

The accurate and sensitive detection of radical oxidants is a central problem in the field of chemical biology.[1] Radical oxidants can be detected in vitro with fluorescent leucodyes such as dihydroethidium (DHE),[2] dihydrorhodamine (DHR),[3] the hydrocyanines,[4] redoxfluor-1,[5a] and their organelle-specific analogues.[5b] Although these probes are widely used in cell culture, their accuracy is compromised by their high levels of background fluorescence,[6] which is caused by their spontaneous oxidation that is catalyzed by light or oxygen. Radical oxidants can be detected by using DHE, DHR, and the hydrocyanines, as they undergo an amine oxidation reaction[7] with cellular oxidants, such as superoxide or hydroxyl radicals. However, the probes also generate background fluorescence by undergoing the same amine oxidation reaction with air and light; this reaction is often attributed to the effects of singlet oxygen (1O2).[8]
The mechanism of the amine oxidation differs significantly for reactions that involve either radical oxidants or singlet oxygen, and in particular, cleavage of the α-amine C–H bond occurs at different points along the reaction coordinate for oxidation with these two oxidants. For example, amine oxidation by singlet oxygen proceeds via an exciplex intermediate, in which the C–H bond cleavage occurs in the rate-determining step. This oxidation reaction therefore exhibits a relatively high kinetic isotope effect (KIE).[9] For instance, the KIE for the oxidation of N,N-dimethyl([D2]benzyl)amine with singlet oxygen is 3.06 ±0.06.[8a] In contrast, a radical mechanism for amine oxidation proceeds through a sequence that involves either an electron transfer (ET)/proton transfer (PT) mechanism or a direct hydrogen atom transfer (HAT) mechanism, and generally has a lower KIE than oxidation with singlet oxygen.[10] For example, the KIE for the oxidation of N,N-dimethyl([D2]benzyl)amine with the tert-butoxy radical (tBuO•) is only 1.4 ± 0.7,[10a–c] and the KIE for the mechanistically similar oxidation of N,N-dimethyl([D2]benzyl)aniline with cytochrome P450 is 1.8 ± 0.2.[10a] This difference in KIEs[11] for the amine oxidation reaction between singlet oxygen and radical oxidants offers the possibility of selectively slowing down the aerial oxidation of radical oxidant probes while maintaining their reactivity with cellular radical oxidants.
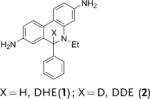
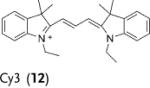
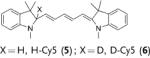
Herein we demonstrate that the efficacy of the commonly used radical oxidant probes, DHE (1), H-Cy3 (3), H-Cy5 (5), H-Cy7 (7), and DHR (9), can be dramatically improved by deuteration at their α-amine C–H bond (Figure 1). Deuterated analogues of DHE (1), H-Cy3 (3), H-Cy5 (5), and H-Cy7 (7) have large KIEs (3.7–4.7) for aerial oxidation; however, their KIEs for oxidation with the superoxide radical anion are only between 2.5–2.8. This difference in KIEs causes the deuterated radical oxidant probes to generate less background fluorescence, but to still generate similar levels of fluorescence in cells that are stimulated to produce radical oxidants. Deuterated radical oxidant probes were significantly more accurate than their hydrogen analogues in the detection of radical oxidants in vitro, in cell culture, and in vivo. For example, the deuterated DHE analogue DDE had several advantages over its hydrogen analogue because of its lower background fluorescence. In particular, DDE had greater storage stability and higher accuracy than DHE, and was also used to detect radical oxidants produced in cell culture from angiotensin II (Ang II) stimulated rat aortic smooth muscle cells (RASM), whereas the oxidants were not detected by using DHE under identical experimental conditions. Similarly, D-Cy7 was also significantly better than its hydrogen analogue H-Cy7 (7) in the detection of radical oxidants in vivo because of its low background fluorescence. Based on these results, we anticipate numerous applications of deuterated radical oxidant probes in biology and medicine.
Figure 1.

a) Nonfluorescent tertiary amines 1–10 that are oxidized to fluorescent iminium cations 11–15 by a radical-mediated amine oxidation (bold arrow) are probes for radical oxidants. However, they also generate background fluorescence by aerial oxidation (dotted arrow, mediated by 1O2). b) Selective suppression of background fluorescence for the deuterated probes leads to a higher signal to noise (S/N) ratio in cell culture and in vivo.
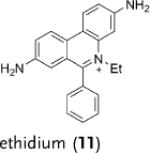
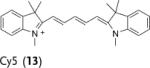
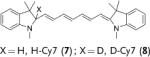
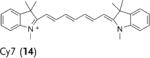
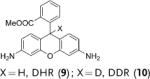
Deuterated analogues of DHE (1), H-Cy3 (3), H-Cy5 (5), H-Cy7 (7), and DHR (9) were synthesized in excellent yields (> 93 %) by reduction of commercially available ethidium bromide (11), Cy3 (12), Cy5 (13), Cy7 (14), and rhodamine (15) dyes, respectively, with sodium borodeuteride (see the Supporting Information). This reduction procedure specifically introduces a deuterium atom at the α-amine carbon atom of ethidium and the cyanines, and at the ε-amine carbon atom of rhodamine. We investigated the stability of the deuterated probes 2, 4, 6, 8, and 10 to deuterium/hydrogen (D/H) exchange, and found them to be resistant to D/H exchange under aqueous conditions (see the Supporting Information).
The rate of aerial oxidation of the deuterated radical oxidant probes 2, 4, 6, 8, and 10 was measured in phosphate-buffered saline (PBS) and compared with the data for the hydrogen analogues (see the Supporting Information). The data in Table 1 show that the deuterated probes 2, 4, 6, 8, and 10 are significantly more stable to aerial oxidation than their hydrogen analogues 1, 3, 5, 7, and 9, respectively. For example, a KIE of 4.7 was observed for DDE and KIEs for the deuterocyanines 4, 6, and 8 were in the range of 3.7–4.7. These large KIE values suggest that C–H bond cleavage is involved in the rate-determining step in aerial oxidation of these probes, and provides a methodology for improving their stability.
TABLE 1.
KIE values (kH/kD) for aerial oxidation and oxidation by superoxide radicals for deuterated radical oxidant probes.
 | |||
|---|---|---|---|
| Probe | Product | kH/kD | |
| Aerial oxidation[a] |
Oxidation by (O2·−)[b] |
||
|
|
4.7 | 2.5 |

|
|
3.7 | 2.6 |
|
|
4.1 | 2.7 |
|
|
4.7 | 2.8 |
|
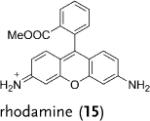
|
2.1 | 1.7 |
Measured by using a 60 μm concentration of 1–10 in PBS at 25 °C (see the Supporting Information).
kH and kD were determined following the method described by Finkelstein et al.[12] (see the Supporting Information). Cy=cyanine dye, DHE=dihydroethidium, DDE=dideuteroethidium, DHR=dihydrorhodamine, DDR=dideuterorhodamine.
Cells contain large amounts of endogenous antioxidants, such as superoxide dismutase (SOD); radical oxidant probes have to compete with these antioxidants in order to detect cellular radical oxidants. A potential limitation of deuterated radical oxidant probes is that they may react slower with cellular radical oxidants such as superoxide radicals, and thus will be unable to compete with cellular antioxidants. We therefore measured the ability of the deuterated probes 2, 4, 6, 8, and 10 to compete with SOD for superoxide radicals and compared their reactivity with their hydrogen analogues 1, 3, 5, 7, and 9 (see the Supporting Information). For these experiments, the deuterated probes, or their hydrogen analogues were incubated with SOD and xanthine/ xanthine oxidase, and the rates of oxidation of the probes 1–10 were measured by fluorescence spectroscopy. Table 1 shows that the deuterated radical oxidant probes 2, 4, 6, 8, and 10 reacted with superoxide radicals 1.7–2.8 times slower than their hydrogen analogues. Importantly, the KIEs observed for oxidation of 2, 4, 6, 8, and 10 with superoxide radicals are lower than their corresponding KIEs for aerial oxidation (1.7–2.8 versus 3.7–4.7). The lower KIEs observed for oxidation with superoxide radicals suggests that the oxidation mechanism for these dyes with superoxide radicals is different than for aerial oxidation. In summary, deuterated radical oxidant probes are stabilized with respect to aerial oxidation, but still maintain a high reactivity toward cellular oxidants.
DHE is currently the most commonly used radical oxidant probe in biology and has been used extensively in cell culture studies.[2] However, its spontaneous aerial oxidation generates high background fluorescence that limits its accuracy, and also makes its storage and handling a challenge. Therefore, we investigated if deuteration would improve the accuracy and storage life of DHE. We compared the performances of DHE and DDE in detecting the hydroxyl radical in vitro at nanomolar concentrations (see the Supporting Information). Figure 2 a shows that DDE has greater accuracy than DHE for the detection of the hydroxyl radical at nanomolar concentrations. For example, DDE could be used to detect nanomolar levels of radical oxidants and had an R2 (linear regression) value of 0.97 in this concentration range. In contrast, we were unable to detect radical oxidants with precision using DHE in the same concentration range. This result is shown by an R2 value of 0.79, and also a much higher standard deviation per measurement than the data for DDE. We further investigated if deuteration would improve the storage life of DHE. Figure 2 b shows that the storage profile of DDE is significantly better than that of DHE. For example, 60 % of solid DHE was oxidized after 10 days storage, whereas only 20 % of solid DDE was oxidized.
Figure 2.

a) Nanomolar concentrations of the hydroxyl radical (generated from Fenton's Reaction) can be detected with DDE but not DHE. b) Solid DDE has a greater storage life than solid DHE (see the Supporting Information).
We performed additional experiments to determine if the greater accuracy of DDE towards the detection of radical oxidants would improve its ability to measure radical oxidants in cell culture. RASMs stimulated with Ang II were used for these experiments because of the importance of Ang II mediated signaling in atherosclerosis.[13] RASMs were isolated as previously described.[13] Cells were then cultured until 80 % confluent, serum starved for 25 hours, stimulated with Ang II or PBS as a control, and then incubated with 10 μm of either DDE or DHE for 10 minutes. The cells were imaged by confocal fluorescence microscopy and the mean fluorescence intensity of the images that contained equal numbers of cells was quantified using Image Pro software (see the Supporting Information). Figure 3 a–h shows that DDE has a 2.5-fold reduction in background fluorescence in comparison to DHE (3.6 vs. 9.2 fluorescence units), but generates only a 30 % lower level of fluorescence than DHE in Ang II stimulated cells (6.8 vs. 9.8 fluorescence units). Importantly, the selective reduction of background fluorescence by DDE allowed the detection of radical oxidants generated by Ang II signaling, whereas detection was not successful when DHE was used under these conditions (compare Figure 3 d and 3 h). To further verify that DDE could be used to detect radical oxidants, the free radical scavenger 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy (TEMPOL) was added to RASMs treated with DHE and Ang II. Figure 3 g shows that TEMPOL significantly reduces the fluorescence of RASMs treated with DHE and Ang II, thus implying that DDE can be used to detect radical oxidants. In summary, the kinetic isotope effect causes DDE to generate low background fluorescence, but still generate similar levels of fluorescence in cells that are stimulated to produce radical oxidants, thus resulting in higher efficacy in comparison to DHE.
Figure 3.

Confocal fluorescence images of RASMs treated with a) 10 μm DHE and PBS; b) 10 μm DHE and 100 nm Ang II; c) 100 nm Ang II for 4 h followed by 5 mm TEMPOL and then 10 μm DHE. d) Quantification of fluorescence intensities from (a), (b), and (c). Confocal fluorescence images of RASMs treated with e) 10 μm DDE and PBS; f) 10 μm DDE and 100 nm Ang II; g) 100 nm Ang II for 4 h followed by 5 μm TEMPOL and then 10 μm DDE. h) Quantification of fluorescence intensities from (e), (f), and (g) (see the Supporting Information). * p < 0.05.
We investigated if the kinetic isotope effect would similarly improve the ability of H-Cy7 (7) to detect radical oxidants in vivo. H-Cy7 is a new radical oxidant probe that can be used to detect radical oxidants in vivo because of its high emission wavelength (765 nm); however, H-Cy7 generates moderate levels of background fluorescence in vivo, which will potentially limit its applications. We compared the ability of H-Cy7 and D-Cy7 (8) to image radical oxidant production in vivo generated by lipopolysaccharide (LPS) stimulated acute inflammation. BALB/c mice were given either an intraperitoneal (I.P.) injection of LPS (1 mg) or saline for 4 hours, treated with either D-Cy7 or H-Cy7 (25 nmol) by I.P. injection, and then imaged in an IVIS imaging system. Figure 4 a–f shows that D-Cy7 is more effective at the imaging of radical oxidants in vivo than H-Cy7. For example, D-Cy7 had a 2.7-fold reduction in background fluorescence in comparison to H-Cy7 (0.7 vs. 1.9 fluorescence units, compare Figure 4d and 4a), but generated a fluorescence level that was only 17 % lower than that of HCy7 in mice stimulated with LPS. As a result, D-Cy7 was significantly better than H-Cy7 for the detection of radical oxidants, and generated a tenfold difference in integrated fluorescence intensity from the I.P. cavity in LPS versus control mice, compared to only a fivefold difference for H-Cy7 (see the Supporting Information). The kinetic isotope effect, therefore, improves the efficacy of D-Cy7 in comparison to H-Cy7.
Figure 4.

Fluorescence images of mice injected with a) PBS and H-Cy7; b) LPS and H-Cy7. c) Quantification of fluorescence intensities from (a) and (b). Fluorescence images of mice injected with d) PBS and D-Cy7; e) LPS and D-Cy7. f) Quantification of fluorescence intensities from (d) and (e).
In summary, we have demonstrated that the oxidation of radical oxidant probes by air, light, and superoxide radicals occurs by different mechanistic pathways, hence leading to different KIEs. The high KIE values for aerial oxidation of the radical oxidant probes 1–10 suggest that they react through a mechanism that involves an exciplex intermediate (see the Supporting Information). In contrast, the low KIE values observed for the oxidation of 1–10 with superoxide radicals suggest that this oxidation occurs via either an ET- PT-ET pathway or an HAT-ET pathway. We have also demonstrated that this difference in KIEs can be used to improve the efficacy of radical oxidant probes. Deuterated radical oxidant probes generate lower background fluorescence than their hydrogen analogues, but generate similar levels of fluorescence in cells and mice stimulated to produce radical oxidants. Based on these results, we anticipate numerous applications of deuterated radical oxidant probes in biology and an increased application of the KIE in biological probe development.
Experimental Section
Determination of the kinetic isotope effects (kH/kD) for aerial oxidation (Table 1): The procedure used to determine the kH/kD values for the probes 1–10 is described below, using DHE and DDE as a representative example. Stock solutions of DDE and DHE in methanol (3.2 mm) were prepared in two separate vials, covered with aluminum foil, and placed in a water bath maintained at 25 °C. At various time points that ranged from 0–25 h, 40 μL aliquots of the stock solutions were diluted in a 3:1 PBS/methanol solution to generate a 60 μm concentration, and the fluorescence intensity was measured (λex = 515 nm, λem = 559 nm). The percentage of oxidized compound was determined by dividing the recorded fluorescence intensity by the fluorescence intensity of a 60 μm ethidium bromide solution. The fluorescence data were plotted as percent oxidized versus time and the rate constants (kH and kD) were calculated assuming first-order kinetics.
Supplementary Material
Footnotes
This work was supported by the Georgia Tech/Emory Center for the Engineering of Living Tissues (funded by NSF-EEC-9731643) (N.M.), NSF-BES-0546962 Career Award (N.M.), NIH UO1 HL80711-01 (N.M.), NIH R21 EB006418 (N.M.) NIH RO1 HL096796-01 (N.M.), NIH RO1 HL090584 (W.R.T.) and J&J/GT Health Care Innovation Seed Grant Proposal (N.M.).
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201002228.
References
- 1.a) Finkel T, Holbrook NJ. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]; b) Forstermann U. Nat. Rev. Cardiol. 2008;2:338–349. [Google Scholar]; c) Hussein SP, Hofseth LJ, Harris CC. Nat. Rev. Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]; d) Barnham KJ, Masters CL, Bush AI. Nat. Rev. Drug Discovery. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]; e) Winterbourn CC. Nat. Chem. Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 2.a) Zielonka J, Vasquez-Vivar J, Kalyanaraman B. Nat. Protoc. 2008;3:8–21. doi: 10.1038/nprot.2007.473. [DOI] [PubMed] [Google Scholar]; b) Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Proc. Natl. Acad. Sci. USA. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Koide Y, Urano Y, Kenmoku S, Kojima H, Nagano T. J. Am. Chem. Soc. 2007;129:10324–10325. doi: 10.1021/ja073220m. [DOI] [PubMed] [Google Scholar]
- 3.Buxser SE, Sawada G, Raub TJ. Methods Enzymol. 1999;300:256–275. doi: 10.1016/s0076-6879(99)00133-0. [DOI] [PubMed] [Google Scholar]
- 4.a) Kundu K, Knight SF, Willett N, Lee S, Taylor WR, Murthy N. Angew. Chem. 2009;121:305–309. doi: 10.1002/anie.200804851. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:299–303. doi: 10.1002/anie.200804851. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lin PW, Myers LE, Ray L, Song S, Nasm TR, Berardinelli AJ, Kundu K, Murthy N, Hansen JM, Neish AS. Free Radical Biol. Med. 2009;47:1205–1211. doi: 10.1016/j.freeradbiomed.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Dickinson BC, Srikun D, Chang CJ. Curr. Opin. Chem. Biol. 2010;14:50–56. doi: 10.1016/j.cbpa.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miller EW, Bian SX, Chang CJ. J. Am. Chem. Soc. 2007;129:3458–3459. doi: 10.1021/ja0668973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Haugland RP. The Handbook: A Guide to Fluorescent Probes and Labeling Technologies. 10th ed. Invitrogen/Molecular Probes; Carlsbad, CA: 2005. [Google Scholar]; b) Halliwell B, Whiteman M. Br. J. Pharmacol. 2004;142:231–255. doi: 10.1038/sj.bjp.0705776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murahashi S, Nakae T, Terai H, Komiya N. J. Am. Chem. Soc. 2008;130:11005–11012. doi: 10.1021/ja8017362. [DOI] [PubMed] [Google Scholar]
- 8.a) Latch DE, Stender BL, Packer JL, Arnold WA, McNeill K. Environ. Sci. Technol. 2003;37:3342–3350. doi: 10.1021/es0340782. [DOI] [PubMed] [Google Scholar]; b) Chen Y, Hu C, Qu J. Environ. Sci. Technol. 2009;43:2760–2765. doi: 10.1021/es803325j. [DOI] [PubMed] [Google Scholar]; c) Cohen SG, Parola AM, Parsons GH. Chem. Rev. 1973;73:141–161. [Google Scholar]
- 9.a) Baciocchi E, Giacco TD, Lapi A. Org. Lett. 2004;6:4791–4794. doi: 10.1021/ol047876l. [DOI] [PubMed] [Google Scholar]; b) Baciocchi E, Giacco TD, Lapi A. Org. Lett. 2006;8:1783–1786. doi: 10.1021/ol0602607. [DOI] [PubMed] [Google Scholar]; c) Baciocchi E, Giacco TD, Lanzalunga O, Lapi A, Raponi D. J. Org. Chem. 2007;72:5912–5915. doi: 10.1021/jo0706980. [DOI] [PubMed] [Google Scholar]
- 10.a) Manchester JL, Dinnocenzo JP, Higgins LA, Jones JP. J. Am. Chem. Soc. 1997;119:5069–5070. [Google Scholar]; b) Goto Y, Watanabe Y, Fukuzumi S, Jones JP, Dinnocenzo JP. J. Am. Chem. Soc. 1998;120:10762–10763. [Google Scholar]; c) Dinnocenzo JP, Karki SB, Jones JP. J. Am. Chem. Soc. 1993;115:7111–7116. [Google Scholar]; d) Dombrowski GW, Dinnocenzo JP, Farid S, Goodman JL, Gould IR. J. Org. Chem. 1999;64:427–431. [Google Scholar]; e) Silverman RB, Hoffman SJ, Catus WB. J. Am. Chem. Soc. 1980;102:7126–7128. [Google Scholar]; f) Rigby SE, Basran J, Combe JP, Mohsen AW, Toogood H, Thiel AV, Sutcliffe MJ, Leys D, Munro AW. Biochem. Soc. Trans. 2005;33:754–757. doi: 10.1042/BST0330754. [DOI] [PubMed] [Google Scholar]; g) Li C, Wu W, Cho K, Shaik S. Chem. Eur. J. 2009;15:8492–8503. doi: 10.1002/chem.200802215. [DOI] [PubMed] [Google Scholar]; h) Chiavarino B, Cipollini R, Crestoni ME, Fornarini S, Lanucara F, Lapi A. J. Am. Chem. Soc. 2008;130:3208–3217. doi: 10.1021/ja077286t. [DOI] [PubMed] [Google Scholar]
- 11.Westheimer FH. Chem. Rev. 1961;61:265–273. [Google Scholar]
- 12.Finkelstein E, Rosen GM, Rauckman EJ. J. Am. Chem. Soc. 1980;102:4994–4999. [Google Scholar]
- 13.Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK. Hypertension. 1998;32:488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.