

Abstract
A major question for gene therapy in brain concerns methods to administer therapeutic genes in a uniform manner over major portions of the brain. A second question in neuroimmunology concerns the extent to which monocytes migrate to the CNS in degenerative disorders. Here we show that CD11b+ cells (largely monocytes) isolated from the bone marrow of GFP (green fluorescent protein)-expressing donors spontaneously home to compacted amyloid plaques in the brain. Injections of these cells as a single pulse show a rapid clearance from circulation (90 min half-life) and tissue residence half-lives of ∼3 d. The uptake into brain was minimal in nontransgenic mice. In transgenic mice containing amyloid deposits, uptake was dramatically increased and associated with a corresponding decrease in monocyte uptake into peripheral organs compared to nontransgenic littermates. Twice weekly infusions of the CD11b+ bone marrow cells transfected with a genetically engineered form of the protease neprilysin completely arrest amyloid deposition in an aggressively depositing transgenic model. Exploiting the natural homing properties of peripherally derived blood cells to deliver therapeutic genes has the advantages of access to the entire CNS, expression largely restricted to sites of injury, low risk of immune reactivity, and fading of expression if adverse reactions are encountered. These observations support the feasibility of testing autologous monocytes for application of therapeutic genes in human CNS disease. Moreover, these data support the results from bone marrow grafts that circulating CD11b+ cells can enter the CNS without requiring the use of lethal irradiation.
Introduction
The role of microglia/macrophages in the amyloid pathology of Alzheimer's disease (AD) is an area of intense investigation (Morgan et al., 2005; Wyss-Coray, 2006). While inflammation associated with the amyloid deposits in AD brain may contribute to the pathogenesis of the disease (Akiyama et al., 2000), macrophages in vitro avidly digest amyloid fibrils (Paresce et al., 1996; Webster et al., 2000). A number of recent studies present evidence that grafted monocytes home to plaques in mouse models of amyloid deposition (Malm et al., 2005; Stalder et al., 2005) and can restrict amyloid deposition, most probably by phagocytosis and digestion of amyloid fibrils (Simard et al., 2006; El Khoury et al., 2007; Takata et al., 2007; Town et al., 2008). However, it has been argued that studies labeling CNS infiltrating monocytes with bone marrow grafts may be confounded by CNS damage produced by the irradiation used to deplete the recipient's bone marrow (Ladeby et al., 2005; Ajami et al., 2007; Mildner et al., 2007). This grafting method of labeling circulating monocytes with grafted donor bone marrow may, thus, exaggerate the true extent of monocyte infiltration.
A separate issue is the challenging problem of exploiting gene therapy to treat neurodegenerative disorders such as Alzheimer's disease. Even using advanced methods such as convection-enhanced delivery (Krauze et al., 2005), it is rare that a gene therapy vector can transfect the entire cerebral cortex of a mouse, a region 1/3000 the size of the human cortex (Burger et al., 2005; Cearley and Wolfe, 2006; Carty et al., 2008). For almost a decade, the potential use of circulating monocytes to home to sites of CNS pathology has been proposed as a solution to this gene delivery problem (Imai et al., 1999). However, to date the utility of this approach to gene therapy for brain disorders has not been demonstrated. The data presented here support the concept that circulating monocytes, and possibly other blood cells, do home to amyloid plaques in the brain and that they can be used as effective gene therapy vectors.
Materials and Methods
Mice.
Doubly transgenic amyloid precursor protein (APP) + presenilin-1 (PS1) mice that are a cross between the mAPP transgenic line Tg2576 (Hsiao et al., 1996) and the mPS1 transgenic line 5.1 (Duff et al., 1996) were used in these studies. This breeding produces both APP+PS1 mice and nontransgenic mice (littermates) used in this study. The mice used in the green fluorescent protein (GFP) transgenic mouse model for bone marrow donors were from The Jackson Laboratory [C57BL/6-Tg(UBC-GFP)30Scha/J (stock #004353)]. These transgenic mice express the GFP under the direction of the human ubiquitin C promoter. Sixteen-month-old transgenic and nontransgenic mice were used for the single injection time course study and 9-month-old APP+PS1 mice were used for the 2 month multiple injection study. All mice were bred and maintained in our animal facility according to institutional guidelines.
Adoptive transfer of monocytes.
CD11b+ cells were collected using minor variations of previously published protocols (Wu et al., 2006; Wang et al., 2008). Transgenic GFP mice were overdosed with pentobarbital. GFP donor mice were killed and their femurs and tibias removed aseptically. Femur and tibia marrow cavities were flushed with RPMI media containing fetal bovine serum (FBS) and HEPES, pH 7.4, using a 25 gauge needle. Single-cell suspensions were prepared by repeat pipetting and the cell preparations passed through a 70 μm nylon mesh to remove particulate matter. Three milliliters per mouse of 1× RBC lysis buffer was added to the cells and incubated at room temperature for 5 min before adding cold PBS. Cells were centrifuged, washed twice in RPMI, and counted using a hemocytometer.
CD11b+ bone marrow cells were collected using Miltenyi Biotec's LS columns and MidiMacs magnet following the manufacturer's instructions. Briefly, 100–150 million bone marrow cells from transgenic mice ubiquitously expressing GFP were suspended in 2.7 ml of PBS+0.5% BSA and incubated for 15 min together with CD11b antibody conjugated to magnetic microbeads at 4°C (Miltenyi Biotec, catalog #130-049-601). These beads can isolate cells while binding only a fraction of the antigenic sites. The cell suspension was applied to the supplied column in a magnetic field and the CD11b+ fraction separated from the unlabeled cells by washing three times with 3 ml of buffer. The column was separated from the magnet and CD11b+ cells were collected.
The purity of immunomagnetically separated and GFP-transfected cells were analyzed using a FACScan (Becton Dickinson) equipped with a 488 nm argon laser. The bone marrow cells from non-GFP C57BL/6 mice were harvested as above. To 107 total cells containing 10 μl of CD11b microbeads (incubated for 15 min at 4°C), we then added 10 μl of anti-CD11b-FITC (a fluorochrome-conjugated antibody, Miltenyi Biotec, #130-081-201) and incubated for 5 min at 4°C. Cells were washed by adding 2 ml of buffer (PBS+0.5% BSA) and centrifuged at 300–400 × g for 10 min. Cells were resuspended in 500 μl of buffer and transported to Flow Cytometry core for analysis.
Based upon the flow cytometry information, 5 × 106 freshly isolated CD11b+ (GFP) cells were then resuspended in 100 μl of saline and injected into the left heart ventricle for the single injection time course studies.
Neprilysin constructs.
Endogenous NEP is a membrane-bound ectoprotease (native neprilysin or NEP-n). Because Aβ accumulates extracellularly in AD, we hypothesized that a secreted diffusible form of the protease would be most effective in degrading Aβ. Consequently, the membrane binding domain was deleted, leaving base pairs 232–2313, and a GDNF signal sequence triggering secretion was added to the construct (NEP-s). As a control, a construct containing neprilysin with a single-point mutation at the catalytic site (E585V) precluding proteolysis was also generated (NEP-m). A hemagglutinin (HA) tag was appended to each construct.
Proteolytic activity of three different NEP constructs was adapted from a previously published fluorometric assay method (Johnson and Ahn, 2000) for a 96-well plate format with slight modifications. HEK 293 cells were transfected with NEP-n, NEP-m, and NEP-s plasmids and control cells were transfected with a GFP plasmid using Lipofectamine 2000 per Invitrogen protocol. Cells were harvested after 72 h and samples were centrifuged at 1000 × g at 4°C for 45 min using Beckman J6-HC Centrifuge (Beckman Instruments) to obtain a culture media fraction and a cell pellet. The cell pellets were resuspended in M-PER mammalian protein extraction reagent buffer (Thermo Scientific) to obtain a cell lysate. Protein concentration was determined from the cell media and cell lysate from each sample using a general BCA assay (Per Pierce Protocol). Aliquots of the cell media and membrane fractions (100 μg of protein) were incubated in NEP ELISA plate containing NEP capture antibody (R&D Systems). The plate was washed with PBS buffer and then incubated with 20 μm (final) of the fluorogenic peptide [(7-methoxycoumarin-4-yl) acetyl (MOCA)-Arg-Pro-Pro-Gly-Phe-Ser-Ala-Phe-Lys(Dnp)-OH, R&D Systems] in Tris-HCl buffer (sodium phosphate, pH 7.4, containing 0.1 m NaCl). MOCA-Arg-Pro-Pro-Gly-Phe-Ser-Ala-Phe-Lys(Dnp)-OH is efficiently quenched by resonance energy transfer to the dinitrophenyl group and the continuous fluorescent intensity is increased upon internal cleavage of the peptide (NEP cleavage between the Ala-Phe bond). The increased fluorescence produced from cleavage of the substrate was measured using a Molecular Devices fMax spectrofluorometer plate reader (MDS Analytical Technologies) with a 60 min time point to normalize independent experiments. A standard curve of MOCA was analyzed along with each assay. Values were calculated and expressed as RFU/min/ml protein.
Implantation of microvascular port.
The microvascular ports (Kent Scientific) are 9 × 9 × 3 mm polyurethane ports that were placed subcutaneously in the nape of the neck of a mouse and were connected via catheter to the right jugular vein. A skin incision was made over the area of the right jugular, a sterile trocar tunneled to the soft tissue between the scapula and an incision placed at the tip of the trocar. The vessel was dissected free from surrounding tissue. Proximal and distal ligatures were placed around the vessel. The distal ligature was closed, and a venotomy was performed and the catheter advanced into the jugular vein 5 mm. The proximal suture was placed around the catheter and secured. A subcutaneous pocket was be made between the scapulae, and the port was placed into the pocket. Both incisions were closed with suture. The catheter was flushed with 100 U/ml heparin in saline solution, approximately three times the dead space of catheter (30 μl). On the day of injection, mice were anesthetized with isoflurane, the catheter was cleared by inserting a 27 gauge needle through the skin into the port and injecting 30 μl of 100U/ml heparin in saline, followed by 100 μl of monocyte cell suspension, followed by another 30 μl of the heparin solution.
Blood collection for time course measurements in circulation.
Mice were anesthetized with pentobarbital and 1 ml of blood was drawn from each mouse via cardiac puncture. Ten milliliters of RBC lysis buffer (eBioscience, catalog #4333) was immediately added to the blood and incubated for 5 min. Each mouse was used for a single determination (we did not serially bleed the same animal). To stop the lysis reaction, 30 ml of 1× PBS was added and the cells were centrifuged at 300 × g at 4°C for 10 min. Cells were resuspended in PBS and evaluated for GFP-labeled cells by flow cytometry analysis.
Tissue collection and histochemical procedures.
On the day the mice were killed, mice were overdosed with pentobarbital (100 mg/kg). The brain, spleen, liver, and lung were removed and bisected, the right half was collected for flow cytometry (see below), and the left half was immersed in freshly prepared 4% paraformaldehyde in 100 mm PO4 buffer, pH 7.4. The organs were postfixed in paraformaldehyde for 24 h. The brain, liver, and spleen tissue were cryoprotected in a series of sucrose solutions, frozen, sectioned in the horizontal plane at 25 μm using a sliding microtome, and stored at 4°C in Dulbecco's PBS for immunocytochemistry and histology. The lung tissue was paraffin embedded before being sectioned using a rotary microtome.
Immunohistochemistry was performed on free-floating sections as described in detail previously (Gordon et al., 1997). A series of eight sections spaced ∼600 μm apart were incubated with primary antibody overnight at 4°C, then incubated in the biotinylated secondary antibody (2 h) followed by streptavidin-peroxidase. Peroxidase reactions consist of 1.4 mm diaminobenzidine with 0.03% hydrogen peroxide in PBS for 5 min.
Single and multiple immunofluorescent labeling were performed as follows: After incubation with the primary antibody, the free-floating sections were incubated for 2 h with the appropriate fluorophore-coupled secondary antibodies [AlexaFluor 594 (1:1500), AlexaFluor 488 (1:1500), AlexaFluor 350 (1:1500) (Invitrogen]. Sections were rinsed in Dulbecco's PBS and coverslipped with VECTASHIELD Mounting Medium.
The following primary antibodies were used for immunohistochemistry: CD11b (rat monoclonal anti-CD11b, Serotec), GFP (chicken anti-GFP, AbCam), 6E10 (mouse monoclonal, Covance), CD68 (rat monoclonal, Serotec), CD45 (rat anti-mouse, Abd Serotec), F4/80 (rat anti-mouse, Abd Serotec), Iba-1 (rabbit anti-IBA-1, WAKO), and hemagglutinin (mouse anti-HA rhodamine, Roche).
Congo red histology was performed using sections mounted on slides and air dried. Rehydrated sections were incubated in an alkaline alcoholic saturated sodium chloride solution (2.5 mm NaOH in 80% alcohol, freshly prepared) for 20 min, then incubated in 0.2% Congo red in alkaline alcoholic saturated sodium chloride solution (freshly prepared and filtered) for 30 min. Sections were rinsed through three rapid changes of 100% ethanol, cleared through three changes of xylene, then coverslipped with DPX.
Image and flow cytometry analysis.
Organs were dissociated by enzymatic digestion followed by centrifugation over a Percoll gradient. GFP fluorescence intensity was measured using a FACScalibur (BD) flow cytometer and analyzed using CellQuest software.
Immunohistochemistry and Congo red staining were quantified with Image Pro Plus (Media Cybernetics) image software. Segmentation of positive stain was performed using RGB identification. Sample numbers are randomized before the start of the tissue processing, and the code is broken only after the analysis is complete. Data were collected from both frontal cortex and hippocampus. All values obtained from a single mouse were averaged together to represent a single value for that animal. Statistical analysis was performed using ANOVA followed by Fischer's LSD post hoc means comparison test (Statview software from SAS).
Stereology.
Stereological analysis was performed as described by West et al. (1991). We used a rare event protocol in which all positive cells in the region analyzed were counted after confirming they were not visible on the upper surface of the section. N = (number of cells counted) × (1/ssf) × (1/asf) × (1/tsf), where ssf is the sampling fraction (i.e., a 1/6 series or 1/17; the fraction of the total sections used), asf is the area sampling fraction (in this case the entire region rather than a dissector), and tsf is the thickness sampling fraction.
Results
We purified CD11b+ bone marrow monocytes from donor mice ubiquitously expressing a GFP marker using magnetic cell sorting. Cells were injected into recipient mice using either intracardiac puncture for single injections, or subcutaneous vascular ports inserted into the jugular vein near the left atrium for repeated injections. In all cases we injected 5 × 106 cells in a volume of 100 μl. In Figure 1 A, blood was collected at multiple times from 5 min to 24 h after a single pulse of cells were injected. GFP+ cells were counted using flow cytometry. We found that the injected CD11b+ cells cleared rapidly from the circulation with a half-life of 90 min. Virtually all injected cells were cleared from the circulation by 24 h after the injection.
Figure 1.

Amyloid pathology increases the migration of infused GFP monocytes into brain. In A, 5 × 106 GFP bone marrow monocytes were injected into the left ventricle of the heart of nontransgenic mice. At the indicated time points, mice blood was collected and the number of GFP monocytes in the blood was measured by flow cytometry (n = 4 at each time point). In B and C, mice were infused with 5 × 106 GFP+ bone marrow monocytes into the left ventricle of the heart, and organs were collected 1, 3, or 7 d following the injection. The organs were bisected and the number of monocytes present in each organ was quantified using flow cytometry (B) or stereology (C). n = 4 for each organ. D–H are representative micrographs of GFP staining in the tissues indicated above each panel. *p < 0.05 by t test. Scale bar, 25 μm.
To identify whether the injected CD11b+ cells migrated to the CNS, we compared the tissue distribution of GFP+ cells after the infusion into both nontransgenic mice and mice with amyloid deposits in their brain [16-month-old APP+PS1 transgenic mice (Holcomb et al., 1998)]. Following exsanguination with saline, the numbers of GFP-labeled cells in liver, spleen, lung, and brain were estimated at 1, 3, and 7 d both by flow cytometry of cell suspensions and by histological cell counts using stereology (Fig. 1 B,C). Nontransgenic mice had few GFP-labeled cells in brain measured either by flow cytometry or stereology (Fig. 1 D,E), yet APP+PS1 transgenic mouse brain had concentrations of GFP-labeled cells in the similar numbers as peripheral organs (liver, spleen, lung). In most peripheral organs (Fig. 1 B,C,F–H), the migration of labeled CD11b+ cells in the transgenic and nontransgenic mice were comparable, with the exception of the liver where a slight but statistically significant reduction of infiltrating cells was detected in transgenic mice, possibly due to increased competition for the circulating cells by the brain tissue. In all tissues, the half-life of the labeled cells found within organs was several days, with only a few cells detected 1 week after the injection.
In the brain, the presence of amyloid deposits greatly enhanced the infiltration of GFP+ cells into the CNS. We found that the GFP-labeled cells homed to the immediate vicinity of the compacted amyloid deposits. The majority of plaques in mice administered twice weekly injections for 2 months had multiple GFP-labeled cells in the surrounding region. We found very few, if any, labeled cells in brain regions that lacked amyloid deposits. Virtually all of these GFP+ cells expressed CD11b (Fig. 2 A–C). A second marker, CD68, expressed by phagocytic cells, labeled largely those microglia/macrophages in the immediate vicinity of the plaque (Fig. 2 E). Double-labeling studies found that some, but not all of the GFP-labeled cells coexpressed CD68, in particular those GFP+ cells that were less ramified tended to coexpress this marker (Fig. 2 F). No GFP signal could be found in APP+PS1 mice that were not injected with labeled monocytes (Fig. 2 G–I).
Figure 2.

GFP-positive monocytes concentrate near plaques in APP+PS1 transgenic animals and exhibit microglial/macrophage phenotype. Imaging of GFP-positive cells was performed using confocal microscopy. Brain sections from APP+PS1 mice collected 1 d after the last of 16 twice weekly injections were stained with fluorescent markers to detect the location and phenotype of the injected monocytes and amyloid plaques. Almost all GFP cells colocalized with CD11b (A–C). Ameboid GFP cells also expressed the phagocyte marker CD68 (D–F), while ramified GFP cells rarely expressed this marker. No GFP-positive cells were detected in uninjected control mice, although CD11b-positive cells were comparable to those in the mice injected with GFP monocytes (G–I). Pictures were taken at 630×. Scale bar, 20 μm.
To further identify the cell type expressing GFP, we evaluated several other markers of brain microglia in double-labeling studies. Figure 3 shows cells labeled with CD45, IBA-1, or F4/80 in conjunction with GFP labeling. As for CD68, not all cells expressing these microglial markers were labeled with GFP, but there were some cells that coexpressed both proteins. Not all GFP cells coexpressed CD45 (Fig. 3 C), nor did all GFP cells coexpress F4/80 (Fig. 3 I), but to a first approximation it appeared that most GFP cells also expressed IBA-1 (Fig. 3 F). Combined with the data from CD68 colocalization, it is certainly the case that the GFP cells assume multiple phenotypes, and the IBA-1 data suggest that to a large extent these are microglial phenotypes.
Figure 3.

Double labeling of GFP-positive cells migrating into the CNS with markers of the microglial phenotype using fluorescence microscopy. Sections were double immunolabeled for GFP and CD45 (A–C), Iba-1 (D–F), or F4/80 (G–I). The left panel in each series used filters specific for the microglial phenotype marker, the middle panel used filters specific to the GFP cells, and the right panel is a superimposition of the left and middle panels. Yellow appears where there is double labeling of the cells. Magnification, 400×.
Neprilysin is an ectoprotease that may be the primary enzyme responsible for Aβ degradation within the brain (Iwata et al., 2000; Leissring et al., 2003; Marr et al., 2003; Hemming et al., 2007). To enhance the likelihood that this enzyme would reach amyloid deposits from transfected microglia, we engineered a secreted form of the enzyme (NEP-s) by deleting the transmembrane domain and substituting the signal peptide for glial-derived neurotrophic factor. We further appended an HA tag to the C terminus of the enzyme to enable specific detection of the recombinant enzyme. As a control gene, we transfected cells with a mutant form of neprilysin containing a single amino acid substitution (E585V) known to inhibit enzyme activity (NEP-m).
The protease activity of these neprilysin constructs was measured by transfecting HEK293 cells and measuring the neprilysin activity in the medium and in the cell homogenates. Cells transfected with GFP showed little activity and were identical to cells transfected with the NEP-m construct (Fig. 4 A), while increased activity was observed in both the NEP-n- and NEP-s-treated samples. As expected, the NEP-n had more activity associated with the cell fraction than NEP-s. In the culture media fraction (Fig. 4 B), the only condition to demonstrate enzyme activity was cells transfected with the NEP-s construct. Thus, both the NEP-m and NEP-s constructs behave as intended with no activity present in either fraction of NEP-m and only the NEP-s construct demonstrating activity in the culture media.
Figure 4.
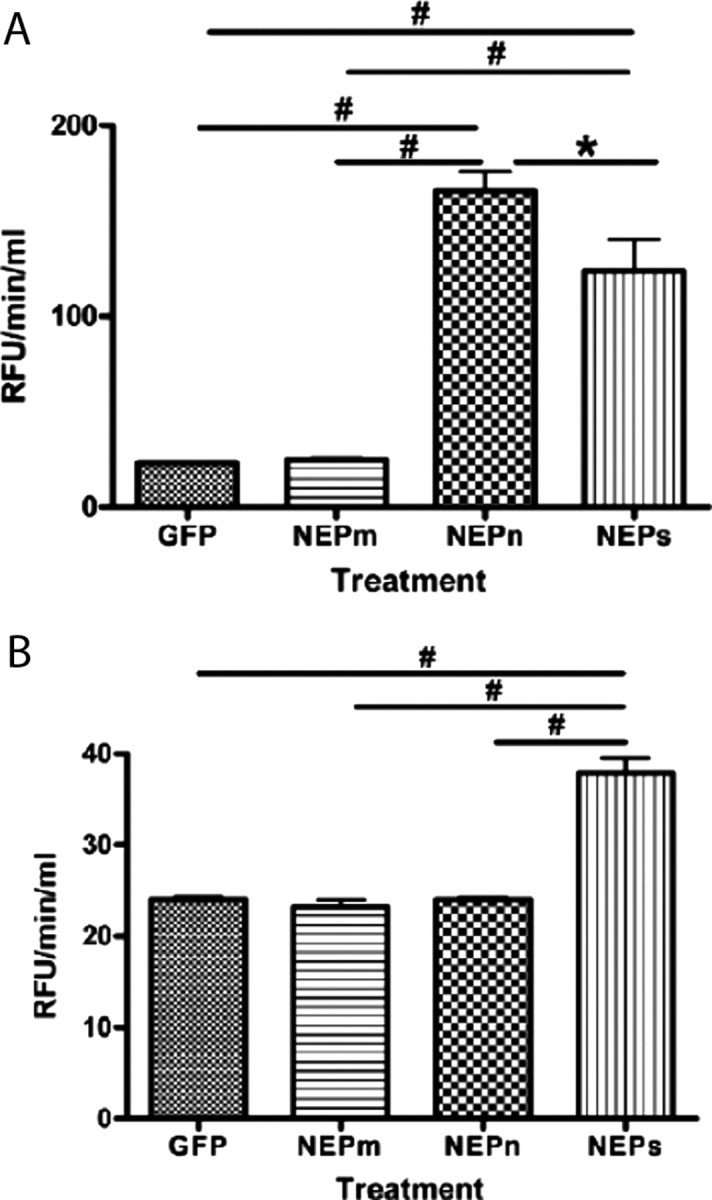
Quantification of neprilysin-specific activity in HEK293-transfected cells. A, Neprilysin-specific activity is significantly higher in the HEK293 cells transfected with NEP-n in the cell lysate/membrane fraction than in NEP-m-, NEP-s-, and GFP-transfected cell lysates. NEP-s-transfected cell lysates showed significantly higher NEP activity than NEP-m- and GFP-expressing cells. B, In the cell media from NEP-s-transfected cells, NEP activity was significantly greater than all other transfected cell groups (NEP-m, NEP-n, and GFP). The asterisk (*) indicates significance with a p value <0.05. The pound sign (#) indicates significance with a p value <0.001. Note differences in y-axis scaling for each panel.
For gene therapy studies, we isolated the CD11b+ monocytes from the bone marrow of GFP donor mice and transfected them by electroporation. A major challenge in working with monocytes is a difficulty in efficiently transfecting them. Transfection efficiencies <10% have been accomplished using traditional transfection techniques, including electroporation, liposomal transfection reagents, and DEAE-dextran (Mayne et al., 2003). By performing electroporation with the Amaxa macrophage protocol, we obtained a 54% transfection efficiency measured by flow cytometry using a GFP-expressing plasmid (in monocytes from non-GFP mice) (Fig. 5 A,B). Staining CD11b+ cells from GFP-expressing mice for HA expression after transfection with NEP plasmids, we show that only the transfected cells are positive for HA when compared to control transfected cells (Fig. 5 C,D). Cells were then infused into the jugular vein of 9-month-old APP+PS1 mice twice weekly using a subcutaneous port. Tissues were collected 2 months later when the mice were 11 months of age. In addition to the mice administered NEP-s and NEP-m monocytes, we also collected tissue from 11-month-old mice that were untreated and from 9-month-old untreated APP+PS1 mice to assess the degree of amyloid deposition when the treatment was initiated.
Figure 5.

Bivariate plots of FACS analysis to analyze cell viability with propidium iodide (PI) and transfection efficiency with GFP fluorescence after electroporation of a GFP-expressing construct. A minimum of 10,000 viable cells were acquired and GFP expression was quantified by gating out the (PI+) dead cells (A). Data are presented as density plots. The second plot shows that 54% of the live cells expressed the transfected GFP plasmid (B). Cells from GFP-expressing donors were transfected with NEP-HA construct and stained for HA. The cells were then visualized for GFP or HA and the images were merged in the rightmost panel (C). A parallel electroporation in the absence of plasmid resulted in no apparent HA staining (D).
In untreated APP+PS1 mice, there was a dramatic increase in the amyloid load detected with Congo red staining between 9 and 11 months, indicating that this was an accelerating phase of amyloid deposition in this model (Fig. 6). Treatment of the mice with NEP-s-transfected cells completely blocked the increase in amyloid deposition found between 9 and 11 months in these mice. However, treatment with CD11b+ cells transfected with the inactive form of the enzyme, NEP-m, showed the same degree of amyloid deposition as the untreated animals.
Figure 6.
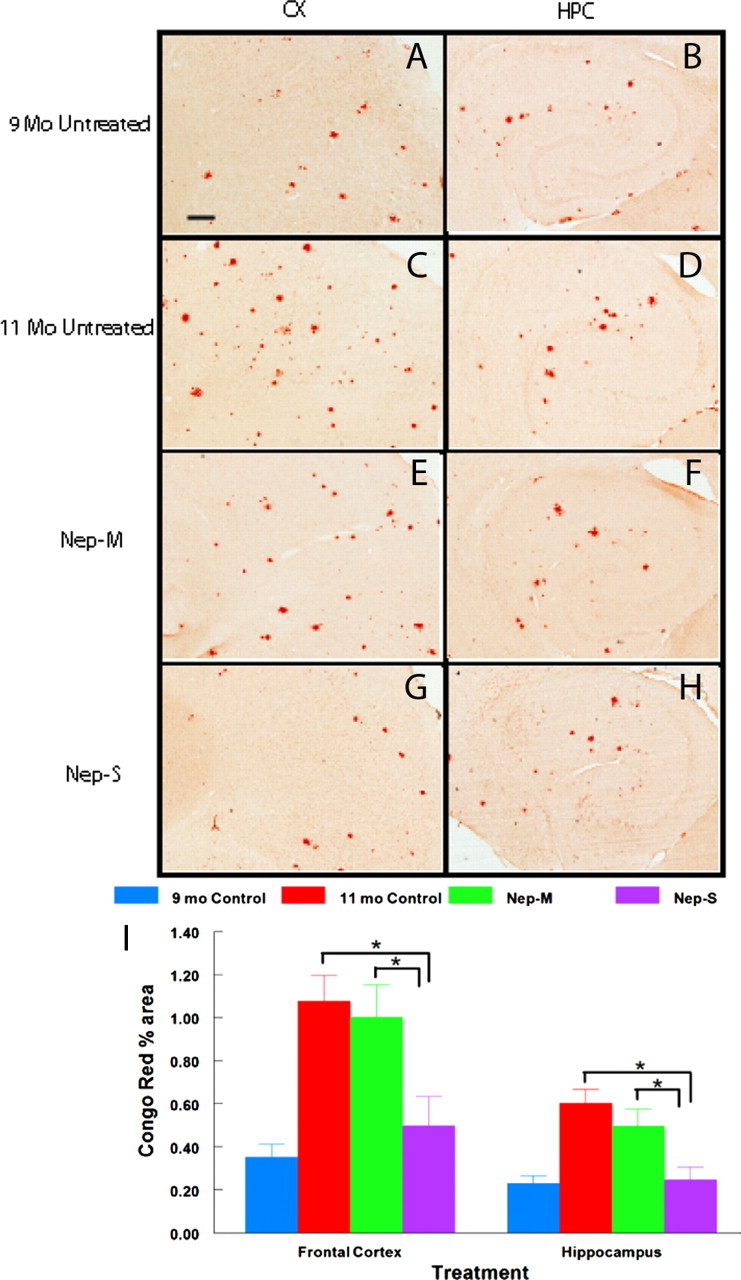
Congophilic amyloid deposition is blocked in APP+PS1 mice infused with GFP monocytes transfected with NEP-s but not NEP-m. Brain sections harvested 1 d after the last infusion were stained with Congo red to estimate the amount of compacted fibrillar plaques. Representative micrographs of staining from frontal cortex (A, C, E, G) or hippocampus (B, D, F, H) are shown for each of the four treatment conditions (indicated to the left of each pair of micrographs). I shows quantification of percentage area of total Congo red staining in the frontal cortex and hippocampus. Sample size per group = 7–9. Statistical analysis was performed using one-way ANOVA with Fisher's LSD multiple-comparison test. Brackets between bars signify statistical significance (p < 0.05). Values are mean ± SEM. Magnification, 40×. Scale bar, 120 μm for all panels.
When amyloid loads were measured with a polyclonal antibody to Aβ, a similar twofold rise in Aβ deposition was obtained between 9 and 11 months of age (Fig. 7). Again, this elevation was abrogated by the administration of cells containing NEP-s, but not NEP-m. There is a trend in the results for the administration of the monocytes with the inactive NEP-m to have less Aβ immunostaining than untreated mice, but this was not statistically significant unless the 9 months control group was excluded from the analysis. Nonetheless, this observation suggests some minor benefit of excess circulating monocytes in slowing Aβ deposition.
Figure 7.
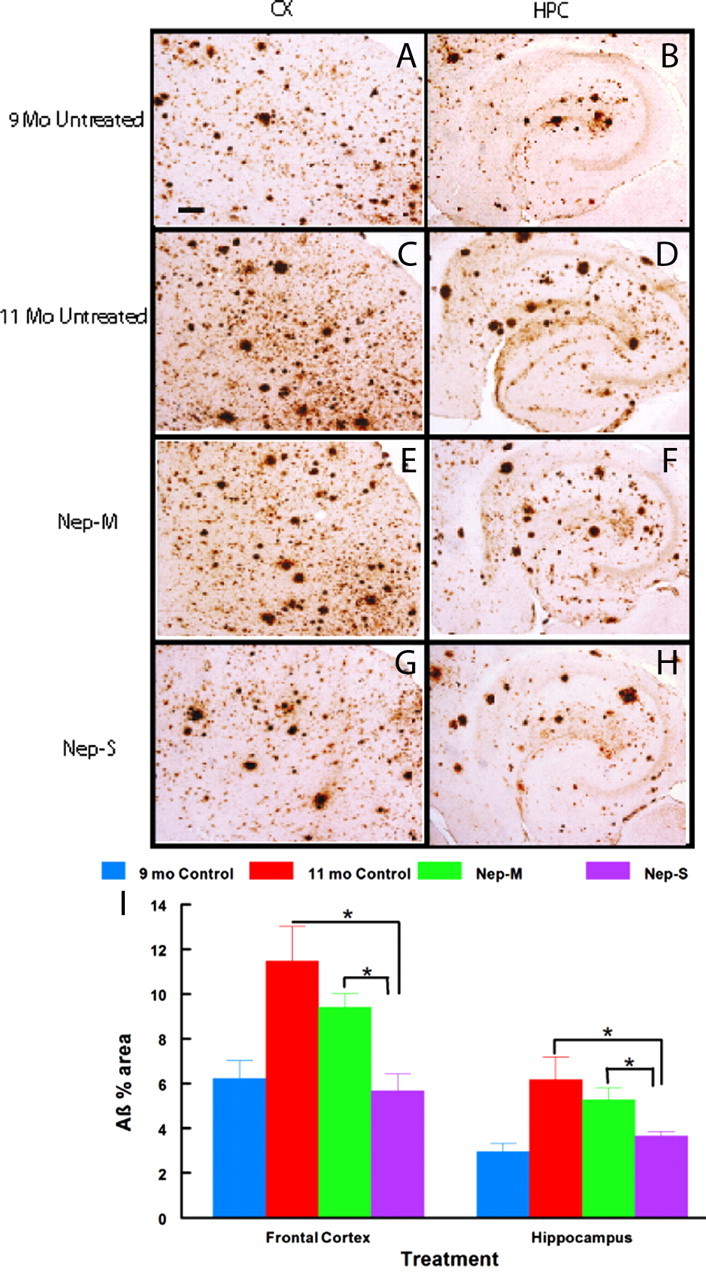
Immunohistochemical Aβ deposition is blocked in APP+PS1 mice infused with GFP monocytes transfected with NEP-s but not NEP-m. Brain sections harvested 1 d after the last infusion were stained with antibody against Aβ to estimate the amount of diffuse and compacted Aβ deposition. Representative micrographs of staining from frontal cortex (A, C, E, G) or hippocampus (B, D, F, H) are shown for each of the four treatment conditions (indicated to the left of each pair of micrographs). I shows quantification of percentage area of Aβ immunostaining in the frontal cortex and hippocampus. Sample size per group = 7–9. Statistical analysis was performed using one-way ANOVA with Fisher's LSD multiple-comparison test. Brackets between bars signify statistical significance (p < 0.05). Values are mean ± SEM (standard error of mean). Magnification, 40×. Scale bar, 120 μm for all panels.
Discussion
These results are the first demonstration that circulating CD11b+ cells derived from bone marrow can be used to deliver therapeutic genes to the CNS. The effect of the cells transfected with NEP-s cannot be simply due to increasing the macrophage population, as the NEP-m-transfected cells had minimal effects on amyloid deposition. Similarly the NEP-m group also controls for the isolation and transfection procedures and expression of an heterologous protein. One minor difference is the NEP-s is secreted while the NEP-m remains membrane bound, Thus it cannot be decisively excluded that secretion of a heterologous protein accounts for the results, regardless of proteolytic activity, but we believe this outcome is unlikely.
At the end of the study (1 d after the final injection), HA expression was detected in brain, but as expected, none could be found in plasma, arguing for a central site of action. Moreover, by measuring the amyloid loads at the beginning of the experiment, we demonstrated that the NEP-s activity totally prevented further amyloid deposition. Few studies have included this treatment initiation control group, and those that have included this group show only slowing of deposition rather than total prevention (Best et al., 2007). The APP+PS1 mouse is an aggressive model of amyloid deposition, with first deposits around 3–4 months of age and amyloid loads at least 3 times greater than the parental singly transgenic APP (Tg2576) mouse at most ages (Holcomb et al., 1998; Gordon et al., 2002). Hence we anticipate that AD patients and other transgenic mouse models with slower rates of Aβ accumulation could benefit from even less exposure to the therapeutic gene than that administered here.
These results add to the increasing evidence that peripheral CD11b+ cells, presumed to be largely monocytes, migrate to sites of neurodegeneration within the brain. In this instance, the migration cannot be accounted for by irradiation damage to the CNS (or other tissues), excess numbers of stem cells in circulation or systemic illness brought about the irradiation. The argument that Aβ deposits stimulate recruitment (Fiala et al., 1998; Giri et al., 2000; Humpel, 2008) is consistent with our observation that very few labeled cells were found in brain regions that lacked amyloid deposits. Moreover, these studies may be the first to estimate the half-life of a circulating CD11b+ cell population and the half-lives of the tissue infiltrating CD11b+ cells after a pulsed administration. Both half-lives were shorter than anticipated. The injected CD11b+ cells initially increase the numbers of these cells in blood by a factor of 2 or more. Hence an elevated monocyte concentration may contribute to the trafficking we find into the CNS of transgenic mice. However, it is unlikely that mechanical or chemical aspects of the intravenous infusion (5% of blood volume in buffered physiological saline) account for the CNS infiltration observed, as very few cells entered the brains of nontransgenic mice. One caveat is that the cells used here were derived from marrow, and thus are immature compared to CD11b+ cells already in circulation. It is conceivable that this immaturity contributes to their CNS migration. We interpret these data as supporting the argument that circulating CD 11b+ cells contribute a meaningful fraction of the activated myeloid cells in the vicinity of compacted amyloid plaques, likely in response to chemotactic factors secreted in the vicinity of the deposits.
This approach to ex vivo gene therapy has multiple applications for demonstrating proof of concept for specific therapeutic genes in human conditions. For trials of human gene therapy, a patient's own monocytes could be elutriated from blood, transfected ex vivo, then reintroduced in a single visit. Given the short half-lives of the monocytes, this would require repeated injections before therapeutic effects might be observed. However, this time-dependent fading of therapeutic gene expression would spontaneously reverse unforeseen adverse effects (much like clearance of a small molecule drug) that could be challenging to treat using more permanent gene therapy approaches. For neurodegenerative disorders, this approach would permit distribution of the therapeutic gene throughout the brain, and deliver the gene precisely at locations undergoing degeneration. Compared to viral vectors, there would be no expression of the gene in unintended cells, or a gradient of expression around the injection site. Moreover, there would be no need for intracranial surgery, the hazards of which were evident in the first gene therapy trial for Alzheimer's disease (Tuszynski et al., 2005). The absence of integration into the host genome by the plasmid and the use of postmitotic cells would diminish the risk for inadvertent transformation into cancerous cells. As a long-term goal, once the efficacy and safety of a gene therapy approach is demonstrated using monocytes, the gene could be transfected into bone marrow stem cells. With appropriate promoters to drive expression of the gene in differentiated tissue macrophages, as opposed to circulating monocytes, this approach might provide a safe, effective, and permanent treatment for some CNS degenerative diseases.
Footnotes
This work is supported by grants from National Institutes of Health (AG-25509, AG-15490, AG04418), from the Johnnie B. Byrd Research Institute and from the Alzheimer's Association. L.L. is the Benjamin Scholar in Alzheimer's Research. We thank the staff of the Division of Comparative Medicine, especially Margaret Baldwin for her assistance with the surgical insertion of microvascular ports. We thank Barbara Nicholson and Bridget Shields for clerical assistance in conducting these studies.
A disclosure has been filed with the University of South Florida Office of Patents and Licensing for possible patent protection of this approach to gene therapy. D.M., L.L., and S.K. are listed as inventors on this patent disclosure.
References
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best JD, Smith DW, Reilly MA, O'Donnell R, Lewis HD, Ellis S, Wilkie N, Rosahl TW, Laroque PA, Boussiquet-Leroux C, Churcher I, Atack JR, Harrison T, Shearman MS. The novel gamma secretase inhibitor N-[cis-4-[(4-chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl)cyclohexyl]-1,1,1-trifluoromethanesulfonamide (MRK-560) reduces amyloid plaque deposition without evidence of notch-related pathology in the Tg2576 mouse. J Pharmacol Exp Ther. 2007;320:552–558. doi: 10.1124/jpet.106.114330. [DOI] [PubMed] [Google Scholar]
- Burger C, Nguyen FN, Deng J, Mandel RJ. Systemic mannitol-induced hyperosmolality amplifies rAAV2-mediated striatal transduction to a greater extent than local co-infusion. Mol Ther. 2005;11:327–331. doi: 10.1016/j.ymthe.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Carty NC, Nash K, Lee D, Mercer M, Gottschall PE, Meyers C, Muzyczka N, Gordon MN, Morgan D. Adeno-associated viral (AAV) serotype 5 vector mediated gene delivery of endothelin-converting enzyme reduces Abeta deposits in APP + PS1 transgenic mice. Mol Ther. 2008;16:1580–1586. doi: 10.1038/mt.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cearley CN, Wolfe JH. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol Ther. 2006;13:528–537. doi: 10.1016/j.ymthe.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Fiala M, Zhang L, Gan X, Sherry B, Taub D, Graves MC, Hama S, Way D, Weinand M, Witte M, Lorton D, Kuo YM, Roher AE. Amyloid-beta induces chemokine secretion and monocyte migration across a human blood–brain barrier model. Mol Med. 1998;4:480–489. [PMC free article] [PubMed] [Google Scholar]
- Giri R, Shen Y, Stins M, Du Yan S, Schmidt AM, Stern D, Kim KS, Zlokovic B, Kalra VK. beta-amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am J Physiol Cell Physiol. 2000;279:C1772–1781. doi: 10.1152/ajpcell.2000.279.6.C1772. [DOI] [PubMed] [Google Scholar]
- Gordon MN, Schreier WA, Ou X, Holcomb LA, Morgan DG. Exaggerated astrocyte reactivity after nigrostriatal deafferentation in the aged rat. J Comp Neurol. 1997;388:106–119. [PubMed] [Google Scholar]
- Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, Connor K, Melachrino J, O'Callaghan JP, Morgan D. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp Neurol. 2002;173:183–195. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- Hemming ML, Patterson M, Reske-Nielsen C, Lin L, Isacson O, Selkoe DJ. Reducing amyloid plaque burden via ex vivo gene delivery of an Abeta-degrading protease: a novel therapeutic approach to Alzheimer disease. PLoS Med. 2007;4:e262. doi: 10.1371/journal.pmed.0040262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Humpel C. Basolateral aggregated rat amyloidβ(1–42) potentiates transmigration of primary rat monocytes through a rat blood-brain barrier. Curr Neurovasc Res. 2008;5:185–192. doi: 10.2174/156720208785425701. [DOI] [PubMed] [Google Scholar]
- Imai F, Sawada M, Suzuki H, Zlokovic BV, Kojima J, Kuno S, Nagatsu T, Nitatori T, Uchiyama Y, Kanno T. Exogenous microglia enter the brain and migrate into ischaemic hippocampal lesions. Neurosci Lett. 1999;272:127–130. doi: 10.1016/s0304-3940(99)00592-3. [DOI] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC. Identification of the major Abeta1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- Johnson GD, Ahn K. Development of an internally quenched fluorescent substrate selective for endothelin-converting enzyme-1. Anal Biochem. 2000;286:112–118. doi: 10.1006/abio.2000.4772. [DOI] [PubMed] [Google Scholar]
- Krauze MT, Saito R, Noble C, Tamas M, Bringas J, Park JW, Berger MS, Bankiewicz K. Reflux-free cannula for convection-enhanced high-speed delivery of therapeutic agents. J Neurosurg. 2005;103:923–929. doi: 10.3171/jns.2005.103.5.0923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, Finsen B. Microglial cell population dynamics in the injured adult central nervous system. Brain Res Rev. 2005;48:196–206. doi: 10.1016/j.brainresrev.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–1093. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Pärepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–142. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Marr RA, Rockenstein E, Mukherjee A, Kindy MS, Hersh LB, Gage FH, Verma IM, Masliah E. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J Neurosci. 2003;23:1992–1996. doi: 10.1523/JNEUROSCI.23-06-01992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne GC, Borowicz RA, Greeneklee KV, Finlay-Jones JJ, Williams KA, Hart PH. Centrifugation facilitates transduction of green fluorescent protein in human monocytes and macrophages by adenovirus at low multiplicity of infection. J Immunol Methods. 2003;278:45–56. doi: 10.1016/s0022-1759(03)00229-1. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol. 2005;64:743–753. doi: 10.1097/01.jnen.0000178444.33972.e0. [DOI] [PubMed] [Google Scholar]
- Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, Coomaraswamy J, Staufenbiel M, Landmann R, Jucker M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci. 2005;25:11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata K, Kitamura Y, Yanagisawa D, Morikawa S, Morita M, Inubushi T, Tsuchiya D, Chishiro S, Saeki M, Taniguchi T, Shimohama S, Tooyama I. Microglial transplantation increases amyloid-beta clearance in Alzheimer model rats. FEBS Lett. 2007;581:475–478. doi: 10.1016/j.febslet.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R, Patel P, Blesch A, Vahlsing HL, Ho G, Tong G, Potkin SG, Fallon J, Hansen L, Mufson EJ, Kordower JH, Gall C, Conner J. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med. 2005;11:551–555. doi: 10.1038/nm1239. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun J, Goldstein H. Human immunodeficiency virus type 1 infection increases the in vivo capacity of peripheral monocytes to cross the blood-brain barrier into the brain and the in vivo sensitivity of the blood-brain barrier to disruption by lipopolysaccharide. J Virol. 2008;82:7591–7600. doi: 10.1128/JVI.00768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Complement component C1q modulates the phagocytosis of Abeta by microglia. Exp Neurol. 2000;161:127–138. doi: 10.1006/exnr.1999.7260. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Wu J, Yang S, Luo H, Zeng L, Ye L, Lu Y. Quantitative evaluation of monocyte transmigration into the brain following chemical opening of the blood-brain barrier in mice. Brain Res. 2006;1098:79–85. doi: 10.1016/j.brainres.2006.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]