

Abstract
Objective
Infiltration of the central nervous system (CNS) by leukemia is a problematic disease manifestation of acute lymphoblastic leukemia (ALL). The mechanisms by which leukocytes interact with brain-derived microvasculature endothelial cells (HBMEnd) and enter the CNS are largely derived from models of inflammation. However, our data indicate that ALL cells do not elicit an inflammatory phenotype by HBMEnd. Our current investigation focuses on the contribution of the unique co-expression of VE-cadherin and PECAM-1 by ALL in mediating leukemic cell interactions with HBMEnd as an in vitro model of the blood-brain-barrier.
Materials and Methods
Primary ALL and ALL cell lines were evaluated for VE-cadherin and PECAM-1 expression. Lentiviral-mediated transduction of VE-cadherin and PECAM-1 into REH cells and antibody neutralization of VE-cadherin and PECAM-1 in SUP-B15 cells was used to delineate the role of these two proteins in mediating ALL adhesion to and migration through HBMEnd monolayers.
Results
While cell line models indicate that VE-cadherin and PECAM-1 expression is found on the surface Ph+ ALL, evaluation of primary ALL demonstrates that VE-cadherin and PECAM-1 are expressed independent of Ph-status. Expression of VE-cadherin and PECAM-1 by ALL enhanced the adhesion of ALL to HBMEnd, while expression of PECAM-1 enhanced ALL adhesion to, and migration through, HBMEnd.
Conclusions
Expression of VE-cadherin and PECAM-1 by ALL cells positions them to interact with HBMEnd. By increasing our understanding of molecular mechanisms through which ALL cells gain entry into the CNS, new strategies may be designed to prevent leukemia cell entry into the CNS.
Introduction
Disease specific prognostic indicators, such as chromosomal translocations and other cytogenetic features, are used to stratify patients with ALL into risk groups for relapse and disease outcomes.[1] In addition to disease specific prognostic indicators, there are also anatomical sites that are therapeutically challenging. Relevant to the current study, infiltration of the CNS by leukemic cells contributes to relapse of disease and predicts poor disease outcome.[2, 3] Risk factors associated with the development of CNS leukemia include age with a higher incidence found in infants and young children, high leukocyte counts, and the presence of high-risk cytogenetics.[4] At diagnosis, less than 5% of children and less than 10% of adults with ALL present with CNS involvement. However, without prophylactic measures, as many as 50%-75% of children and 33% of adults with ALL would develop CNS manifestations.[4] The use of prophylaxis significantly decreases the rates of CNS involvement, but treatments targeted for action in the CNS produce unique toxicities including seizure, dementia, intellectual dysfunction, leukoencephalopathy, and growth retardations.[5, 6] While prophylaxis reduces the rate of CNS involvement, the implications of CNS directed therapeutic toxicities in a pediatric population, the persistence of CNS relapse in some patients despite prophylactic measures, and the dismal prognosis surrounding CNS relapse highlight the need to better understand the biology involved in the communication between ALL cells and the CNS.
Circulating leukemic cells are carried by the internal carotid arteries or the vertebral arteries to the blood-brain-barrier (BBB), the interface of general circulation and the CNS.[7] The BBB, which serves to isolate the parenchyma of the brain from general circulation and to tightly regulate movement of material into and out of the CNS, has classically been regarded as the most logical site for immune cells to enter the CNS.[8] The BBB is composed of microvascular endothelial cells joined together by relatively impermeable and highly developed tight and adherens junctions.[7, 8] Tight junctions are composed of transmembrane proteins, including occludin and claudin-5, which interact homotypically with adjacent endothelial cells and are linked to the cytoskeleton through the ZO family of proteins.[9, 10] The transmembrane proteins of adherens junctions, VE-cadherin and PECAM-1, also bind homotypically to adjacent endothelial cells and are linked to the cytoskeleton through beta-catenin.[9] Together these structures form the anatomical basis of the BBB, which restrict the paracellular migratory pathway for circulating cells into the CNS.[7, 11]
Much of what is known about leukocyte migration into the CSN was discovered using the murine experimental autoimmune encephalomyelopathy model of human multiple sclerosis. In this model, self-reactive T-and B-lymphocytes as well as monocytes enter the CNS under inflammatory conditions.[12] Our data, however, indicate that the leukemic blasts of ALL do not induce the inflammatory phenotype of brain microvascular endothelial cells associated with classical extravasation. Based on these observations we have investigated migration of ALL across monolayers of brain-derived microvascular endothelial cells, focusing on the contribution of ALL VE-cadherin and PECAM-1 expression. Through the use of lentiviral-mediated expression of these two proteins and neutralization of protein function with specific antibodies, we demonstrate that expression of VE-cadherin and PECAM-1 by ALL confers an advantage to the leukemic cells with respect to adhering to, and migrating through, human brain derived microvascular endothelial cell monolayers.
Materials and Methods
Cell culture
The ALL cell lines JM-1 (CRL-10423), REH (CRL-8286), and SUP-B15 (CRL-1929) were obtained from ATCC (Manassas, VA). Nalm-27 cells were provided by the Fujisaki Cancer Center (Okayama, Japan). Leukemic cells were maintained at a density of 1×106 cells/mL in Iscove's DMEM (Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), 2 mM l-glutamine (Mediatech), 0.05 μM 2-mercaptoethanol (Sigma-Aldrich, St. Louis, Missouri), 100 U/mL penicillin (Sigma-Aldrich), and 0.1 mg/mL streptomycin (Sigma-Aldrich). Primary ALL cells included de-identified samples from leukaphoresis products, bone marrow aspirates, or cerebrospinal fluid as indicated. Mononuclear lymphocytes were isolated from all primary samples using Accu-Prep Lymphocytes (Accurate Chemical & Scientific Corporation, Westbury, NY) according to the manufacturer's instructions. Positively selected peripheral blood CD19+ B-cells from a healthy donor were purchased from AllCells (Emeryville, CA). Human brain-derived microvascular endothelial cells (HBMEnd) were obtained from Angioproteomie (Boston, MA) and maintained in complete EGM-2MV media (Lonza, Basel, Switzerland).
Production of stable VE-cadherin and PECAM-1 expressing REH
Cloning of human VE-cadherin (CDH5) into pLenti6.2-DEST/V5 (Invitrogen, Carlsbad, CA), as well as generation of the pLenti6.2-DEST/V5 empty vector control has been reported previously.[13] Human PECAM-1 full length cDNA in the pENTR vector was obtained from Invitrogen and cloned into pLenti6.2-DEST/V5 using Gateway cloning technology. Production of lentiviral particles and titer determination in this system has been previously described.[13] To generate REH cell lines stably expressing empty vector (REH VECT), VE-cadherin (REH CDH5), or PECAM-1 (REH PECAM1), cells were infected with the lentivirus particles with a multiplicity of infection of 1. Clones stably expressing the genes of interest were selected by blasticidin (3 μg/mL) and gene expression was confirmed by real-time reverse transcriptase (RT) PCR and flow cytometry.
Real-time RT-PCR
Total cellular RNA was isolated using the RNEasy RNA isolation kit (Qiagen, Valencia, CA). Real-time RT-PCR was performed using 50 ng RNA per reaction according to the manufacturer's specifications using the QuantiTech SYBR Green RT-PCR kit (Qiagen). Primers specific for human CDH5 and PECAM1 were obtained from SABiosciences (Frederick, MD). Primers specific for the housekeeping gene PPIA were from Real Time Primers, LLC (Elkins Park, PA). Samples were prepared in triplicate and analyzed using the Applied Biosystems 7500 Real-time PCR system (Foster City, CA). Relative gene expression was determined using the Comparative Ct method.[14]
Antibodies
The following antibodies were used for flow cytometry, confocal microscopy, and western blot analysis: rabbit IgG and mouse IgG1 isotypes (Southern Biotechnology Associates, Birmingham, AL), rabbit anti-human VE-cadherin (Axorra, San Diego, CA), mouse anti-human PECAM-1 (Santa Cruz Biotechnologies, Santa Cruz, CA), mouse anti-human GAPDH (Fitzgerald, Concord, MA) mouse anti-human ICAM-1 (R&D systems, Minneapolis, MN), and mouse anti-human VCAM-1 (BD Biosciences, San Jose, CA). Alexa Fluor conjugated secondary antibodies were obtained from Invitrogen. For neutralization experiments, mouse IgG1 isotype (Southern Biotechnology Associates), mIgG2a kappa (BD Pharmingen, San Diego, CA), mouse anti-human CD31 (PECAM-1, Ancell, Bayport, MN), and mouse anti-human VE-cadherin (clone BV9, Santa Cruz Biotechnologies) were utilized where indicated.
Immunofluorescence, flow cytometry, and confocal microscopy
Flow cytometric detection of cell surface adhesion molecules was carried out by incubating 1×106 cells with primary antibody (1 μg) or matched isotype control on ice for 20 min, washing with 1X phosphate buffered saline (PBS), and incubating with a fluorochrome-labeled secondary antibody (1 μg) on ice for an additional 20 min. Immunofluorescence was evaluated using the BD FACSCalibur flow cytometer and CellQuest Pro software (BD Biosciences). Data were analyzed using WinMidi software version 2.8. To determine expression and localization of proteins in HBMEnd, cells were grown to confluence on 0.2% gelatin coated glass coverslips. Samples were fixed in 4% paraformaldehyde and subsequently permeabilized with 0.5% Triton-X-100 at room temperature for 15 min. After blocking in 5% BSA/1X PBS, cells were incubated with primary antibody (1 μg) or matched isotype control antibody. Samples were washed in 1X PBS and incubated with a fluorochrome-labeled secondary antibody (1 μg). Coverslips were mounted on glass slides using ProLong Gold plus DAPI (Invitrogen). Confocal images were acquired using a Zeiss LSM510 confocal system connected to an AxioImage ZI microscope. The images were analyzed using the Zeiss LSM510 software (Carl Zeiss, Thornwood, NY). Images were prepared using Adobe Photoshop CS2 version 9.0.2 and Adobe Illustrator CS2 version 12.0.1.
Immunohistochemistry
High-grade lymphoma biopsy samples were formalin-fixed and paraffin-embedded. Five micron sections were stained using ducal breast carcinoma and tonsil as control tissue for VE-cadherin and PECAM-1, respectively. For the detection of VE-cadherin the primary antibody (mouse monoclonal anti-human VE-cadherin, clone BV6; Millipore Billerica, MA) was used at a dilution of 1:10 for 24 h. For the detection of PECAM-1 the primary antibody (mouse monoclonal anti-human PECAM-1, clone JC/70A; Abcam Cambridge, MA) was applied at a dilution of 1:25 for 6 h. The slides were then incubated with a conjugated secondary antibody and counterstained with hematoxylin. The slides were then analyzed using light microscopy and all images shown are 400×.
Antibody neutralization of VE-cadherin and PECAM-1
To inhibit the function of ALL expressed VE-cadherin and PECAM-1, Nalm-27 and SUP-B15 cells (1×106 cells/mL) were treated with (3 μg/mL) anti-VE-cadherin antibodies, anti-PECAM-1 antibodies, or matched isotype controls for 15 min prior to the initiation of incubation with HBMEnd and antibody remained in cultures for the duration of functional assays.
Leukemic cell adhesion assay
Leukemic cells were fluorescently labeled according to the manufacturer's specification using CellTracker Green CMFDA (Invitrogen). The fluorescently labeled cells (1×106 cells/mL in 500 μL of complete culture media) were incubated with confluent HBMEnd monolayers for 15 min (neutralization assay), 30 min, 1 h, or 4 h. Following incubations, the culture media was removed and wells were rinsed three times with 1X PBS. Samples were collected by trypsinization and the number of adherent leukemic cells was determined by flow cytometry by counting the number of fluorescent events collected during 30 s of high flow rate. Samples were evaluated in triplicate.
Transendothelial migration assay
HBMEnd were grown to confluence on 0.2% gelatin coated polycarbonate inserts of a 24-well transwell system (5 μm pore size, Corning, Lowell, MA). Fluorescently labeled leukemic cells (1×106 cells/mL in 150 μL media) were added to the top chamber of the transwell and allowed to migrate through the HBMEnd layer toward media supplemented with 100 ng/mL CXCL12 for 4 h. Samples were collected from the bottom chamber and were evaluated by flow cytometry. Migration is expressed as the number of fluorescent positive events acquired during 30 s of high flow rate. Samples were evaluated in triplicate.
Microarray analysis
Human extracellular matrix and adhesion molecule pathway-focused Hytube GEArrays and reagents were obtained from SABiosciences and used according to the manufacturer's instructions. RNA isolated from JM-1, REH, Nalm-27, and SUP-B15 cells was used to synthesize cDNA. cDNA (3 μg) was used to produce biotin-labeled cRNA, which was hybridized with microarray membranes overnight. The hybridized microarray membrane was incubated with alkaline phosphatase-streptavidin, followed by CDP-Star and subsequently exposed to x-ray film. The resulting images were analyzed using GEArray Expression Analysis Suite 2.0. Gene expression was normalized to GAPDH. (GEO accession: GSE18516)
Electric cell substrate impedance sensing (ECIS)
To evaluate changes in HBMEnd barrier function induced by ALL cells, ECIS assays were performed as described previously.[15] Briefly, HBMEnd were grown to confluence on gelatin-coated 8-well-10-electrode ECIS cultureware (Applied BioPhysics, Troy, NY). Two hours prior to the assay, HBMEnd culture media was replaced with leukemic cell culture media. Baseline resistance readings were collected from the HBMEnd monolayer for 30 min. REH, Nalm-27, or SUP-B15 cells were added to the HBMEnd monolayers at 1×106 cells/mL. Addition of media alone served as a negative control, while addition of thrombin (4U/mL, Enzyme Research Laboratories, South Bend, IN). Data were collected every 30 sec for 5 h and are indicated as resistance normalized to the initial resistance reading.
Statistical analysis
Where appropriate statistical significance was determined using Student t test and indicates p<0.05.
Results
ALL cells do not elicit an inflammatory response by HBMEnd
To evaluate potential inflammation-induced phenotypic changes caused by endothelial cell exposure to leukemia cells, immunofluorescent staining to detect endothelial cell VCAM-1 and ICAM-1 was performed. Upon treatment of HBMEnd with TNF-α, a known inflammatory cytokine, VCAM-1 and ICAM-1 expression is increased on the surface of HBMEnd (Figure 1A). Exposure of HBMEnd to REH, Nalm-27, or SUP-B15 cells does not result in upregulation of cell surface VCAM-1 or ICAM-1.
Figure 1. ALL does not elicit an inflammatory response by HBMEnd.
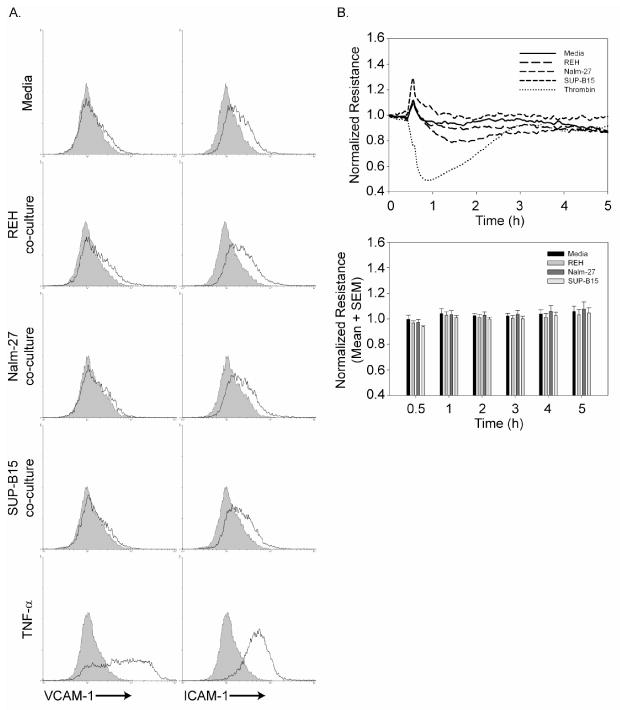
A. HBMEnd were grown to confluence in 6-well plates and exposed to ALL co-culture (1×106 leukemia cells/mL), media (negative control) or TNF-α (100ng/mL) for 4h. Following treatment, leukemia cells were rinsed away and samples were collected, immunostained with antibodies to detect VCAM-1 or ICAM-1 (solid line) or matched isotype control (shaded histogram), and analyzed by flow cytometry. B. (Top panel) HBMEnd were grown to confluence on ECIS electrodes (8-well, 10 electrode). After collecting baseline resistance data for 30 min, leukemia cells (1×106 cells/mL), media negative control, or thrombin positive control (4U/mL) were added to each ECIS well. Resistance data were collected for a total of 5h and are normalized to the initial resistance reading for each condition. (Bottom panel) Normalized resistance readings for each treatment are shown at discrete time points following addition of each condition. Data are expressed as mean normalized resistance+SEM, N=3.
As alteration in endothelial barrier integrity is a component of inflammatory models, we performed ECIS experiments to determine whether exposure of HBMEnd to REH, Nalm-27, and SUP-B15 cells disrupted endothelial barrier function. As expected, when HBMEnd are challenged with thrombin, a known vasoactive agent, barrier function as measured by normalized resistance decreases (Figure 1B, top). However, following exposure of HBMEnd to ALL cell lines, resistance across the HBMEnd monolayer does not decrease (Figure 1B) implying that ALL cells do not overtly disrupt HBMEnd barrier function. Taken together, these data show that ALL cells do not induce classical inflammatory changes in the endothelial cells including upreguation of VCAM-1 and ICAM-1 and disruption of endothelial barrier integrity.
ALL cells co-express the adherens junction proteins VE-cadherin and PECAM-1
Previously published observations from our laboratory have indicated that Ph+ ALL cell lines express the adhesion molecule VE-cadherin, while Ph- ALL cell lines do not.[13] The unique expression of this classical endothelial marker in a hematologic malignancy prompted us to further characterize the expression of adhesion molecules and extracellular matrix proteins by ALL cell lines. Using a pathway-focused cDNA microarray, we compared the expression of extracellular matrix proteins and adhesion molecule genes between the Ph- ALL cell lines JM-1 and REH and the Ph+ ALL cell lines Nalm-27 and SUP-B15. Consistent with the pattern of VE-cadherin expression in cell line models, another adherens junction protein, PECAM-1, was expressed to a greater extent by Nalm-27 and SUP-B15 than JM-1 or REH cells (Figure 2A). Gene expression results generated by the microarray were confirmed using real-time RT-PCR and cell surface immunofluorescent staining and demonstrate that VE-cadherin and PECAM-1 are expressed by Nalm-27 and SUP-B15 cells (Figure 2B-C).
Figure 2. Ph+ ALL cell lines co-express cell-surface VE-cadherin and PECAM-1.
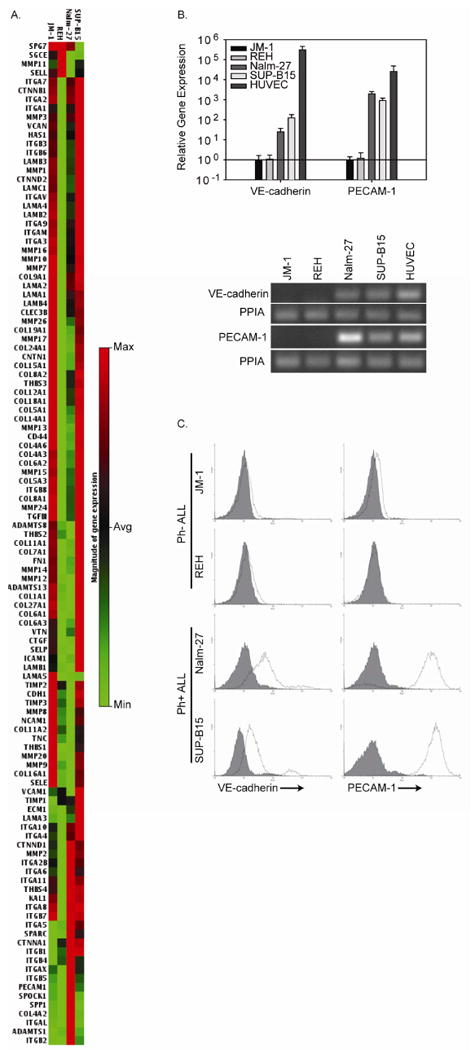
A. RNA isolated from the Ph-ALL cell lines, JM-1 and REH, and the Ph+ ALL cell lines, Nalm-27 and SUP-B15, was evaluated by a pathway specific cDNA microarray focused on expression of human extracellular matrix proteins and adhesion molecules. For each gene evaluated, the gene expression map compares gene expression across the cell lines evaluated and shows low level gene expression in light green and high level gene expression in red. B. (Top) RNA isolated from ALL cell lines was subject to real-time RT-PCR to confirm VE-cadherin and PECAM-1 expression. The expression level of VE-cadherin and PECAM-1 for each cell line is compared to the expression levels of JM-1 cells. (Bottom) Gel electrophoresis of the PCR products demonstrates that VE-cadherin and PECAM-1 is expressed by Nalm-27 and SUP-B15 cells with PPIA used as a loading control and HUVEC RNA used as a positive control. C. JM-1, REH, Nalm-27, and SUP-B15 cells were evaluated for cell surface VE-cadherin and PECAM-1 expression by immunostaining and flow cytometric analysis (solid line represents specific primary antibody while shaded histograms represents the isotype matched control).
While cell lines provide researchers with invaluable models for in vitro manipulations including gain-of-function and loss-of-function studies, we next sought to expand our understanding of VE-cadherin and PECAM-1 expression using primary ALL clinical samples. Primary ALL cells derived from leukaphoresis products and bone marrow aspirates were evaluated for VE-cadherin and PECAM-1 expression using real-time RT-PCR and cell surface immunofluorescent staining. In contrast to expression patterns observed in cell line models, VE-cadherin and PECAM-1 are expressed on the surface of both Ph- ALL and Ph+ ALL patient samples (Figure 3A and B). Flow cytometric evaluation of ALL cells derived from cerebrospinal fluid (CSF) demonstrates cell surface expression of VE-cadherin and PECAM-1 (Figure 3B). Peripheral blood CD19+ B-cells, which would be found in the circulation of healthy individuals, however, do not demonstrate cell surface VE-cadherin or PECAM-1 expression (Figure 3B). Consistent with peripheral B-cells, high-grade B-cell lymphomas also lacked expression of VE-cadherin and PECAM-1 (Figure 3C). As adherens junction proteins interact homotypically, demonstrating the unique cell surface co-expression of VE-cadherin and PECAM-1 by ALL cells prompted us to hypothesize that the expression of these two proteins by ALL may enhance the interaction between leukemia cells and HBMEnd that also express VE-cadherin and PECAM-1 (Figure 3D).
Figure 3. Primary ALL expresses cell surface VE-cadherin and PECAM-1.
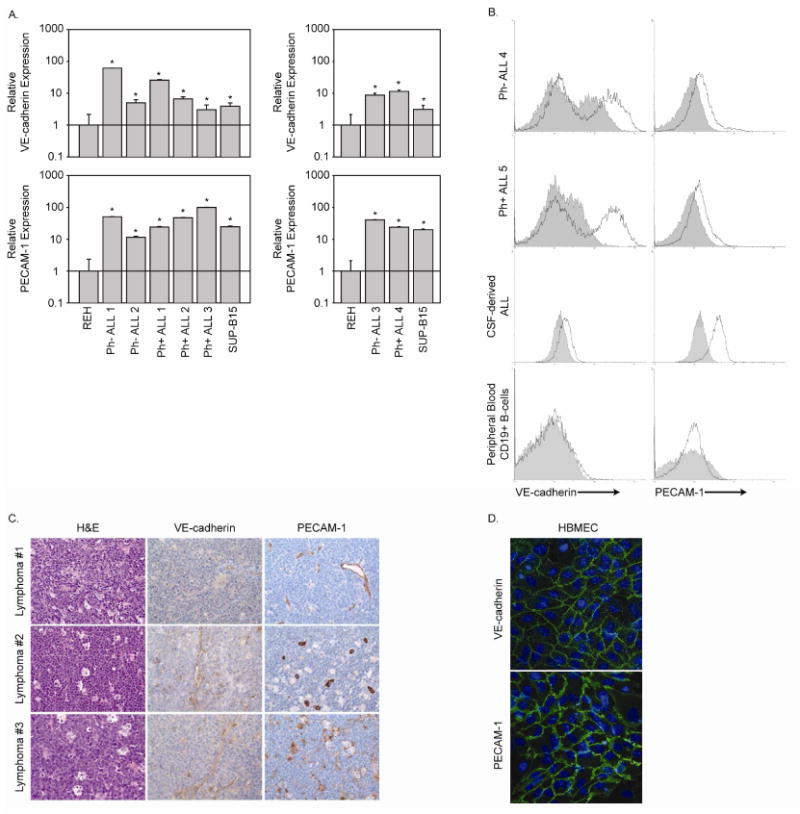
A. RNA from leukemic cells isolated from leukaphoresis (Ph- ALL1, Ph- ALL 2, Ph- ALL 3, and Ph+ ALL 1) and bone marrow aspirates (Ph+ ALL 2, Ph+ ALL 3, and Ph+ ALL 4) was examined for VE-cadherin and PECAM-1 expression by real-time RT-PCR. RNA from REH and SUP-B15 cells was included as negative and positive controls, respectively. Data are expressed as fold increase over the gene expression levels in REH cells. B. Leukemic cells derived from bone marrow aspirates (Ph- ALL 4 and Ph+ ALL 5) and cerebrospinal fluid (CSF) as well as peripheral blood CD19+ B-cells from a healthy donor were evaluated for cell surface VE-cadherin and PECAM-1 expression by immunostaining and flow cytometric analysis (solid line represents specific primary antibody while shaded histograms represents the isotype matched control). C. High-grade B-cell lymphoma biopsy samples were evaluated for VE-cadherin and PECAM-1 expression by immunohistochemistry. Samples were also stained with hematoxylin and eosin. Photomicrographs were taken at 400× magnification. D. HBMEnd grown to confluence on glass coverslips were fixed and immunostained to detect the adherens junction proteins VE-cadherin and PECAM-1. Cell nuclei were stained using DAPI.
Expression of VE-cadherin by ALL cells enhances leukemia cell adhesion to HBMEnd, while expression of PECAM-1 enhances leukemia cell adhesion to HBMEnd and tumor transendothelial migration
To examine the contribution of VE-cadherin and PECAM-1 to mediating ALL cell interaction with HBMEnd, REH cells were transduced with the human VE-cadherin gene (REH CDH5), PECAM-1 gene (REH PECAM1), or empty vector control (REH VECT) using lentiviral-mediated gene delivery. Flow cytometric analysis of immunofluorescent staining in Figure 4 demonstrates that transduction of REH cells with the VE-cadherin or PECAM-1 gene results in cell-surface expression of VE-cadherin or PECAM-1, respectively, while the empty vector control expresses neither protein. To evaluate the role of VE-cadherin and PECAM-1 expression in mediating ALL adhesion to HBMEnd, REH cells expressing VE-cadherin, PECAM-1, or the vector control were used in an adhesion assay. Compared to the REH cells transduced with the empty vector, REH CDH5 and REH PECAM1 cells were better able to adhere to HBMEnd at each of the time points evaluated (Figure 5A). In support of this, when Nalm-27 cells were treated with VE-cadherin (Figure 5B) or PECAM-1 (Figure 5C) neutralizing antibodies their ability to adhere to HBMEnd was decrease compared to cells treated with isotype control antibodies by 24.7% and 33.8%, respectively. Treatment of SUP-B15 cells with neutralizing antibodies against VE-cadherin (Figure 5B) and PECAM-1 (Figure 5C) resulted in inhibition of adhesion by 26.6% and 27.4%, respectively. Combining VE-cadherin and PECAM-1 inhibition by treating Nalm-27 cells with both neutralizing antibodies only modestly reduced the adhesion over single inhibition (38.9% vs. 24.7% and 33.8%, data not shown). Interestingly, when these same cells were compared using a different functional readout, transendothelial migration (TEM), the REH PECAM1 cells were better able to cross HBMEnd monolayers than REH VECT or REH CDH5 cells (Figure 6A) and only neutralization of PECAM-1 was able to blunt SUP-B15 migration (Figure 6B). Taken together these data suggest that the expression of VE-cadherin and PECAM-1 by ALL cells can enhance the ability of the tumor to interact with HBMEnd.
Figure 4. Lentiviral mediated transduction of human CDH5 or PECAM1 into REH cells results in surface expression of VE-cadherin and PECAM-1, respectively.
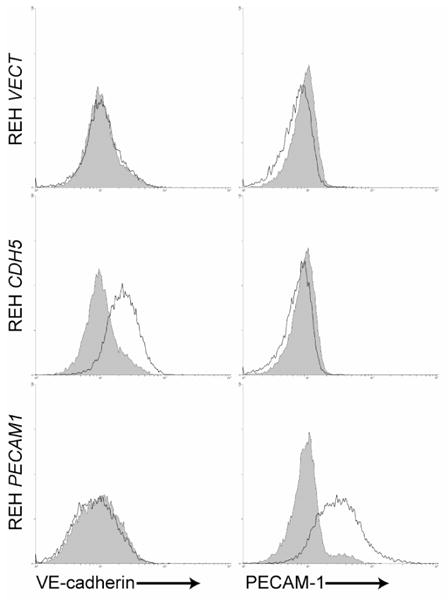
REH VECT, REH CDH5, and REH PECAM-1 were immunostained with antibodies specific for VE-cadherin or PECAM-1 (solid lines) or matched isotype control antibodies (shaded histograms). Samples were evaluated using flow cytometry.
Figure 5. ALL expression of VE-cadherin and PECAM-1 enhances leukemia cell adhesion to HBMEnd.
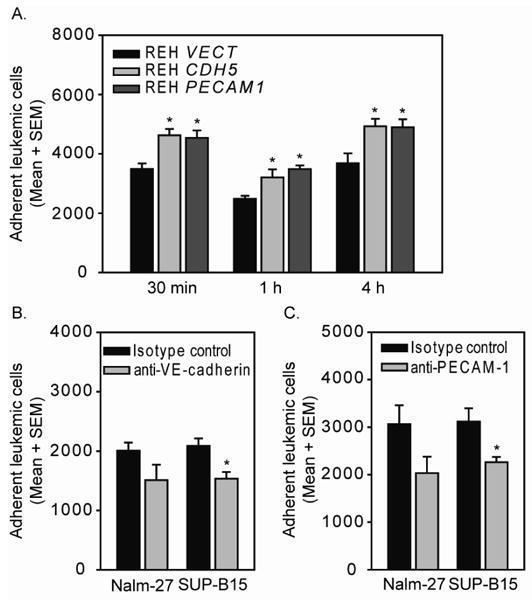
A. REH VECT, REH CDH5, and REH PECAM1 were labeled with CellTracker green dye then incubated HBMEnd for the indicated times. B and C. Nalm-27 and SUP-B15 cells were labeled with CellTracker green dye then treated with neutralizing antibodies against VE-cadherin (B), PECAM-1 (C), or matched isotype control antibodies for 15 min. Following this pretreatment, Nalm-27 and SUP-B15 cells were incubated with HBMEnd for 15 min. Following incubation, non-adherent leukemia cells were removed and the remaining adherent population was recovered by trypsinization. The number of adherent leukemic cells was determined using flow cytometry by collecting the number of fluorescently labeled events that occurred during 30 s of high flow rate. Data are expressed as mean number of adherent cells + standard error of the mean (SEM, N=3) and are representative of at least three independent experiments. *p<0.05
Figure 6. ALL expression of PECAM-1 promotes leukemia cell migration through HBMEnd monolayers.
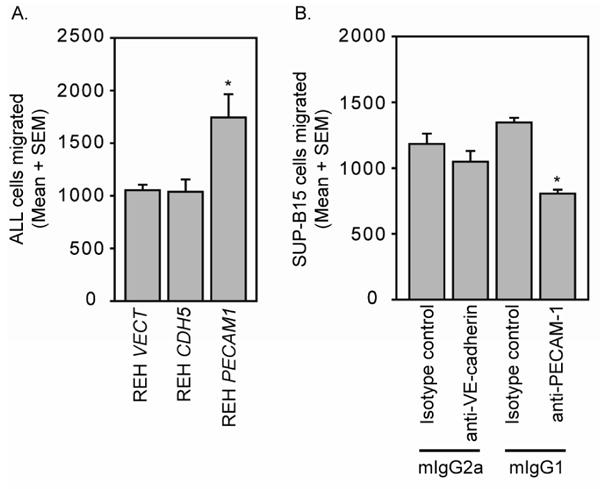
HBMEnd were grown to confluence on transwell inserts. A. REH VECT, REH CDH5, and REH PECAM1 were labeled with CellTracker green dye then added to the top chamber of the transwell system and allowed to migrate for 4h through the HBMEnd toward SDF-1 (100ng/mL). B. SUP-B15 cells were labeled with CellTracker green dye then treated with neutralizing antibodies against VE-cadherin, PECAM-1, or matched isotype control antibodies for 15 min. Following this pretreatment the cells were added to the top chamber of the transwell system and allowed to migrate for 4h through the HBMEnd toward SDF-1. Samples were collected and enumerated using flow cytometry by collecting fluorescently labeled events in 30 s of high flow rate. Data are expressed as mean number of cells migrated + standard error of the mean (SEM, N=3) and are representative of at least three independent experiments. *p<0.05
Discussion
In the current study, we investigated the interaction between ALL and HBMEnd, an in vitro model of the BBB, to understand the functional significance of coincident VE-cadherin and PECAM-1 expression by ALL. In vitro models of the BBB have been used with success to elucidate mechanisms of immune cell and tumor cell adhesion to brain microvascular endothelial cells and TEM.[16-18] Consistent with the phenotype of endothelial cells widely used to represent the BBB, the low passage HBMEnd used in our current studies express tight junction markers, including occludin, claudin-5, and ZO-1 (data not shown), as well as the adherens junction proteins VE-cadherin and PECAM-1.[19] This particular model system, combined with lentiviral-mediated expression of VE-cadherin and PECAM-1 and the use of specific neutralizing antibodies for VE-cadherin and PECAM-1 elucidated the specific role of each of these proteins in mediating ALL cell interaction with HBMEnd.
It is recognized that, when compared with other hematologic malignancies such as acute non-lymphoblastic leukemia, Hodgkin lymphoma, non-Hodgkin lymphoma, and AML, ALL is marked by more prevalent involvement of the CNS.[13, 20, 21] As adhesion of a circulating leukemic cell to the endothelial cells that compose the BBB may represent the first step of invasion into the CNS and models of immune cell invasion of the CNS are derived from inflammatory settings, we first explored the role of inflammation in our model. While exposure of HBMEnd to TNF-α and thrombin increased ICAM-1 and VCAM-1 expression and disrupted endothelial barrier resistance as is seen in inflammation, respectively, exposure of HBMEnd to REH, Nalm-27, or SUP-B15 cells did not promote upregulation of HBMEnd ICAM-1 or VCAM-1 and disrupt barrier function as measured by ECIS (Figure 1A and B). Collectively, these observations suggest that interaction of ALL cells with HBMEnd may be distinct from signaling that is central to inflammatory dogma.
Based on our observation that induction of adhesion molecules that are typically increased subsequent to inflammation did not occur following interaction of ALL cells with endothelial cells, we explored adhesion molecules expressed constitutively by ALL cell lines that could enhance leukemic cell adhesion to HBMEnd. Evaluation of primary ALL samples, including leukemic cells isolated from CSF, demonstrated that VE-cadherin and PECAM-1 are co-expressed on the tumor cell surface (Figure 3A and B) in contrast to high-grade B-cell lymphoma (Figure 3C) which was negative for both proteins of interest. Several reports have documented that VE-cadherin and PECAM-1 are expressed early in normal B-cell development with both down-regulated through maturation.[22-24] Therefore, while VE-cadherin and PECAM-1 may be expressed on healthy, or malignant, pro-pre-B lineage cells resident in the bone marrow, the only circumstance in which co-expression of these proteins would be expected in a peripherally circulating hematopoietic cell of comparable differentiation stage, pro- or pre-B, would be unique to patients with leukemia. Subsequently, the circumstance in which co-expression of these proteins is likely to facilitate CNS infiltration of a hematopoietic cell in the circulation is quite limited and likely unique to malignant progenitor cells. Consistent with earlier studies, we confirmed that these two proteins are not expressed on peripheral blood CD19+ B-cells from a healthy donor and are not expressed by more mature B-cell neoplasm (Figure 3B and C). The lack of co-expression of VE-cadherin and PECAM-1 on mature B lineage cells that are routinely in the periphery of healthy individuals again suggests the phenotype is restricted and does not represent a common mechanism by which healthy mature B-cells interact with endothelial barriers. Based on the classical role of VE- cadherin and PECAM-1 mediating homotypic interactions between adjacent endothelial cells, we hypothesized that expression of these two proteins by ALL cells would enhance their interaction with HBMEnd.
The expression of VE-cadherin in non-endothelial cells has been documented in fetal cytotrophoblast cells, as well as several cancers, where it is notably associated with an aggressive phenotype.[25-28] Through the study of human placental development, Zhou et. al. described that as cytotrophoblasts differentiate, they adopt a vascular phenotype, which includes the expression of VE-cadherin.[25] Subsequent studies by Bulla et. al. demonstrated that VE-cadherin was required for cytotrophoblast adhesion to and invasion into endothelial cell monolayers. In the setting of cancer, work by Hendrix et. al. has shown that VE-cadherin is expressed by aggressive melanoma and that its expression contributes to vascular mimicry.[27] Of note then are the observations that aggressive melanoma and gestational trophoblastic neoplasia are marked by frequent CNS metastasis.[29, 30] Concordant with the findings of Bulla et. al., forced expression of VE-cadherin in REH cells resulted in enhanced adhesion to HBMEnd, while neutralization of endogenous VE-cadherin resulted in diminished adhesion of Nalm-27 and SUP-B15 cells to HBMEnd (Figure 5).
Like VE-cadherin, the expression of PECAM-1 has also been documented during differentiation of cytotrophoblasts and development of the placenta.[25] In a study by Bulla et. al., cytotrophoblast treatment with a neutralizing antibody to PECAM-1 resulted in modestly diminished adhesion to endothelial cells and decreased transendothelial migration.[26] In addition to its role in normal physiology, the expression of PECAM-1 has been documented in various cancer models and has been implicated in mediating tumor cell adhesion to endothelial cells.[31, 32] We also observed enhanced adhesion and transendothelial migration following lentiviral mediated expression of PECAM-1 in REH cells, while neutralization of PECAM-1 resulted in diminished ALL adhesion and transendothelial migration (Figure 6). These findings are consistent with the documented necessity for PECAM-1 in mediating leukocyte transendothelial migration.[33-35]
In conclusion, we have demonstrated that subsets of ALL cells have the potential to express a unique combination of the adherens junction proteins, VE-cadherin and PECAM-1. Overexpression of VE-cadherin and PECAM-1 enhances the interaction of ALL cells with HBMEnd with VE-cadherin and PECAM-1 increasing adhesion and PECAM-1 augmenting adhesion and transendothelial migration. Cancers having central nervous system involvement place patients at high risk for poor disease outcomes. In the setting of ALL, implementation of CNS-directed prophylaxis, including chemotherapy and radiotherapy, dramatically enhanced patient survival, however not without associated toxicities.[1, 5, 6] Therefore, understanding the molecular mechanisms through which ALL cells initially gain entry into the CNS remains invaluable for designing strategies to prevent leukemia cell entry into the CNS to minimize the need to treat aggressive leukemia in this unique anatomical site.
Acknowledgments
The authors acknowledge support of this project by the WVU Microscope Imaging Facility (P20 RR016440) and thank Dr. Kathy Brundage for her assistance with experiments completed in the West Virginia University Flow Cytometry Core Facility (P20 RR016440 and S10 RR020866). This work was supported in part by NIH R01 HL056888 (LFG), NIH R01 CA134573 (LFG), and P20 RR016440 (LFG).
Footnotes
Conflict of Interest Disclosure: No financial interest/relationships with financial interest relating to the topic of this article have been declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pui CH, Evans WE. Acute lymphoblastic leukemia. N Engl J Med. 1998;339:605–615. doi: 10.1056/NEJM199808273390907. [DOI] [PubMed] [Google Scholar]
- 2.Pui CH. Central nervous system disease in acute lymphoblastic leukemia: prophylaxis and treatment. Hematology Am Soc Hematol Educ Program. 2006:142–146. doi: 10.1182/asheducation-2006.1.142. [DOI] [PubMed] [Google Scholar]
- 3.Lazarus HM, Richards SM, Chopra R, et al. Central nervous system involvement in adult acute lymphoblastic leukemia at diagnosis: results from the international ALL trial MRC UKALL XII/ECOG E2993. Blood. 2006;108:465–472. doi: 10.1182/blood-2005-11-4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cortes J. Central nervous system involvement in adult acute lymphocytic leukemia. Hematol Oncol Clin North Am. 2001;15:145–162. doi: 10.1016/s0889-8588(05)70203-3. [DOI] [PubMed] [Google Scholar]
- 5.Butler RW, Haser JK. Neurocognitive effects of treatment for childhood cancer. Ment Retard Dev Disabil Res Rev. 2006;12:184–191. doi: 10.1002/mrdd.20110. [DOI] [PubMed] [Google Scholar]
- 6.Cole PD, Kamen BA. Delayed neurotoxicity associated with therapy for children with acute lymphoblastic leukemia. Ment Retard Dev Disabil Res Rev. 2006;12:174–183. doi: 10.1002/mrdd.20113. [DOI] [PubMed] [Google Scholar]
- 7.Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3:569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- 8.Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 2005;26:485–495. doi: 10.1016/j.it.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 10.Vorbrodt AW, Dobrogowska DH. Molecular anatomy of interendothelial junctions in human blood-brain barrier microvessels. Folia Histochem Cytobiol. 2004;42:67–75. [PubMed] [Google Scholar]
- 11.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 12.Hickey WF. Migration of hematogenous cells through the blood-brain barrier and the initiation of CNS inflammation. Brain Pathol. 1991;1:97–105. doi: 10.1111/j.1750-3639.1991.tb00646.x. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, O'Leary H, Fortney J, Gibson LF. Ph+/VE-cadherin+ identifies a stem cell like population of acute lymphoblastic leukemia sustained by bone marrow niche cells. Blood. 2007;110:3334–3344. doi: 10.1182/blood-2007-01-068122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 15.Xu M, Waters CL, Hu C, Wysolmerski RB, Vincent PA, Minnear FL. Sphingosine 1-phosphate rapidly increases endothelial barrier function independently of VE-cadherin but requires cell spreading and Rho kinase. Am J Physiol Cell Physiol. 2007;293:C1309–C1318. doi: 10.1152/ajpcell.00014.2007. [DOI] [PubMed] [Google Scholar]
- 16.Alter A, Duddy M, Hebert S, et al. Determinants of human B cell migration across brain endothelial cells. J Immunol. 2003;170:4497–4505. doi: 10.4049/jimmunol.170.9.4497. [DOI] [PubMed] [Google Scholar]
- 17.Man S, Ubogu EE, Williams KA, Tucky B, Callahan MK, Ransohoff RM. Human brain microvascular endothelial cells and umbilical vein endothelial cells differentially facilitate leukocyte recruitment and utilize chemokines for T cell migration. Clin Dev Immunol. 2008;2008:384982. doi: 10.1155/2008/384982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li B, Zhao WD, Tan ZM, Fang WG, Zhu L, Chen YH. Involvement of Rho/ROCK signalling in small cell lung cancer migration through human brain microvascular endothelial cells. FEBS Lett. 2006;580:4252–4260. doi: 10.1016/j.febslet.2006.06.056. [DOI] [PubMed] [Google Scholar]
- 19.Weksler BB, Subileau EA, Perriere N, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 20.Law IP, Blom J. Adult acute leukemia: frequency of central system involvement in long term survivors. Cancer. 1977;40:1304–1306. doi: 10.1002/1097-0142(197709)40:3<1304::aid-cncr2820400346>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 21.Pfeifer H, Wassmann B, Hofmann WK, et al. Risk and prognosis of central nervous system leukemia in patients with Philadelphia chromosome-positive acute leukemias treated with imatinib mesylate. Clin Cancer Res. 2003;9:4674–4681. [PubMed] [Google Scholar]
- 22.Kim I, Yilmaz OH, Morrison SJ. CD144 (VE-cadherin) is transiently expressed by fetal liver hematopoietic stem cells. Blood. 2005;106:903–905. doi: 10.1182/blood-2004-12-4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baumann CI, Bailey AS, Li W, Ferkowicz MJ, Yoder MC, Fleming WH. PECAM-1 is expressed on hematopoietic stem cells throughout ontogeny and identifies a population of erythroid progenitors. Blood. 2004;104:1010–1016. doi: 10.1182/blood-2004-03-0989. [DOI] [PubMed] [Google Scholar]
- 24.Jackson DE, Gully LM, Henshall TL, Mardell CE, Macardle PJ. Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) is associated with a naive B-cell phenotype in human tonsils. Tissue Antigens. 2000;56:105–116. doi: 10.1034/j.1399-0039.2000.560201.x. [DOI] [PubMed] [Google Scholar]
- 25.Zhou Y, Fisher SJ, Janatpour M, et al. Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J Clin Invest. 1997;99:2139–2151. doi: 10.1172/JCI119387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bulla R, Villa A, Bossi F, et al. VE-cadherin is a critical molecule for trophoblast-endothelial cell interaction in decidual spiral arteries. Exp Cell Res. 2005;303:101–113. doi: 10.1016/j.yexcr.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 27.Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Molecular plasticity of human melanoma cells. Oncogene. 2003;22:3070–3075. doi: 10.1038/sj.onc.1206447. [DOI] [PubMed] [Google Scholar]
- 28.Labelle M, Schnittler HJ, Aust DE, et al. Vascular endothelial cadherin promotes breast cancer progression via transforming growth factor beta signaling. Cancer Res. 2008;68:1388–1397. doi: 10.1158/0008-5472.CAN-07-2706. [DOI] [PubMed] [Google Scholar]
- 29.Marchetti D, Denkins Y, Reiland J, et al. Brain-metastatic melanoma: a neurotrophic perspective. Pathol Oncol Res. 2003;9:147–158. doi: 10.1007/BF03033729. [DOI] [PubMed] [Google Scholar]
- 30.Rustin GJ, Newlands ES, Begent RH, Dent J, Bagshawe KD. Weekly alternating etoposide, methotrexate, and actinomycin/vincristine and cyclophosphamide chemotherapy for the treatment of CNS metastases of choriocarcinoma. J Clin Oncol. 1989;7:900–903. doi: 10.1200/JCO.1989.7.7.900. [DOI] [PubMed] [Google Scholar]
- 31.Aroca F, Renaud W, Bartoli C, Bouvier-Labit C, Figarella-Branger D. Expression of PECAM-1/CD31 isoforms in human brain gliomas. J Neurooncol. 1999;43:19–25. doi: 10.1023/a:1006233816724. [DOI] [PubMed] [Google Scholar]
- 32.Tang DG, Chen YQ, Newman PJ, et al. Identification of PECAM-1 in solid tumor cells and its potential involvement in tumor cell adhesion to endothelium. J Biol Chem. 1993;268:22883–22894. [PubMed] [Google Scholar]
- 33.Rijcken E, Mennigen RB, Schaefer SD, et al. PECAM-1 (CD 31) mediates transendothelial leukocyte migration in experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G446–G452. doi: 10.1152/ajpgi.00097.2007. [DOI] [PubMed] [Google Scholar]
- 34.Duncan GS, Andrew DP, Takimoto H, et al. Genetic evidence for functional redundancy of Platelet/Endothelial cell adhesion molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J Immunol. 1999;162:3022–3030. [PubMed] [Google Scholar]
- 35.Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178:449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]