

SUMMARY
The widespread resistance of malaria parasites to all affordable drugs has made the identification of new targets urgent. Dipeptidyl aminopeptidases (DPAPs) represent potentially valuable new targets that are involved in hemoglobin degradation (DPAP1) and parasite egress (DPAP3). Here we use activity-based probes to demonstrate that specific inhibition of DPAP1 by a small molecule results in the formation of an immature trophozoite that leads to parasite death. Using computational methods we designed stable, non-peptidic covalent inhibitors that kill Plasmodium falciparum at low nanomolar concentrations. These compounds show signs of slowing parasite growth in a murine model of malaria, which suggests that DPAP1 might be a viable anti-malarial target. Interestingly, we found that re-synthesis and activation of DPAP1 after inhibition is rapid, suggesting that effective drugs would need to sustain DPAP1 inhibition for a period of 2–3h.
INTRODUCTION
Malaria remains one of the most devastating infectious diseases, with more than a quarter billion clinical cases and close to a million deaths per year (Aregawi et al., 2008). Yet, the most dramatic aspect of the disease is the widespread resistance of Plasmodium species to all affordable front line drugs. Multi-drug resistant strains are commonly identified in field isolates (Chaijaroenkul et al., 2005; Wilairatana et al., 2002; Wongsrichanalai et al., 2002), and the first signs of resistance to artemisinin-based combination therapy, the current gold standard for treatment, are starting to appear in south East Asia (Dondorp et al., 2009; Noedl et al., 2009; Rogers et al., 2009). It is therefore urgent to develop new strategies to combat malaria and especially to identify new drug targets.
The success of protease inhibitors for the treatment of HIV and hypertension has put this class of enzymes at the forefront of drug development. In a wide range of pathologies such as cancer, diabetes, or hepatitis C, protease inhibitors have reached an advanced stage of clinical development (Fear et al., 2007). The central role of proteases in parasitic diseases (McKerrow et al., 2006; McKerrow et al., 2008) and the wealth of knowledge about protease inhibitors have made these enzymes one of the target families for neglected diseases. For example, inhibitors of cruzain, a Trypanosoma cruzi cysteine protease, are currently in the advanced stages of pre-clinical trials for the treatment of Chagas disease (McKerrow et al., 2009).
Although there are multiple species of parasites that cause malaria, Plasmodium falciparum is the most virulent and accounts for more than 90% of all malarial related deaths. Proteases are essential throughout the erythrocytic cycle of P. falciparum and are involved in a variety of biological processes such as hemoglobin degradation (Goldberg, 2005), protein trafficking (Binder and Kim, 2004), rupture (Blackman, 2008; Roiko and Carruthers, 2009), and red blood cell invasion (Dowse et al., 2008). Furthermore, inhibition of cysteine proteases results in the disruption of parasite growth, egress, and invasion. However, the study of cysteine proteases in P. falciparum has mainly focused on the falcipains (FPs). FP2, 2’ and 3 are active in the food vacuole (FV) and are involved in hemoglobin degradation (Rosenthal, 2004), the main source of amino acids during parasite growth. FP1 is expressed at the later stages of the erythrocytic cycle and is likely involved in host cell invasion (Greenbaum et al., 2002).
Dipeptidyl aminopeptidases (DPAPs) were recently identified as key regulators of the erythrocytic cycle of P. falciparum. DPAP1 (PF11_0174) is localized to the FV and is believed to be involved in the final stages of the hemoglobin degradation pathway (Klemba et al., 2004). This protease degrades oligopeptide products of upstream proteases into dipeptides that can then be converted into single amino acids by aminopeptidases (Dalal and Klemba, 2007; Klemba, 2009; Ragheb et al., 2009). Although a GFP-tagged DPAP1 had been successfully cloned into 3D7 P. falciparum, DPAP1 has been refractory to genetic deletion suggesting that it is an essential protease (Klemba et al., 2004).
DPAP3 (PFD0230c) is also expressed in an active form during the erythrocytic cycle of P. falciparum. We recently identified this protease as a key regulator of parasite egress (Arastu-Kapur et al., 2008). Because DPAPs are involved at distinct stages of the parasite life cycle, they have the potential to be valuable drug targets. Since each DPAP is likely to be essential, a drug that is able to target all of these proteases may be less likely to induce resistance because it disrupts multiple biological processes. Additionally, DPAP2 (PFL2290w), the last member of the DPAP family, is mainly expressed in gametocytes (Young et al., 2005), where its function remains largely unknown.
DPAPs are also potentially good drug targets because the closest human homologue is cathepsin C (hCat C), a protease that is not essential in mammals. Although hCatC directly activates a family of serine proteases involved in inflammation and the immune response (Adkison et al., 2002; Pham and Ley, 1999), a recent study in rats showed that more than 90% of Cat C activity needs to be inhibited in order to see any significant decrease in the levels of serine protease activities (Methot et al., 2008; Methot et al., 2007). Moreover, continuous inhibition of Cat C for several weeks is not toxic to rats (Methot et al., 2008). Finally, Cat C knockout mice are healthy and reproduce normally (Pham and Ley, 1999). DPAPs differ from other endopeptidases in that they recognize the N-terminal free amine of substrate proteins and cleave N-terminal dipeptides. Structurally, in addition to the heavy and light chains of a papain-fold catalytic domain, DPAPs also possess an exclusion domain positioned such that it occludes access to the active site beyond the P2 position, thus forming a heterotrimer (Olsen et al., 2001; Turk et al., 2001). The exclusion domain N-terminal aspartate directly interacts with the N-terminal amino group of substrates. Therefore, the active sites of DPAPs are relatively small compared to that of other endopeptidases, and most of their substrate specificity is dictated by the N-terminal residue (P2). This smaller active site is more amenable to binding of low molecular weight drug leads and should be more suitable for computational docking and in-silico design of inhibitors.
Given the lack of readily available techniques to conditionally disrupt gene expression in P. falciparum it will be necessary to use highly specific compounds to demonstrate that DPAPs are viable drug targets. In this study, we demonstrate that a highly selective inhibitor of DPAP1 causes a block in progression of the blood stage life cycle and subsequently kills parasites. While this selective lead compound was a valuable tool for in vitro studies, its overall lack of stability prevented its use for in vivo studies. Therefore, we used computational methods to design potent non-peptidic inhibitors of DPAP1 that could be used in mouse models of malaria. Our most potent lead compounds kill P. falciparum at single digit nanomolar concentrations in culture, are stable in mouse serum, and although toxic in vivo, cause a decrease in parasite load in a mouse model of malaria. Furthermore, our studies demonstrate that effective parasite killing by DPAP1 inhibitors requires sustained inhibition of its protease as the result of rapid recovery of activity after inhibition.
RESULTS
Selective inhibition of DPAP1 kills P. falciparum in culture
In order to validate DPAP1 as a drug target, we needed to identify selective inhibitors. Specifically, we needed to avoid inhibition of the FPs or DPAP3 since these are also essential papain-fold cysteine proteases. Ala-4(I)Phe-DMK (Figure 1A) was initially developed by Merck as an irreversible inhibitor of hCat C (Guay et al., 2009; Methot et al., 2007). The diazomethyl ketone (DMK) reactive group, which selectively targets cysteine proteases (Powers et al., 2002), covalently modifies the catalytic active site cysteine of hCat C. In order to assess the specificity of Ala-4(I)Phe-DMK for DPAP1, DPAP3, and the FPs, we performed competition assays using several activity-based probes (ABPs). In these assays, the inhibitor is added to total parasite extracts and inhibition is determined by labeling of residual active proteases with the ABP (Arastu-Kapur et al., 2008). We found that Ala-4(I)Phe-DMK efficiently blocked labeling of DPAP1 but not FPs or DPAP3 (Figure 1B), indicating that it is DPAP1 selective.
Figure 1. Selective inhibition of DPAP1 results in parasite death.

(A) Structure of Ala-4(I)Phe-DMK. (B) DPAP and Falcipain (FP) inhibition by Ala-4(I)Phe-DMK. Trophozoites or merozoites lysates were incubated for 30 min with 10 µM of Ala-4(I)Phe-DMK followed by 1h labeling with FY01 or 125I-DCG04. Labeling was analyzed by fluorescent scan of SDS-PAGE gels or autoradiography, respectively. Loss of labeling indicates inhibition of the indicated protease. (C) Ala-4(I)Phe-DMK acts as an irreversible inhibitor. Trophozoite lysates were incubated with inhibitor for 0.1–2h in acetate buffer. Residual activity was measured as the turnover rate of the DPAP1 specific substrate (Pro-Arg)2-Rho. The ki was obtained by fitting the data to eq 1. Standard errors of the fitted parameters are shown in parentheses. (D) Ala-4(I)Phe-DMK kills P. falciparum at low nM concentrations. Ring stage parasites were treated with inhibitor and cultured for ~75 h. Parasitemia was quantified by FACS, and the EC50Pot determined by fitting the data to a dose response curve. (E) Phenotypic effects due to Ala-4(I)Phe-DMK treatment. Rings (upper pictures) or schizonts (lower pictures) were cultured for 24h in the presence of inhibitor. Phenotypic effects were visualized in Geimsa-stained thin blood smears.
See also Figure S1.
To confirm that Ala-4(I)Phe-DMK acts as a covalent, irreversible inhibitor, we measured the dose response for DPAP1 inhibition at various time points using the (Pro-Arg)2-Rho fluorogenic substrate. We have previously demonstrated that this substrate is only processed by DPAP1 in parasite lysates (Deu et al., 2010). As expected for an irreversible inhibitor, the IC50 values decreased in a time dependent manner (Figure 1C). Furthermore, these data confirm that Ala-4(I)Phe-DMK is a potent inhibitor of DPAP1 with a second order rate constant of inhibition (ki) of 35000 ± 3000 M−1s−1.
To test whether DPAP1 inhibition results in parasite death, we treated ring stage parasites for ~ 75h with Ala-4(I)Phe-DMK and measured the fraction of infected RBCs by FACS (Figure 1D). The half maximal parasite growth dose (EC50Pot) for Ala-4(I)Phe-DMK (2.8 nM) was similar to the half maximal inhibition dose for a 2h treatment (IC50DPAP1,2h) of parasite lysates (2.0 nM; Figure 1C), which suggests that specific inhibition of DPAP1 directly correlates with parasite death. Moreover, the EC50Pot measured after 1h treatment at trophozoite stage (16 nM) is similar to the IC50DPAP1,1h measured in lysates (6.4 nM; Figure 2).
Figure 2. Stability requirements for DPAP1 inhibitors.

(A) Trophozoites are more susceptible to DPAP1 inhibition. Parasites at the indicated life stages were treated for 1h with Ala-4(I)Phe-DMK. Decrease in parasitemia was determined by FACS. Error bars show the standard error of 3 independent measurements. (B) Structure of SAK2 (C) Peptidic inhibitors are not stable in mouse serum. The stability of SAK2 and Ala-4(I)Phe-DMK for DPAP1 was determined by measuring their ability to block labeling of DPAP1 by FY01 either before (top) or after (bottom) overnight incubation in mouse serum. (D) DPAP1 activity is rapidly restored in living parasites. A culture of mixed-stage parasites was treated with 0.16–100 µM of SAK2 or 0.8–2500 nM of Ala-4(I)Phe-DMK for 0.5–7h. After cell lysis, DPAP1 activity was measured using the DPAP1 specific substrate (Pro-Arg)2-Rho. Representative data are shown in the upper graph. The IC50DPAP1 values for SAK2 and Ala-4(I)Phe-DMK at each treatment time are shown in the bar graph. The first two bars indicate the EC50Pot for each compound measured in the standard 75 h replication assay. Error bars represent the standard deviation of the fitted parameter.
See also Figure S2.
Trophozoite stage parasites are most sensitive to DPAP1 inhibition
Since Ala-4(I)Phe-DMK is a DPAP1 specific inhibitor, it could be used to determine whether a specific phenotype is associated with DPAP1 inhibition in live parasites. We treated a synchronous culture at either ring or schizont stage for 24h with Ala-4(I)Phe-DMK and monitored the parasites by thin blood smears (Figure 1E). Control treated ring stage parasites developed into mature schizonts that occupy most of the RBC volume. With increasing concentrations of the inhibitor, we observed a decrease in the size of the trophozoites. This same phenotype was observed for other classes of DPAP1 selective inhibitors such as SAK2(Arastu-Kapur et al., 2008) suggesting that this effect is likely due to specific target inhibition rather than off-target effects (Figure S1). These observations suggest that DPAP1 is involved in trophozoite growth, which agrees with its gene expression profile (Bozdech et al., 2003; Le Roch et al., 2003; Llinas et al., 2006) and its proposed role in hemoglobin degradation (Klemba et al., 2004). The lack of a FV defect when ring parasites were treated with Ala-4(I)Phe-DMK confirms that this inhibitor does not target FP2, 2’ or 3. In addition, we treated schizonts with Ala-4(I)Phe-DMK for 24 h and found that at 200 nM it causes a significant decrease in the number of rings and an increase in schizonts (Figure 1E). This suggests that DPAP1 inhibition might also delay egress. At higher concentrations (≥ 1 µM), it also induces schizont arrest and a FV defect, which might be a result from cross-reactivity with DPAP3 and the FPs, but that occurs at concentrations that are 1000-fold higher than those required to inhibit DPAP1 or kill parasites.
Although the peak of DPAP1 expression occurs at trophozoite stage, the absolute level of DPAP1 expression remains very high throughout the erythrocytic cycle (Bozdech et al., 2003; Llinas et al., 2006) and we have observed DPAP1 activity at all blood staged using FY01 (data not shown). To determine when DPAP1 activity is most essential, we treated highly synchronous parasites for 1h at different points of the blood stage lifecycle with Ala-4(I)Phe-DMK and monitored parasite growth by FACS (Figure 2A). This compound kills parasites at all stages of the lifecycle but shows a nearly ten-fold increase in potency against trophozoite stage parasites.
Peptidic inhibitors of DPAP1 are not stable in mouse serum
Although Ala-4(I)Phe-DMK is a highly DPAP1 selective inhibitor, it is not likely to be useful in vivo because it has very poor pharmacokinetic (PK) properties (personal communication, David Percival, Merck). Similarly, the poor PK properties of JCP-410, a peptidic inhibitor discovered in our lab (Arastu-Kapur et al., 2008), explains why it has no effect in vivo (Figure S2). To determine the stability of peptidic inhibitors we measured the ability of Ala-4(I)Phe-DMK and the peptide vinyl sulfone SAK2 (Figure 2B) to inhibit DPAP1 after overnight incubation at 37 °C in mouse serum. Both inhibitors lost virtually all of their activity after pre-incubation in mouse serum (Figure 2C).
SAK2 and Ala-4(I)Phe-DMK inhibit DPAP1 in parasite lysates at low nanmolar concentrations (Figure 2C), however only Ala-4(I)Phe-DMK is a potent inhibitor of parasite growth (Figure 1). SAK2 is 1000-fold less active against parasites in culture (Figure 2D). To further understand the discrepancy between the poor efficacy of SAK2 and the high potency of Ala-4(I)-Phe-DMK against live parasites, we measured DPAP1 activity using our fluorogenic assay at various time points after treatment with a range of SAK2 and Ala-4(I)Phe-DMK concentrations (Figure 2D). These results indicated that at > 0.8 µM SAK2, most DPAP1 activity was inhibited within the first 30 minutes of treatment. However, over the next 7 h, DPAP1 activity recovered substantially. This recovery of activity is consistent with the lack of stability of SAK2 inside parasites, but, because it is a covalent inhibitor, it also indicates that DPAP1 levels are replenished by expression or activation of new enzyme. However, this recovery of activity is not observed with Ala-4(I)Phe-DMK, which indicates that although unstable in mouse serum, this inhibitor is stable inside parasites.
Because of the restoration of DPAP1 activity after SAK2 treatment, the IC50DPAP1 increases up to a plateau of ~ 45 µM at 3 h, which corresponds to the EC50Pot for SAK2. This suggests that DPAP1 activity needs to be inhibited for at least 3h in order to kill P. falciparum. Similar dynamics are observed for Ala-4(I)Phe-DMK where the IC50DPAP1,2h is very similar to the EC50Pot (Figure 1).
In-silico design of potent non-peptidic inhibitors of DPAP1
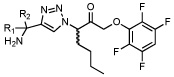
To overcome the stability issues of peptidic inhibitors, we screened several recently reported non-peptidic scaffolds from a large library of compounds that was initially designed to target cysteine cathepsins in mammals (Brak et al., 2008; Brak et al., 2010; Inagaki et al., 2007; Patterson et al., 2006; Wood et al., 2005). These inhibitors use a 2,3,5,6-tetrafluorophenoxyarylmethyl ketone (TFPAMK) reactive group, and the P1 and P2 residues are linked through a triazole ring (see Table 1 for structures). We tested only the molecules in the library containing an N-terminal free amine (first 7 compounds in Table 1) since this is one of the major determinants of substrate specificity for DPAPs. HN3019 was the most potent inhibitor against DPAP1 (Table 1). We then tested whether an acyloxymethylketone (AOMK) or a nitrile (ML4091 or ML4101, respectively) reactive group would provide any benefits over the TFPAMK reactive group of HN3019. We found that the TFPAMK group is 18 and 380-fold more potent than the AOMK or the nitrile group, respectively. Therefore, the TFPAMK was chosen for the synthesis of further non-peptidic inhibitors.
Table 1.
Specificity, potency, and toxicity parameters of non-peptidic inhibitors of DPAP1.
 | |||||
|---|---|---|---|---|---|
| Compound Name |
Structure | ki (M−1s−1) | IC50DPAP1 (nM) | EC50Pot (nM) | EC50Tox (nM) |
| ML4057 | 520 (30) | 700 (40) | 3000 (1300) | 6500 (1500) | |
| ML4066 | 310 (30) | 1100 (100) | 1000 (500) | 8000 (3000) | |
| ML4046A | 500 (20) | 700 (40) | 60 (30) | 2100 (300) | |
| ML4046B | < 20 | > 20000 | 3800 (3000) | 10500 (500) | |
| ML4053 | 1.6 (0.2) | > 100000 | 35000 (30000) | 8300 (400) | |
| ML4068 | 4.9 (0.5) | 74000 (9000) | 3000 (2000) | > 100000 | |
| HN3019 | 630 (30) | 560 (40) | 70 (30) | > 100000 | |
| ML4161 | 810 (50) | 410 (30) | 35000 (30000) | 90000 (20000) | |
| ML4123 | 910 (50) | 410 (10) | 20 (10) | 5500 (2500) | |
| ML4124 | 510 (40) | 700 (50) | 140 (60) | > 100000 | |
| ML4118 | 2070 (30) | 186 (2)* | 19 (2) | 9300 (600) | |
| ML4118S | 16500 (1200) | 19 (1) | 5.2 (0.5) | 8500 (1500) | |
| ML4118R | 28 (3) | 14000 (1500)* | 890 (520) | 7000 (2000) | |
| ML4162 | < 20 | > 20000 | 220 (160) | 68000 (4000) | |
| ML4163 | < 20 | > 20000 | > 100000 | > 100000 | |
| ML4154 | < 20 | > 20000 | 45000 (26000) | 30000 (5000) | |
| ML5004 | 50 (2) | 7700 (200)* | n.d. | n.d. | |
| ML6076 | 920 (70) | 420 (30) | 930 (130) | n.d. | |
| ML4091 | 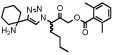 |
34 (2) | 11000 (1000) | 6000 (3000) | 12000 (3000) |
| ML4101 | 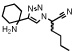 |
1.7 (0.2) | > 100000 | > 100000 | > 100000 |
DPAP1 inhibition was measured using our DPAP1 specific fluorogenic assay. IC50DPAP1 were determined after 30 min incubation of parasite lysates with 5 nM to 100 µM inhibitor. ki values were obtained by fitting the data to eq 1. EC50Pot values were measured using our standard 75 h replication assay. Cell toxicity (EC50Tox) was determined by measuring ATP production after 24 h treatment of HFF cells with 0.01–100 µM of compound. All IC50 and EC50 were obtained by fitting the data to a dose response curve. n.d., Not Determined.
IC50DPAP1 and ki values obtained using the competition assay between inhibtor and probe rather than the fluorogenic assay. The standard errors of the fitted paramaters are shown in parentheses.
To guide the synthesis of new compounds, we built a homology model of DPAP1 based on the crystal structure of hCat C covalently bound to the Gly-Phe-diazomethane inhibitor (PDB 2DJF; (Molgaard et al., 2007); See Figure S3 for the validation of this model). HN3019 was covalently linked to DPAP1 as the final product of the reaction between the catalytic cysteine and the inhibitor (Figure 3A). These models suggested that there was space on the back side of the S2 pocket that is not filled by the P2 cyclohexane ring of HN3019. In order to better fill the S2 pocket, we tried opening the cyclohexane ring (compounds ML4161, ML4123, ML4124, and ML4118S) or adding bulk at the distal carbon of the cyclohexane ring (compounds ML4162, ML4163 and ML4154). The latter strategy failed (Table 1) probably due to steric clashes of the piperidine group with S2 pocket residues (Figure S3). On the other hand, the former strategy resulted in compounds with improved potency towards DPAP1 (Table 1).
Figure 3. In silico design of non-peptidic inhibitors for DPAP1.

(A) Docking of HN3019 and ML4118S into the active site of the DPAP1 model. HN3019 (green) and ML4118S (cyan) were docked as the final covalent product of the reaction between the inhibitor and the catalytic cysteine. The structure of the DPAP1 model bound to HN3019 is shown as an electrostatic surface. (B) Docking of ML4118S (cyan) and ML4118R (orange) into the active site of the DPAP1 model.
See also Figure S3.
ML4118 was initially synthesized as a mixture of the R and S diastereomers. Based on the docked structures of these two compounds (Figure 3B), ML4118S would be expected to be much more potent because its cyclopentane moiety is buried deep in the S2 pocket while the R isomer leaves this group partially exposed to the solvent. To test this hypothesis we synthesized ML4118S and ML4118R (Figure 4A). We measured the specificity of these isomers as well as the original racemate towards different P. falciparum cysteine proteases. As expected, ML4118S was substantially more potent than ML4118R, while ML4118 showed intermediate potency (Figure 4B & C). Interestingly, unlike Ala-4(I)-Phe-DMK, ML4118S was also active against DPAP3 and FP2/3 (although with reduced potency compared to DPAP1) indicating that it is a more broad spectrum inhibitor (Figure 4B & E). In order to correlate target inhibition with potency against the parasite, we performed inhibition studies with the ML4118 analogs in intact parasites. These compounds are cell permeable as they show the same pattern of DPAP1 inhibition in intact parasites or in lysates (Figure 4C). Additionally, the same trend in potency observed in the competition studies in lysates was observed in the replication assay (Figure 4D). To further correlate parasite death with DPAP1 inhibition, we compared the IC50 values against DPAP1, DPAP3 and FP2/3 with potency in a 1h replication assay since the IC50 were obtained after 0.5h incubation of parasite lysates with compound prior to probe labeling for 1h (Figure 4E). For both ML4118 and ML4118S, we observed a direct correlation between specificity and parasite death, with only the IC50 values for DPAP1 inhibition matching the EC50Pot,1h values in the replication assay. Because ML4118R is much less potent against DPAP1, its EC50Pot,1h may reflect some off target effects since this compound is also toxic to human foreskin fibroblast (HFF) cells at high concentrations (Figure 4E). Importantly, the fact that ML4118S is more than 500-times more potent than ML4118R at killing parasites, yet both have similar general toxicity profiles in HFF cells (EC50Tox ~ 10 µM) suggests that potency in parasite killing is due to target inhibition rather than non-specific toxic effects.
Figure 4. ML4118S is 500-fold more potent than ML4118R.

(A) Structures. ML4118 is a mixture of the R and S diastereomers. (B) Specificity towards DPAPs and flacipains (FPs) measured using the competition assay. (C) DPAP1 Inhibition in intact parasites vs. lysates. Purified schizonts (filled symbols) or lysates (empty symbols) were treated with ML4118S (triangles), ML4118 (diamonds), or ML4118R (circles) followed by labeling with FY01 for 1h. (D) Potency of killing parasites determined using a 75h replication assay. (E) Specificity vs. potency vs. toxicity. IC50 values were obtained by quantifying the gel bands shown in B. EC50Pot values were determined by FACS after treating schizonts for 1h with compound, followed by 3 washes with media, and culture of the parasites for another 48h. Cell toxicity was determined by incubating HFF cells for 24h with inhibitor and measuring ATP production using the CellTiter-Glo® assay. Error bars represent the standard deviation of the fitted parameter.
To determine if switching to a non-peptidic scaffold results in increased overall stability, we measured the potency of ML4118S towards DPAP1 before and after overnight incubation in mouse serum at 37 °C (Figure 5A). Unlike the peptide inhibitors (Figure 2C), ML4118S shows only minimal loss of potency after serum treatment. This increased stability also allowed ML4118S to induce sustained DPAP1 inhibition in intact parasites (Figure 5B). Interestingly, the kinetic inhibition studies in intact parasites suggest that 1 nM of ML4118S is sufficient to induce a prolonged inhibition of 70% of DPAP1 activity without killing parasites (i.e. virtually no effect at 1 nM on overall parasite replication – Figure 4D). These results, combined with our results using SAK2, indicate that efficient parasite killing requires at least 95% inhibition of DPAP1 activity for ~2h.
Figure 5. ML4118S is stable in mouse serum and sustains DPAP1 inhibition.

(A) Serum stability of ML4118S. The stability was determined by measuring their ability of the ML41118S to block labeling of DPAP1 by FY01 either before (top) or after (bottom) overnight incubation in mouse serum. (B) Kinetics of DPAP1 inhibition by ML4118S in intact parasites. DPAP1 activity in a mix-stage parasite culture was measured after treatment with ML4118S over a range of times. DPAP1 activity was measured using the DPAP1 specific substrate (Pro-Arg)2-Rho.
Evaluation of the anti-parasitic activity of ML4118S in a mouse model of malaria
Because ML4118S is a potent a cell permeable DPAP1 inhibitor that is stable in mouse serum and has overall low toxicity in HFF cells, we decided to test its in vivo efficacy using a Plasmodium berghei mouse model of malaria. Before testing ML4118S in vivo, we wanted to make sure that this compound also targets the DPAP1 homologue in P. berghei (PbDPAP1). We measured ML4118S specificity towards PbDPAP1 and the FPs homologues, berghipains 1 and 2 (BP1 and BP2), in P. berghei lysates (Figure 6A). ML4118S shows inhibition of PbDPAP1 starting at 20 nM with no inhibition of BP2 at concentrations as high as 20 µM. It also has only weak activity towards BP1 at concentrations above 1 µM. Thus, ML4118S is a potent and selective PbDPAP1 inhibitor in P. berghei.
Figure 6. Non-peptidic DPAP1 inhibitors decrease parasite load in vivo.

(A&B) Specificity of ML4118S (A) and KB16 (B) for PbDPAP1, BP1, and BP2 using competition assays in P. berghei lysates. (C) Parasite growth in mice after treatment with ML4118S and KB16. Treatment with 20 mg/Kg of compound via IP is indicated with black (KB16, n=4) or grey (ML4118S, n=2–5) dots. Parasitemia was quantified by Geimsa-stained thin blood smears (days 2–6) or by FACS (days 7–15). Control mice were treated either following the ML4118S (n=2) or the KB16 (n=3) treatment schedule. No difference in parasitemia was observed between the two sets of control mice. The value reported for the vehicle-treated mice is the average of the 5 control mice. Asterisks indicate significant differences between vehicle and drug-treated mice (p-values were determined using the Student T-test: *, p > 0.95; **, p > 0.99; ***, p > 0.999).
We initially planned to treat mice infected with NK65 P. berghei parasites with 20 mg/Kg of ML4118S twice a day for a period of 10 days. We chose this time course for treatment based on the amount of time it takes for P. berghei parasites to start growing exponentially in vivo (Figure 6C). In our studies, we found that parasite levels increase from day 1–3 and then temporarily drop from day 3–6. While we cannot explain the reason for this behavior, we found that it was consistent among all animals in the trail and therefore does not effect our conclusions regarding effects of inhibitor treatment. Five mice were treated with a single dose of ML4118S on the day of infection followed by two doses (8h apart) the next day. Clear signs of compound toxicity were observed in two mice that were therefore euthanized. On day 2, we decreased the dosage to a single and final treatment. A third mouse had to be euthanized by day 5 due to compound toxicity issues. Although we had to discontinue dosing due to toxicity of the compound, we collected blood from day 2 through 15 for the surviving animals and estimate the percentage parasitemia by Geimsa-stained thin blood smear or FACS (Figure 6C). Due to the toxicity of the compound, we were unable to repeat the studies with a larger group of animals, thus the in vivo data must be interpreted with caution. Regardless, we observed a significant decrease in parasitemia on the days following treatment with ML4118S (days 2–4; inset Figure 6C).
Because the toxicity of ML4118S limited the utility of the compound, we decided to shift our efforts to a related compound (KB16) known to have low toxicity in mice (no toxicity at 20 mg/kg/day for three weeks). KB16 was designed to target the cysteine protease cruzain from Trypanosoma cruzi (Brak et al., 2008) and has a reasonably long half-life in blood (3–4 h). Unfortunately, KB16 is not a very potent inhibitor of PbDPAP1 (IC50DPAP1 ~1 µM; Figure 6B). However, because it is an irreversible inhibitor with a long half-life in blood, we thought it might have a chance to sustain PbDPAP1 inhibition in vivo. When infected mice were treated from day 0 to 9 with 20 mg/Kg/day of KB16, we observed a significant decrease of parasitemia up to day 14. Given that KB16 also targets BP1 (IC50BP1 ~ 0.5 µM), it is possible that the decrease in parasitemia is due to a combined effect of BP1 and DPAP1 inhibition. Taken together, our in vivo results suggest that inhibition of DPAP1 may be a viable means to reduce parasite growth in vivo.
DISCUSSION
The hemoglobin degradation pathway has been pursued as a potential site for intervention with antimalarial drugs for the last 20 years (Francis et al., 1997). However, most of the efforts to block this essential pathway have focused on inhibition of the FPs (Rosenthal et al., 2002) and plasmepsins (Ersmark et al., 2006) in the FV. All of these proteases are important for the initial stages of hemoglobin degradation but their biological roles in this pathway have been shown to be redundant not only within a protease family but also between families (Bonilla et al., 2007a; Bonilla et al., 2007b). Single and multiple genetic knockouts of FV plasmepsins (Liu et al., 2005; Omara-Opyene et al., 2004), and deletion of FPs 1 and 2 are not lethal (Sijwali et al., 2006). On the other hand, there is only one DPAP in this pathway, and efforts to knock out this enzyme suggest that it is essential (Klemba et al., 2004). Therefore, DPAP1 is likely to be a more suitable target for drug development than the FV FPs or plasmepsins.
In this study we show that selective inhibition of DPAP1 by Ala-4(I)Phe-DMK results in parasite death in culture. This inhibitor has at least 1000-fold selectivity towards DPAP1 over the other papain fold cysteine proteases of P. falciparum (DPAP3 and the FPs). Several lines of evidence suggest that Ala-4(I)Phe-DMK effects on parasites are specifically due to inhibition of DPAP1 rather than the result of off target effects. First, DPAP1 inhibition closely correlates with parasite death (IC50DPAP1,2h = 2.0; EC50Pot = 2.8 nM). Second, parasites are 10-times more sensitive to Ala-4(I)Phe-DMK at the trophozoite stage consistent with the role of DPAP1 in hemoglobin degradation, a pathway that is most essential at trophozoite stage. Third, the phenotypic effect observed for this inhibitor, (i.e. formation of an underdeveloped trophozoite), is in agreement with DPAP1 being the primary target since the same phenotype was observed in parasite treated with SAK2, P-hF-AOMK and A-4(I)F-AOMK, which are selective covalent DPAP1 inhibitors whose reactive groups are structurally unrelated to Ala-4(I)Phe-DMK (Figure S1). Finally, the correlation between DPAP1 inhibition and potency in the replication assay was also observed for our library of non-peptidic inhibitors. The ki and EC50Pot values of Table 1 show that the stronger an inhibitor is against DPAP1, the more potent it is at killing parasites. More specifically, the dramatic difference in potency between the ML4118S and ML4118R diastereomers indicates that these compounds are not killing parasites by non-specific toxic effects.
Although the original peptidic vinylsulfone inhibitors were not suitable for use in vivo due to their overall lack of stability, they allowed us to study the dynamics of DPAP1 expression/activation in living parasites (Figure 2). Studies of DPAP1 inhibition over time showed that DPAP1 activity recovers rapidly after it has been irreversibly inhibited. This implies that either DPAP1 is constantly expressed and trafficked into the FV to replenish the DPAP1 activity that is lost due to protease turnover, or that P. falciparum tightly regulates the levels of DPAP1 activity and is able to respond to a drop in activity.
We also provide several lines of evidence to show that sustained inhibition of DPAP1 for 2–3h is required to prevent the recovery of activity and induce parasite death. The potency of Ala-4(I)Phe-DMK in the parasite replication assay corresponds to the IC50DPAP1 after 2h of treatment of parasite lysates (Figure 1). Furthermore, the IC50DPAP1 for SAK2 reaches a plateau after 3h of treatment that corresponds to its EC50Pot in the replication assay (Figure 2). Therefore, an effective drug against DPAP1 either needs to have a long half-life in blood or be designed such that it will be retained inside the FV for several hours. In theory this could be achieved by taking advantage of the acidic environment inside the FV or by using a pro-drug strategy to enhance stability in blood.
We were able to overcome the stability problems of peptidic DPAP1 inhibitors by switching to a non-peptidic scaffold. Moreover, homology modeling and computational docking methods allowed us to improve the specificity of our initial hit, HN3019, by more than 25-fold. This suggests that in-silico methods may be valuable for the design of DPAP inhibitors. Analysis of the structure activity relationship (SAR) of our two classes of compounds, dipeptide vinylsulfone (Table S1) and non-peptidic TFPAMK (Table 1) inhibitors indicates that short amino acid side chains are preferred at the S2 pocket (See supplementary material for a detailed SAR analysis). For peptidic inhibitors, Pro in P2 or 4(I)Phe in P1 enhances inhibitor selectivity towards DPAP1 with respect to the other papain fold cysteine proteases (Figure S1). Interestingly, a Pro in P2 within the context of the non-peptidic scaffold (ML6076) does not result in a DPAP1 selective inhibitor (Figure S1). Finally, our results with KB16 suggest that secondary amines in P2 might improve the PK and toxicity properties of non-peptidic inhibitors.
In this study, we also showed that DPAP1 inhibitors have effects on parasite growth in vivo. In P. berghei, ML4118S is a much more specific inhibitor of DPAP1 than in P. falciparum. Although this inhibitor has toxicity issues in mice at 20 mg/Kg, we were able to observe a significant decrease in parasitemia 2 days after treatment. Unfortunately, the relatively small sample size that was caused by compound toxicity, coupled with overall animal-to-animal variability in parasite growth makes our data with ML4118S still less than conclusive. However, we did find that the non-toxic KB16 resulted in a significant decrease in parasitemia during the 10 days of treatment. Although these results suggest that DPAP1 inhibition can lead to a decrease in parasitemia in vivo, additional studies using less toxic and more selective inhibitors will be required to confirm our findings. We are currently working towards the development of selective, non-toxic DPAP1-specific compounds that have long blood half-lives in blood. These compounds should allow us to confirm that DPAP1 inhibition can clear parasitemia in vivo.
In general the papain-fold proteases are likely to be interesting as potential anti-malarial drug targets. Members of this family of proteases play essential roles in various aspects of Plasmodium biology, and inhibitors that target multiple family members may be beneficial over compounds that target only one member. Furthermore, the fact that the closest related human homologue to DPAPs can be inhibited without any significant toxic effects suggests that parasite specific targeting may not need to be a priority in drug design for this target family. While the compounds described in this study are certainly not optimal as drug leads due to lack of potency, poor PK and/or toxicity issues, they do provide the first direct evidence that DPAP1 is a viable anti-malarial drug target. Based on these promising results, we have recently completed a screen of more than 100,000 small molecules using a DPAP1 specific fluorogenic substrate assay in parasite lysates. This screen has identified many new inhibitor scaffolds that we are currently evaluating for their viability as potential drug leads.
SIGNIFICANCE
The emergence of drug resistance in malaria species continues to be the main hurdle to treat this devastating disease. In order to control this pandemic disease it is crucial to identify new drug targets that can expand the portfolio of anti-malaria drugs. This work demonstres the potential of targeting DPAP1 with small molecules. We showed that DPAP1 inhibition impairs parasite development, thus validating this cysteine protease as a new antimalarial target. Additionally, we showed that in silico methods can be successfully applied to design potent DPAP inhibitors. Finally, we demonstrated that the non-covalent inhibitors presented in this work have improved stability compared to more classical, peptidic-based, protease inhibitors. Overall, our results validate DPAP1 as an antimalarial target but also indicate that effective DPAP drugs will require either long half-lives or high retention in infected RBCs to sustain DPAP1 inhibition for several hours.
EXPERIMENTAL PROCEDURES
Protease inhibitor synthesis and characterization
Ala-4(I)Phe-DMK was a valuable gift from David Percival (Merck); its synthesis and characterization was described in (Guay et al., 2009). The vinyl sulfone inhibitors used in this study were described in (Arastu-Kapur et al., 2008), and KB16 was characterized in (Brak et al., 2008). The AOMK inhibitors were synthesized as described in (Kato et al., 2005). The synthesis and characterization of all other non-peptidic inhibitor is described in the Supplementary Methods.
Parasite culture, harvesting and lysate preparation
3D7 and D10 P. falciparum clones were cultured as described in (Arastu-Kapur et al., 2008). To tightly synchronized D10 parasites within a 1h window, schizonts were harvested 6h prior to rupture using a 70% percol gradient and cultured until they were rupturing. Blood was then added to this culture, and merozoites were allowed to invade uninfected RBCs for 1 h. Newly formed rings were purified with a sorbitol treatment. 3D7 parasites were grown asynchronously and were used when a mixed-stage parasite culture was needed. Parasite pellets and lysates were obtained as described in (Arastu-Kapur et al., 2008).
Labeling of cysteine protease activity with ABPs and competition of labeling by protease inhibitors
FY01, a cell permeable fluorescently-tagged probe, and radiolabeled 125I-DCG04 were used to label the activity of DPAPs and FPs, respectively, in intact schizonts or parasite lysates (in acetate buffer: 50 mM sodium acetate, 5 mM MgCl2, and 5 mM DTT at pH 5.5). Labeling and competition assays were performed as described in (Arastu-Kapur et al., 2008).
ki determination using a fluorogenic activity assay for DPAP1
(Pro-Arg)2-Rho is a DPAP1 specific substrate in parasite lysates (Deu et al., 2010). DPAP1 activity was measured at 25 °C in acetate buffer containing 1% of parasite lysates and 10 µM (Pro-Arg)2-Rho. Substrate turnover was measured for 5 min in a 96-well plate at 523 nm (λexcitation = 492 nm and an emission cutoff filter at 515 nm) in a Spectramax M5 plate-reader (Molecular Devices).
Accurate ki values (second order rate constant of inhibition) were obtained by treating parasite lysates in acetate buffer with inhibitor for a given period of time (t). Residual DPAP1 activity was measured using our fluorogenic assay. The rates of substrate turnover relative to DMSO controls (v/v0) were fitted to a simple irreversible inhibitor model (eq. 1).
| eq 1 |
Kinetics of DPAP1 inhibition and activation in intact parasite
A mix-stage culture of 3D7 parasites at ~ 20% parasitemia was treated with different concentrations of inhibitor or DMSO. 200 µl aliquots of culture were taken after 0.5–7h of treatment, and the RBCs were lysed in 100 µL of acetate buffer by three cycles of freezing in liquid nitrogen and thawing at 37 °C. DPAP1 activity was measured using the fluorogenic assay.
P. falciparum replication assay
200 µL synchronous D10 parasites at ring stage (~ 2% parasitemia and 0.5% hematocrit) were treated with compound and cultured in 96-well plates for ~75 h, when the DMSO controls reach schizont stage. Infected and uninfected RBCs were quantified by FACS as described in (Arastu-Kapur et al., 2008). All FACS measurements were taken on a BD FACScan flow cytometer (Becton, Dickinson and Co.). All EC50Pot values for parasite death were obtained by fitting the parasitemia values to a dose response curve.
Susceptibility of the different life cycle stages to DPAP1 inhibition
Tightly synchronized parasites were treated for 1h with Ala-4(I)Phe-DMK or DMSO at early rings (1h post invasion- p.i.), rings (10h p.i.), late rings (18h p.i.), trophozoites (30h p.i.), schizonts (38h p.i.), or late schizonts (46h p.i.). Cells were then washed 3 times with media and cultured until the DMSO control parasites progressed through egress, and newly infected RBCs reached schizont stage (~ 80h p.i.). Parasitemia was quantified by FACS.
Cell toxicity
200 µL of an HFF culture was incubated in a 96-well plate for 24h with inhibitor or DMSO. Cell viability was measured using the CellTiter-Glo® Assay (Promega) according the manufacturer’s instructions. EC50Tox values were obtained by fitting the data to a dose response curve.
Homology modeling and docking
The homology model of DPAP1 was built based on the crystal structure of hCatC covalently bound to the Gly-Phe-diazomethane inhibitor (PDB 2DJF; (Molgaard et al., 2007)) using the default parameters of the Molecular Operating Environment (MOE) software (Chemical Computing group). To dock HN3019 in the homology model of DPAP1, the inhibitor backbone was oriented according to the structure of human cathepsin S bound to a non-peptidic chloromethyl ketone inhibitor (PDB 2H7J;(Patterson et al., 2006)) that has the same inhibitor backbone as HN3019. We decided to dock the structure of HN3019 as the product of the reaction between the warhead and the active site, i.e. the catalytic cysteine was covalently linked to the α-carbon of the inhibitor after displacement of the tetrafluorophenoxy group.
Energy minimizations of the P2, P1 and covalently modified cysteine were performed sequentially followed by an energy minimization of the full molecule and the catalytic cysteine side chain. A final energy minimization including HN3019 and all side chains within 4.5 Å yielded our final model of DPAP1 bound to HN3019. All energy minimizations were performed using the default parameters in MOE. To dock other non-peptidic inhibitors, the P2 position of HN3019 bound to the DPAP1 model was modified to that of the new inhibitors, and the structure of the inhibitor-protein complex was energy minimized as described above.
Infection of mice with P. berghei parasites and treatment with DPAP1 inhibitors
All mouse experiments were approved by the Stanford Administration Panel on Laboratory Animal Care and strictly followed their specific guidelines. Balb/c female mice (20–24 g) were infected via tail vein (day 0) with 1 million of freshly collected parasites from an infected mouse. All treatments were administered via IP in 45% polyethylene glycol (M.W. 400), 35% propylene glycol, 10% ethanol, 10% DMSO, and 10% w/v 2-hydroxypropyl β-cyclodextrin. Mice treated with ML4118S (n = 5) were administered a dose of 20 mg/Kg in 200 µL of vehicle on day 0, two on day 1, and a last one on day 2. KB16 treatment was administered once a day from day 0 to 9 at 20 mg/Kg (n=4). Control mice were treated with vehicle following either the ML4118S (n=2) or the KB16 (n=3) treatment schedules. Blood was collected from the tail and analyzed by FACS and Geimsa-stained thin blood smears from days 2–15. Mice were euthanized on day 15.
Determination of parasitemia in mouse blood
Parasitemia was quantified by FACS when it reached more than 2% (days 6–15). From days 2–5, the parasite load was too low to get accurate quantification by FACS, and parasitemia was quantified from Geimsa-stained thin blood smears. Parasitemia was estimated by counting all infected RBCs in 15–20 optical fields containing 200–1000 cells. For each field the total number of RBCs was roughly estimated. The final parasitemia for each mouse and day was determined from an estimate of more than 10,000 RBCs per slide.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank David Percival (Merck) for providing Ala-4(I)Phe-DMK and sharing information about its PK properties. We thank Michael Klemba for valuable information about the DPAPs and for sharing his results about DPAP1 substrate specificity before its publication. We thank A. Guzetta for the mass-spec analysis of the PK studies. This work was supported by funding by NIH grants R01 EB005011 and R01 AI078947 and a New Investigator Award in Pathogenesis from the Burrough Wellcome Fund (All to MB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–371. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arastu-Kapur S, Ponder EL, Fonovic UP, Yeoh S, Yuan F, Fonovic M, Grainger M, Phillips CI, Powers JC, Bogyo M. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nature chemical biology. 2008;4:203–213. doi: 10.1038/nchembio.70. [DOI] [PubMed] [Google Scholar]
- Aregawi M, Cibulskis R, Otten M, Williams R, Dye C, Programme WGM. World Malaria Report 2008. World Health Organization; 2008. p. 190. [Google Scholar]
- Binder EM, Kim K. Location, location, location: trafficking and function of secreted proteases of Toxoplasma and Plasmodium. Traffic. 2004;5:914–924. doi: 10.1111/j.1600-0854.2004.00244.x. [DOI] [PubMed] [Google Scholar]
- Blackman MJ. Malarial proteases and host cell egress: an 'emerging' cascade. Cell Microbiol. 2008;10:1925–1934. doi: 10.1111/j.1462-5822.2008.01176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla JA, Bonilla TD, Yowell CA, Fujioka H, Dame JB. Critical roles for the digestive vacuole plasmepsins of Plasmodium falciparum in vacuolar function. Mol Microbiol. 2007a;65:64–75. doi: 10.1111/j.1365-2958.2007.05768.x. [DOI] [PubMed] [Google Scholar]
- Bonilla JA, Moura PA, Bonilla TD, Yowell CA, Fidock DA, Dame JB. Effects on growth, hemoglobin metabolism and paralogous gene expression resulting from disruption of genes encoding the digestive vacuole plasmepsins of Plasmodium falciparum. International journal for parasitology. 2007b;37:317–327. doi: 10.1016/j.ijpara.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Bozdech Z, Llinas M, Pulliam BL, Wong ED, Zhu J, DeRisi JL. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003;1:E5. doi: 10.1371/journal.pbio.0000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brak K, Doyle PS, McKerrow JH, Ellman JA. Identification of a new class of nonpeptidic inhibitors of cruzain. J Am Chem Soc. 2008;130:6404–6410. doi: 10.1021/ja710254m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brak K, Kerr ID, Barrett KT, Fuchi N, Debnath M, Ang K, Engel JC, McKerrow JH, Doyle PS, Brinen LS, et al. Nonpeptidic tetrafluorophenoxymethyl ketone cruzain inhibitors as promising new leads for Chagas disease chemotherapy. J Med Chem. 2010;53:1763–1773. doi: 10.1021/jm901633v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaijaroenkul W, Bangchang KN, Mungthin M, Ward SA. In vitro antimalarial drug susceptibility in Thai border areas from 1998–2003. Malaria journal. 2005;4:37. doi: 10.1186/1475-2875-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal S, Klemba M. Roles for two aminopeptidases in vacuolar hemoglobin catabolism in Plasmodium falciparum. J Biol Chem. 2007;282:35978–35987. doi: 10.1074/jbc.M703643200. [DOI] [PubMed] [Google Scholar]
- Deu E, Yang Z, Wang F, Klemba M, Bogyo M. Use of activity-based probes to develop high throughput screening assays that can be performed in complex cell extracts. PLoS One In Submission. 2010 doi: 10.1371/journal.pone.0011985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowse TJ, Koussis K, Blackman MJ, Soldati-Favre D. Roles of proteases during invasion and egress by Plasmodium and Toxoplasma. Subcell Biochem. 2008;47:121–139. doi: 10.1007/978-0-387-78267-6_10. [DOI] [PubMed] [Google Scholar]
- Ersmark K, Samuelsson B, Hallberg A. Plasmepsins as potential targets for new antimalarial therapy. Med Res Rev. 2006;26:626–666. doi: 10.1002/med.20082. [DOI] [PubMed] [Google Scholar]
- Fear G, Komarnytsky S, Raskin I. Protease inhibitors and their peptidomimetic derivatives as potential drugs. Pharmacol Ther. 2007;113:354–368. doi: 10.1016/j.pharmthera.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis SE, Sullivan DJ, Jr, Goldberg DE. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol. 1997;51:97–123. doi: 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- Goldberg DE. Hemoglobin degradation. Curr Top Microbiol Immunol. 2005;295:275–291. doi: 10.1007/3-540-29088-5_11. [DOI] [PubMed] [Google Scholar]
- Greenbaum DC, Baruch A, Grainger M, Bozdech Z, Medzihradszky KF, Engel J, DeRisi J, Holder AA, Bogyo M. A role for the protease falcipain 1 in host cell invasion by the human malaria parasite. Science. 2002;298:2002–2006. doi: 10.1126/science.1077426. [DOI] [PubMed] [Google Scholar]
- Guay D, Beaulieu C, Jagadeeswar Reddy T, Zamboni R, Methot N, Rubin J, Ethier D, David Percival M. Design and synthesis of dipeptidyl nitriles as potent, selective, and reversible inhibitors of cathepsin C. Bioorg Med Chem Lett. 2009;19:5392–5396. doi: 10.1016/j.bmcl.2009.07.114. [DOI] [PubMed] [Google Scholar]
- Inagaki H, Tsuruoka H, Hornsby M, Lesley SA, Spraggon G, Ellman JA. Characterization and optimization of selective, nonpeptidic inhibitors of cathepsin S with an unprecedented binding mode. J Med Chem. 2007;50:2693–2699. doi: 10.1021/jm070111+. [DOI] [PubMed] [Google Scholar]
- Kato D, Boatright KM, Berger AB, Nazif T, Blum G, Ryan C, Chehade KA, Salvesen GS, Bogyo M. Activity-based probes that target diverse cysteine protease families. Nature chemical biology. 2005;1:33–38. doi: 10.1038/nchembio707. [DOI] [PubMed] [Google Scholar]
- Klemba M. On the location of the aminopeptidase N homolog PfA-M1 in Plasmodium falciparum. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:E55. doi: 10.1073/pnas.0903493106. author reply E56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemba M, Gluzman I, Goldberg DE. A Plasmodium falciparum dipeptidyl aminopeptidase I participates in vacuolar hemoglobin degradation. J Biol Chem. 2004;279:43000–43007. doi: 10.1074/jbc.M408123200. [DOI] [PubMed] [Google Scholar]
- Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, Haynes JD, De La Vega P, Holder AA, Batalov S, Carucci DJ, et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science. 2003;301:1503–1508. doi: 10.1126/science.1087025. [DOI] [PubMed] [Google Scholar]
- Liu J, Gluzman IY, Drew ME, Goldberg DE. The role of Plasmodium falciparum food vacuole plasmepsins. J Biol Chem. 2005;280:1432–1437. doi: 10.1074/jbc.M409740200. [DOI] [PubMed] [Google Scholar]
- Llinas M, Bozdech Z, Wong ED, Adai AT, DeRisi JL. Comparative whole genome transcriptome analysis of three Plasmodium falciparum strains. Nucleic Acids Res. 2006;34:1166–1173. doi: 10.1093/nar/gkj517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerrow JH, Caffrey C, Kelly B, Loke P, Sajid M. Proteases in parasitic diseases. Annu Rev Pathol. 2006;1:497–536. doi: 10.1146/annurev.pathol.1.110304.100151. [DOI] [PubMed] [Google Scholar]
- McKerrow JH, Doyle PS, Engel JC, Podust LM, Robertson SA, Ferreira R, Saxton T, Arkin M, Kerr ID, Brinen LS, et al. Two approaches to discovering and developing new drugs for Chagas disease. Mem Inst Oswaldo Cruz. 2009;104 Suppl 1:263–269. doi: 10.1590/s0074-02762009000900034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerrow JH, Rosenthal PJ, Swenerton R, Doyle P. Development of protease inhibitors for protozoan infections. Curr Opin Infect Dis. 2008;21:668–672. doi: 10.1097/QCO.0b013e328315cca9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot N, Guay D, Rubin J, Ethier D, Ortega K, Wong S, Normandin D, Beaulieu C, Reddy TJ, Riendeau D, et al. In vivo inhibition of serine protease processing requires a high fractional inhibition of cathepsin C. Mol Pharmacol. 2008;73:1857–1865. doi: 10.1124/mol.108.045682. [DOI] [PubMed] [Google Scholar]
- Methot N, Rubin J, Guay D, Beaulieu C, Ethier D, Reddy TJ, Riendeau D, Percival MD. Inhibition of the activation of multiple serine proteases with a cathepsin C inhibitor requires sustained exposure to prevent pro-enzyme processing. J Biol Chem. 2007;282:20836–20846. doi: 10.1074/jbc.M702615200. [DOI] [PubMed] [Google Scholar]
- Molgaard A, Arnau J, Lauritzen C, Larsen S, Petersen G, Pedersen J. The crystal structure of human dipeptidyl peptidase I (cathepsin C) in complex with the inhibitor Gly-Phe-CHN2. Biochem J. 2007;401:645–650. doi: 10.1042/BJ20061389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noedl H, Socheat D, Satimai W. Artemisinin-resistant malaria in Asia. N Engl J Med. 2009;361:540–541. doi: 10.1056/NEJMc0900231. [DOI] [PubMed] [Google Scholar]
- Olsen JG, Kadziola A, Lauritzen C, Pedersen J, Larsen S, Dahl SW. Tetrameric dipeptidyl peptidase I directs substrate specificity by use of the residual pro-part domain. FEBS Lett. 2001;506:201–206. doi: 10.1016/s0014-5793(01)02911-8. [DOI] [PubMed] [Google Scholar]
- Omara-Opyene AL, Moura PA, Sulsona CR, Bonilla JA, Yowell CA, Fujioka H, Fidock DA, Dame JB. Genetic disruption of the Plasmodium falciparum digestive vacuole plasmepsins demonstrates their functional redundancy. J Biol Chem. 2004;279:54088–54096. doi: 10.1074/jbc.M409605200. [DOI] [PubMed] [Google Scholar]
- Patterson AW, Wood WJ, Hornsby M, Lesley S, Spraggon G, Ellman JA. Identification of selective, nonpeptidic nitrile inhibitors of cathepsin s using the substrate activity screening method. J Med Chem. 2006;49:6298–6307. doi: 10.1021/jm060701s. [DOI] [PubMed] [Google Scholar]
- Pham CT, Ley TJ. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8627–8632. doi: 10.1073/pnas.96.15.8627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers JC, Asgian JL, Ekici OD, James KE. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- Ragheb D, Bompiani K, Dalal S, Klemba M. Evidence for catalytic roles for Plasmodium falciparum aminopeptidase P in the food vacuole and cytosol. J Biol Chem. 2009;284:24806–24815. doi: 10.1074/jbc.M109.018424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers WO, Sem R, Tero T, Chim P, Lim P, Muth S, Socheat D, Ariey F, Wongsrichanalai C. Failure of artesunate-mefloquine combination therapy for uncomplicated Plasmodium falciparum malaria in southern Cambodia. Malaria journal. 2009;8:10. doi: 10.1186/1475-2875-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roiko MS, Carruthers VB. New roles for perforins and proteases in apicomplexan egress. Cell Microbiol. 2009;11:1444–1452. doi: 10.1111/j.1462-5822.2009.01357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PJ. Cysteine proteases of malaria parasites. International journal for parasitology. 2004;34:1489–1499. doi: 10.1016/j.ijpara.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Rosenthal PJ, Sijwali PS, Singh A, Shenai BR. Cysteine proteases of malaria parasites: targets for chemotherapy. Curr Pharm Des. 2002;8:1659–1672. doi: 10.2174/1381612023394197. [DOI] [PubMed] [Google Scholar]
- Sijwali PS, Koo J, Singh N, Rosenthal PJ. Gene disruptions demonstrate independent roles for the four falcipain cysteine proteases of Plasmodium falciparum. Molecular and biochemical parasitology. 2006;150:96–106. doi: 10.1016/j.molbiopara.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Turk D, Janjic V, Stern I, Podobnik M, Lamba D, Dahl SW, Lauritzen C, Pedersen J, Turk V, Turk B. Structure of human dipeptidyl peptidase I (cathepsin C): exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 2001;20:6570–6582. doi: 10.1093/emboj/20.23.6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilairatana P, Krudsood S, Treeprasertsuk S, Chalermrut K, Looareesuwan S. The future outlook of antimalarial drugs and recent work on the treatment of malaria. Arch Med Res. 2002;33:416–421. doi: 10.1016/s0188-4409(02)00371-5. [DOI] [PubMed] [Google Scholar]
- Wongsrichanalai C, Pickard AL, Wernsdorfer WH, Meshnick SR. Epidemiology of drug-resistant malaria. Lancet Infect Dis. 2002;2:209–218. doi: 10.1016/s1473-3099(02)00239-6. [DOI] [PubMed] [Google Scholar]
- Wood WJ, Patterson AW, Tsuruoka H, Jain RK, Ellman JA. Substrate activity screening: a fragment-based method for the rapid identification of nonpeptidic protease inhibitors. J Am Chem Soc. 2005;127:15521–15527. doi: 10.1021/ja0547230. [DOI] [PubMed] [Google Scholar]
- Young JA, Fivelman QL, Blair PL, de la Vega P, Le Roch KG, Zhou Y, Carucci DJ, Baker DA, Winzeler EA. The Plasmodium falciparum sexual development transcriptome: a microarray analysis using ontology-based pattern identification. Molecular and biochemical parasitology. 2005;143:67–79. doi: 10.1016/j.molbiopara.2005.05.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.