not detected in these reactions. This observation is in contrast to that of Bubnov (ref. 10) who notes that a 2:1 mixture of O-allylation and N-allylation products is obtained from nitrosobenzene and triallylborane. In the reaction of tricrotylborane, Bubnov also notes allylic rearrangement.
- 14. Ramachary DB, Barbas CF., III Org. Lett. 2005;7:1577. doi: 10.1021/ol050246e. See also: Becker AR, Sternson LA. J. Org. Chem. 1980;45:1708.
- 15.Bubnov also notes N-O bond cleavage but claims a different mechanism that is light-promoted. We find that the reactions of allyl boronates proceed equally well in the dark.
- 16.(a) Fleming I, Lawrence NJ. J. Chem. Soc., Perkin Trans. 1. 1992;24:3309. [Google Scholar]; (b) Fleming I, Lawrence NJ. Tetrahedron Lett. 1988;29:2073. [Google Scholar]; (c) Fleming I, Lawrence NJ. Tetrahedron Lett. 1988;29:2077. [Google Scholar]; (d) Pelz NF, Morken JP. Org. Lett. 2006;8:4557. doi: 10.1021/ol0616891. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.