

Abstract
The dystrophin-associated protein complex (DAPC) links the cytoskeleton to the extracellular matrix, is essential for muscle cell survival, and is defective in a wide range of muscular dystrophies. The DAPC contains two transmembrane subcomplexes - the dystroglycans and the sarcoglycans. Although several extracellular binding partners have been identified for the dystroglycans, none have been described for the sarcoglycan subcomplex. Here we show that the small leucine-rich repeat proteoglycan biglycan binds to α– and γ– sarcoglycan as judged by ligand blot overlay and co-immunoprecipitation assays. Studies with biglycan-decorin chimeras show that α– and γ– sarcoglycan bind to distinct sites on the polypeptide core of biglycan. Both biglycan proteoglycan as well as biglycan polypeptide lacking glycosaminoglycan side chains are components of the dystrophin glycoprotein complex isolated from adult skeletal muscle membranes. Finally, immunohistochemical and biochemical studies with biglycan null mice show that the expression of α- and γ- sarcoglycan is selectively reduced in muscle from young (P14-P21) animals, while levels in adult muscle (≥P35) are unchanged. We conclude that biglycan is ligand for two members of the sarcoglycan complex and regulates their expression at discrete developmental ages.
Introduction
The dystrophin glycoprotein complex (DAPC) links the cytoskeleton to the extracellular matrix and is necessary for muscle cell survival. The core constituents of the DAPC include the cytoskeletal scaffolding molecule dystrophin, the dystroglycan and sarcoglycan transmembrane subcomplexes and sarcospan (Dalkilic and Kunkel, 2003; Hack et al., 2000a; Straub and Campbell, 1997). The importance of the DAPC for maintaining muscle cell viability is underscored by its role in disease. Mutations in dystrophin lead to Duchenne and Becker muscular dystrophy, while mutations in any of the sarcoglycans result in limb-girdle muscular dystrophies. Therefore, understanding DAPC structure and function is essential for elucidating the pathogenesis of these dystrophies as well as for designing therapies to combat them (Campbell and Stull, 2003).
Dystrophin was originally discovered as the product of the gene mutated in Duchenne muscular dystrophy (DMD) and was subsequently established as the cytoskeletal cornerstone of the DAPC (Hoffman et al., 1987). Dystrophin binds actin, syntrophins, dystrobrevins and the cytoplasmic tail of β-dystroglycan (Adams et al., 1993; Blake et al., 2002; Jung et al., 1995; Nawrotzki et al., 1998; Peters et al., 1998; Rybakova et al., 1996). The extracellular domain of β-dystroglycan associates with α-dystroglycan, which in turn binds to the ECM molecules agrin, laminin, and perlecan (Bowe et al., 1994; Gee et al., 1994; Hemler, 1999). This association requires glycosylation of α–dystroglycan, and some muscular dystrophies with strong CNS involvement are due to defects in the addition of these carbohydrates (Haliloglu and Topaloglu, 2004; Moore et al., 2002; Muntoni et al., 2002). α–Dystroglycan also binds to biglycan, but via a structurally distinct mechanism (Bowe et al., 2000; and see below).
In contrast to the dystrophin-dystroglycan-basal lamina axis of the DAPC, the function and molecular associations of the sarcoglycan subcomplex are much less clear. In mature muscle this subcomplex consists of four transmembrane proteins (α, β, γ and δ), each having a large extracellular and a small cytoplasmic domain joined by a single transmembrane span (Holt and Campbell, 1998; Ozawa et al., 2005; Yoshida et al., 1994). Biochemical and genetic evidence indicates that the sarcoglycans can be tightly associated with one another in mature muscle (Chan et al., 1998; Liu and Engvall, 1999). However, there are no known extracellular binding partners for this subcomplex, nor is it understood how the sarcoglycans associate with either the extracellular matrix or with the rest of the DAPC.
These gaps in our understanding are especially significant in view of the central role of sarcoglycans in muscular dystrophy. In DMD, the sarcoglycans, dystrophin and the dystroglycans are lost from the muscle membrane. However, loss of the sarcoglycan complex alone causes Limb-Girdle Muscular Dystrophies (LGMD) which have a milder phenotype than DMD… Four limb-girdle muscular dystrophies, LGMD2 -D,-E,-C and -F, arise from mutations in α-, β-, γ- and δ- sarcoglycan, respectively (reviewed in) (Durbeej et al., 2003; Hack et al., 2000b; Ozawa et al., 2001). Thus, the loss of sarcoglycans is likely to be particularly important in the pathogenesis of both DMD and LGMDs.
In previous work we have shown that the small leucine-rich repeat proteoglycan biglycan binds to α-dystroglycan and is expressed on the muscle cell surface. This interaction requires biglycan's chondroitin sulfate side chains and the carboxyl-terminal third of the α-dystroglycan polypeptide (Bowe et al., 2000). Biglycan is a member of a family of small proteoglycans that includes its closest relation decorin as well as asporin, fibromodulin, lumican, keratocan, PRELP, osteoadherin, epiphycan and osteoglycin (Fisher et al., 1989; Henry et al., 2001; Hocking et al., 1998; Iozzo, 1998; Lorenzo et al., 2001). Biglycan's 38 kD polypeptide core harbors 10 leucine-rich repeats (LRRs), two flanking cysteine-rich domains and two glycosaminoglycan attachment sites at the amino-terminus (Fig. 3a). LRRs are protein-protein interaction domains which in biglycan are involved in binding to collagens I, VI and TGF-ß family members such as BMP-4 (Hildebrand et al., 1994; Moreno et al., 2005; Wiberg et al., 2001). The collagen VI binding is of particular interest since mutations in this molecule result in both Bethlem myopathy and Ullrich's congenital muscular dystrophy in humans. Biglycan is highly expressed in bone and biglycan null mice exhibit a late-onset osteoporosis (>6 months of age) (Ameye and Young, 2002; Xu et al., 1998). Biglycan is highly expressed during myogenesis and plays a role in muscle regeneration (Casar et al., 2004)(Lechner et al., 2006). Finally, biglycan can induce the formation of stress fibers in cultured cells in a manner that is dependent on small GTPases (Tufvesson and Westergren-Thorsson, 2003).
Figure 3. Distinct binding sites for α- and γ- sarcoglycan on the biglycan core polypeptide.
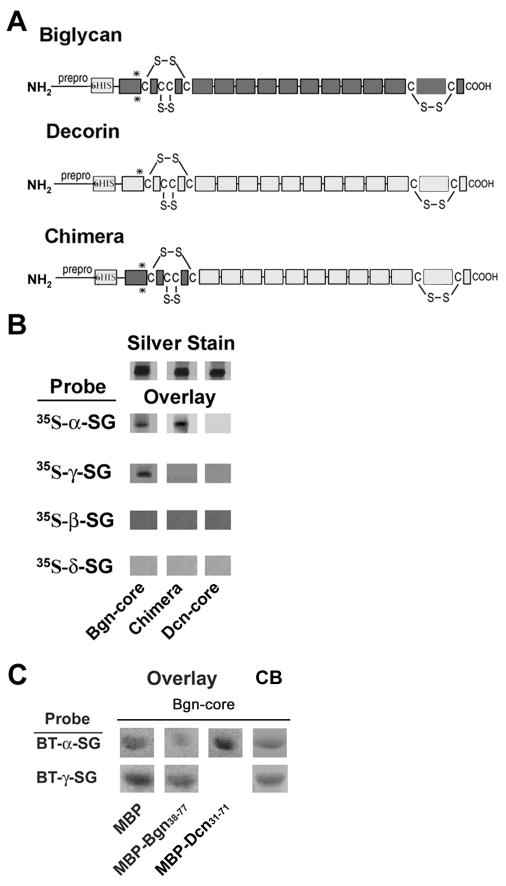
A. Domain structure of biglycan, decorin and a biglycan-decorin chimera. The location of the pre-pro peptide (‘prepro’), 6-His tag, cysteine-rich amino- and carboxyl- domains, LRRs (ten open rectangles in the central domain; some schemes predict an 11th in the carboxyl-terminal cysteine-rich region) and GAG attachment sites (asterisks) are indicated. Note that these sites are present in the recombinant proteins used in this experiment, but they are not substituted with GAGs. B. Binding of sarcoglycans to biglycan, decorin and a chimera. One microgram of each of the purified recombinant proteins was separated by SDS-PAGE and either directly stained (‘silver’) or blotted and probed with 35S-methionine-labelled, in vitro-translated sarcoglycans as indicated. Both α- and γ- sarcoglycan bind to the immobilized biglycan core but not to decorin core. In contrast, only α-sarcoglycan binds to the biglycan-decorin chimeric protein. Thus the first 30 amino acids of biglycan is necessary for its binding to α-sarcoglycan. Neither β- nor δ- sarcoglycan bind to biglycan, decorin or the chimera. C. Competition studies. Sarcoglycan binding to purified recombinant biglycan core polypeptide in the presence of excess MBP-Bgn38-77 (amino acids 38 to 77 of biglycan) or MBP-Dcn31-71 (amino acids 31 to 71 of decorin). These sequences correspond to the first 40 amino acids of the mature biglycan and decorin polypeptides, respectively. Four micrograms of biglycan were separated by SDS-PAGE and either directly stained with Coomassie Blue (CB) or blotted and probed with biotinylated (BT), in vitro-translated α- or γ-sarcoglycan as indicated. Binding of α-sarcoglycan to full-length biglycan was inhibited in the presence of MBP-Bgn38-77, while binding between γ-sarcoglycan and biglycan binding remained unchanged. The decorin fusion protein did not inhibit this interaction.
In view of the binding of biglycan to α-dystroglycan, we wondered whether it might associate with additional DAPC components, be a component of the DAPC in muscle, and regulate the expression of DAPC elements. Here we show that biglycan binds α– and γ– sarcoglycan, but not β– or δ– sarcoglycan. The polypeptide core of biglycan is sufficient for these interactions and the binding sites on biglycan for α– and γ– sarcoglycan are distinct. Both biglycan core and proteoglycan forms are expressed in muscle and are components the DAPC. Finally, the expression of α– and γ– sarcoglycan is reduced in muscle from young (P14-P21) biglycan null animals, while levels in adult muscle (≥P35) are unchanged. We conclude that biglycan is a ligand for two members of the sarcoglycan complex and regulates their expression at discrete developmental ages.
Results
α- and γ- Sarcoglycan bind to biglycan
In earlier work we showed that the CSPG biglycan purified from Torpedo postsynaptic membranes binds to α-dystroglycan (Bowe et al., 2000). Here we asked whether biglycan binds to the sarcoglycans, which together comprise the other major transmembrane subcomplex of the DAPC. As a first step we used a ligand blot overlay assay to test the binding of in vitro-translated α–, β–, γ– and δ– sarcoglycan to preparations of purified Torpedo postsynaptic membranes. As we reported previously, α-dystroglycan binds to biglycan, which in these fractions migrates as a heavily glycosylated, polydisperse band whose position is centered at ∼125kD (Fig. 1a). We observed that α– and γ– sarcoglycan bind to a polypeptide whose appearance and migration are indistinguishable from that of Torpedo biglycan (Fig. 1a). In contrast, no binding of β– or δ– sarcoglycan to this band was detected.
Figure 1. Biglycan binds to α- and γ- sarcoglycan.
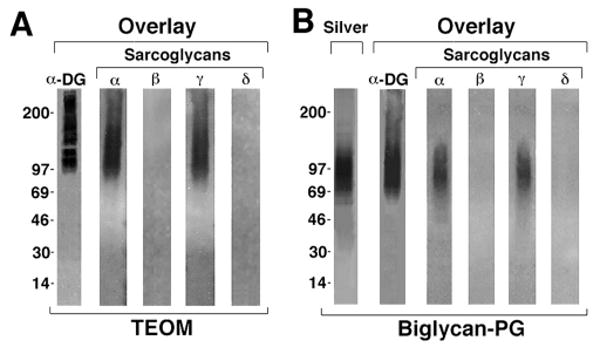
A. Sarcoglycan binding to native biglycan. Postsynaptic membrane fractions from Torpedo electric organ (TEOM; 0.8 μg) were separated on SDS-PAGE gels, blotted onto nitrocellulose then probed with either 35S-methionine-labelled in vitro translated α-dystroglycan or sarcoglycans (α, β, γ, or δ) and analyzed by autoradiography. α-Dystroglycan as well as α- and γ-sarcoglycan bound to a polydisperse band whose center of migration was ∼125kD. In previous work a polypeptide with identical mobility, appearance and α–dystroglycan binding capacity was purified from these fractions and shown to be the proteoglycan biglycan (Bowe et al., 2000). No binding of β– or δ– sarcoglycan to this or any other polypeptide in these fractions was detected. B. Binding of α-dystroglycan and sarcoglycans to purified recombinant biglycan proteoglycan (Biglycan-PG). One microgram of biglycan was separated by SDS-PAGE and either stained with silver or blotted onto nitrocellulose (‘Overlay’) and probed as described above. α-Dystroglycan and α- and γ-sarcoglycan bind to this recombinant, GAG-containing biglycan proteoglycan, while no binding of β– or δ– sarcoglycan is detected.
We next confirmed these results using purified biglycan. Recombinant biglycan proteoglycan was produced in osteosarcoma cells, which yields a product bearing chondroitin sulfate side chains and whose migration is centered at ∼90kd (Fig 1b; Hocking et al., 1996). In agreement with previous results, α-dystroglycan binds to this biglycan CSPG. Figure 1b also shows that α– and γ–sarcoglycan bind to this purified biglycan. However, no binding of β– or δ– sarcoglycan to biglycan was observed.
The binding of α– and γ– sarcoglycan to biglycan could require this proteoglycan's polypeptide core, its GAG side chains, or both domains. To distinguish among these possibilities, we tested the binding of the sarcoglycans to purified recombinant biglycan lacking GAG side chains (“biglycan core”). Fig. 2a shows that the biglycan polypeptide core is sufficient for binding both α– and γ– sarcoglycan. However, we observed no α–dystroglycan binding to the biglycan core. This result is in agreement with our previous work showing that α–dystroglycan binds to neither bacterially-produced recombinant biglycan (non-glycosylated) nor chondroitinase-treated native biglycan (Bowe et al., 2000). Neither β- nor δ- sarcoglycan bound the biglycan polypeptide. Thus the sarcoglycans (α– and γ–) and α-dystroglycan bind to biglycan via distinct mechanisms (GAG -independent and -dependent, respectively).
Figure 2. The biglycan core polypeptide is sufficient for binding to both immobilized and soluble α- and γ-sarcoglycan.
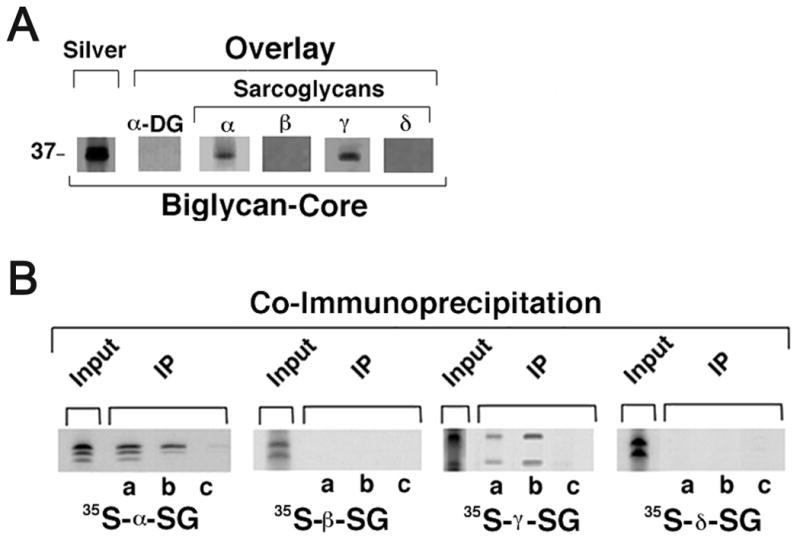
A. Purified recombinant biglycan core polypeptide (1μg) was separated by SDS-PAGE and either silver stained or blotted and probed as described above. α-Dystroglycan did not bind to this GAG-free biglycan. In contrast, both α- and γ- sarcoglycan bind to the biglycan core polypeptide. B. Co-immunoprecipitation of purified recombinant biglycan to recombinant sarcoglycans. His-tagged biglycan core polypeptide (0.5μg/ml) was incubated with the indicated 35S-methionine-labeled, in vitro translated sarcoglycan for 1 hr followed by either anti-biglycan (a), anti-poly-His (b) or normal rabbit Ig (c). Immune complexes were then precipitated with protein G beads and analyzed by SDS-PAGE and autoradiography. Note that both α- and γ- sarcoglycan co-immunoprecipitate with biglycan, while β- and δ- sarcoglycan do not. The labelling of the various sarcoglycans is shown by direct autoradiography of SDS-PAGE-separated in vitro translated polypeptides (‘Input’).
The experiments described above were based on a blot overlay method where denatured and immobilized biglycan was used to assess the binding of in vitro-translated sarcoglycan. To confirm and extend these findings we tested the binding of biglycan to sarcoglycans in solution. Biglycan produced in osteosarcoma cells is properly folded as judged by a number of biochemical and biophysical criteria including far-UV CD and fluorescence emission spectroscopy (Krishnan et al., 1999). We mixed the purified biglycan core with each of the in vitro-translated sarcoglycans and then immunoprecipitated the biglycan. Fig. 2b shows that both α– and γ– sarcoglycan co-immunoprecipitated with biglycan. In contrast, neither β– nor δ- sarcoglycan was observed to associate with biglycan. Together, these results indicate that α– and γ–sarcoglycan bind to biglycan in solution.
α– and γ– Sarcoglycan bind to distinct sites on the biglycan polypeptide
We next characterized the binding of α– and γ– sarcoglycan to the biglycan core polypeptide in more detail. Decorin is a small leucine-rich repeat proteoglycan that is ∼55% identical to biglycan, exhibits a virtually identical domain organization and is also expressed in muscle (Brandan et al., 1992). We find that none of the sarcoglycans bind to purified recombinant decorin (Fig. 3b). Since the primary structures of biglycan and decorin are most divergent at the cysteine-rich domains, we tested sarcoglycan binding to a biglycan-decorin chimera where only the amino-terminal, cysteine-rich domain (30 aa) is derived from biglycan (Fig. 3a). α–Sarcoglycan binds to this chimeric protein; in contrast, γ–sarcoglycan does not interact with this hybrid. Thus, the amino-terminal cysteine-rich domain of biglycan is necessary for mediating its interaction with α–sarcoglycan. Further, the failure of γ–sarcoglycan to bind to this chimera indicates that the binding sites for α– and γ– sarcoglycan on biglycan are likely to be distinct.
To determine whether the amino-terminal domain of biglycan in isolation interacts with α-sarcoglycan, we utilized a recombinant protein comprising amino acids 38 to 77 of human biglycan precursor (corresponding to amino acids 1-40 of the mature polypeptide) fused to the C-terminus of maltose binding protein (MBP-Bgn38-77). Ligand blot overlay showed that biotinylated α– or γ– sarcoglycan bind to purified recombinant biglycan (Fig. 3c). However, binding between α–sarcoglycan and biglycan was inhibited in the presence of MBP-Bgn38-77, demonstrating that the N-terminal cysteine-rich region of biglycan can compete for binding with α-sarcoglycan. MBP-biglycan did not affect binding to γ-sarcoglycan, confirming that the amino-terminal cysteine-rich domain of biglycan is not necessary for its interaction with γ-sarcoglycan. Finally, a recombinant decorin-MBP fusion protein comprising amino acids 31 to 71 of human decorin precursor (MBP-Dcn31-71; amino acids 1-41 of the mature polypeptide) did not affect biglycan binding to α– or γ– sarcoglycan.
Biglycan expression in skeletal muscle
Previous in situ hybridization and immunohistochemical studies have shown that biglycan is expressed by muscle and is localized at the cell surface (Bianco et al., 1990; Bowe et al., 2000). To characterize the biglycan protein expressed in mature muscle we analyzed membrane preparations by immunoblotting (Fig. 4a). Two forms of biglycan, both of which can be solubilized by digitonin, are detected in these preparations. One is a polydisperse band whose migration ranges between ∼60-100kD and is centered at ∼75kD. The second form is a discrete band that migrates at ∼40 kD. The identification of both of these polypeptides as biglycan is confirmed by their absence in muscle membrane fractions prepared from biglycan null mice. The faster-migrating band is only slightly larger than the predicted molecular weight of the biglycan polypeptide core (38 kD) and is likely to represent the polypeptide chain that is N-glycosylated but devoid of GAG chains. The mobility and appearance of the ∼75 kD polypeptide indicates that it is the proteoglycan form of biglycan. Thus, membrane fractions from mature muscle contain two forms of biglycan.
Figure 4. Biglycan is a component of the native DAPC.
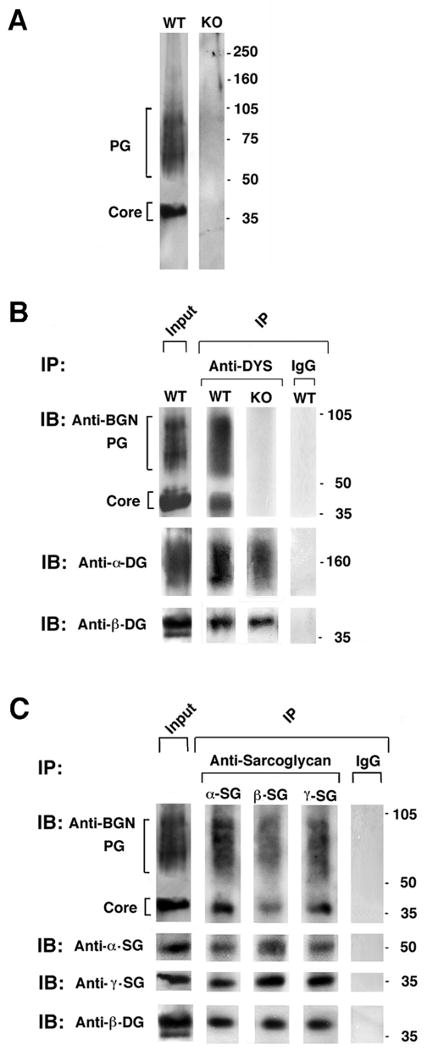
A. Characterization of biglycan expressed in skeletal muscle membranes. Digitonin-solubilized, KCl-washed skeletal muscle membranes (3μg) from either wild type or biglycan null mice (KO; littermates, see methods) were separated on a 3-12% gradient SDS-PAGE. Western blotting for biglycan reveals two polypeptides, one migrating as a discrete band at ∼40kD (core) and a second polydisperse band with migration ranging from 60-100kD (PG). Both forms are absent in membranes prepared from biglycan null muscle. Identical results were obtained when intact membranes were used as starting material for the Western blot (not shown). B. Endogenous biglycan co-immunoprecipitates with dystrophin. Digitonin solubilized membranes were incubated with anti-dystrophin antisera DYS; or control IgG and immunoprecipitates IP were probed for dystroglycan (α and β) and biglycan by Western blotting. As expected, α– and β- dystroglycan co-immunoprecipitate with dystrophin. Notably, both the core and the proteoglycan forms of biglycan were present in these immune complexes. Neither biglycan form was detected in immunoprecipitates from biglycan null mice. C. Co-immunoprecipitation of biglycan with sarcoglycans. Digitonin-solubilized membranes were incubated with antibodies to α–, ß- or γ– sarcoglycan. Both the core and the proteoglycan forms of biglycan are detected in the immunoprecipitates. Equivalent levels of the three sarcoglycans are detected in the immunoprecipitates from wild type and biglycan null muscle membranes.
Biglycan is associated with the native DAPC
The experiments presented above show that biglycan binds by distinct mechanisms to three elements of the DAPC. To determine whether endogenous biglycan is associated with the native DAPC, digitonin-solubilized membrane fractions from mature muscle were immunoprecipitated with antibodies to various components of the complex. Fig. 4b shows that both the core and the proteoglycan forms of biglycan co-immunoprecipitate with dystrophin. Moreover, biglycan also co-immunoprecipitated with α–, β– or γ– sarcoglycan (Fig. 4c). Since biglycan does not bind directly to either dystrophin or to β-sarcoglycan, its presence in these immunoprecipitates indicates that biglycan associates with the mature DAPC expressed in muscle cell membranes. Finally, these biochemical data also indicate that in mature muscle neither sarcoglycan nor dystroglycan expression is affected by the absence of biglycan. Immunostaining for the sarcoglycans also showed no difference in the expression of these DAPC components in mature muscle (Table I.)
Table I. Expression of DAPC components during development in biglycan null as compared to wild type muscle*.
| Component | P14 | P21 | P35 |
|---|---|---|---|
| Dystrophin | ⇔ | ⇔ | ⇔ |
| α-sarcoglycan | ⇓ | ⇔ | ⇔ |
| γ-sarcoglycan | ⇔ | ⇓ | ⇔ |
| β-sarcoglycan | ⇔ | ⇔ | ⇔ |
| δ-sarcoglycan | ⇔ | ⇔ | ⇔ |
| β-dystroglycan | ⇔ | ⇔ | ⇔ |
No change
Reduced in biglycan null compared to controls
α- and γ- Sarcoglycan expression is reduced in immature biglycan null mice
Biglycan expression is strongly developmentally regulated, with maximal levels observed in the first two to three weeks of postnatal development in the mouse (Casar et al., 2004)(Lechner et al., 2006). We therefore examined whether the absence of biglycan can affect α- and/or γ- sarcoglycan expression at these ages. Quadriceps femoris muscles from P14 and P21 wild type and biglycan null mice were harvested, sectioned, and immunolabeled for dystrophin, α-, β–, γ– and δ– sarcoglycan. The level of dystrophin (Fig. 5a) as well as β–dystroglycan was unchanged at both ages (Table I). However, sarcoglycan expression showed age-dependent expression changes in biglycan null mice. In P14 mice there was a selective reduction in the expression of α–sarcoglycan as judged by both immunostaining and western blotting (Fig. 5). By P21, α-sarcoglycan levels had recovered and were comparable to controls at (Fig. 5B). In wild type muscle there was a dramatic increase in the level of γ-sarcoglycan expression between P14 and P21. However, γ-sarcoglycan levels remained low at P21 in the biglycan null muscle. In contrast, the expression of β– and δ– sarcoglycans were unchanged at any of the ages examined (Fig. 5C). The results are summarized in Table I. Thus, α- and γ- sarcoglycan show distinctive, selective, and developmentally-dependent decreases in expression levels in biglycan null skeletal muscle.
Figure 5. Reduced α- and γ- sarcoglycan expression in immature biglycan null mice.
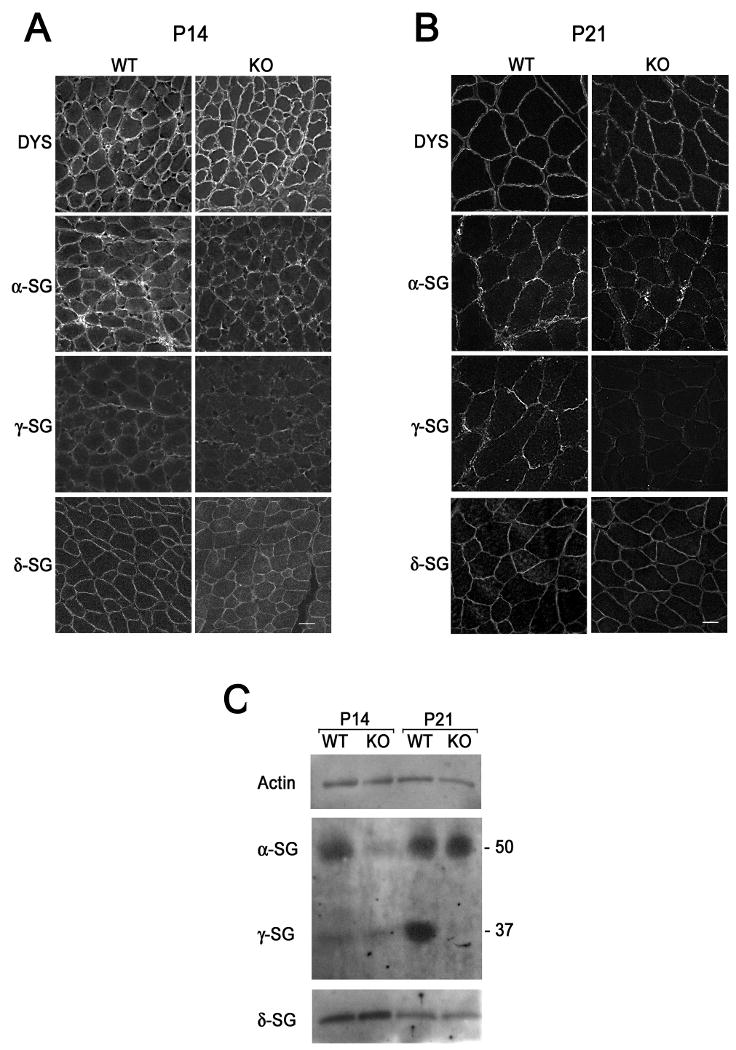
Immunohistochemical analysis of P14, A., and P21, B., mouse muscle. Sections of quadriceps femoris from congenic P14 wild type and biglycan null (KO) mice were sectioned, mounted on the same slides and immunolabelled for dystrophin or α-, γ-, or δ- sarcoglycan. A. α-Sarcoglycan levels at the sarcolemma of P14 biglycan null muscle are selectively reduced in the biglycan null as compared to wild type muscle. B. γ-Sarcoglycan expression is reduced in P21 biglycan null mice, while α-sarcoglycan is unchanged. The level of dystrophin and δ-sarcoglycan is the same at both ages. Scale bars = 10 μm. C. Biochemical analysis of sarcoglycan expression. α- and γ- Sarcoglycan expression is reduced at distinct postnatal ages in skeletal muscle membranes from immature mice. KCl-washed skeletal muscle membranes (3 μg) from P14 or P21 congenic wild type or biglycan null mice (KO) were separated via SDS-PAGE. Western blotting was performed for α-, γ-, or δ- sarcoglycan or actin (loading control). Western blotting for α- and γ- sarcoglycan was performed on the same gel. Western blotting for δ-sarcoglycan and actin was performed in parallel on the same gel. α- Sarcoglycan levels are reduced in P14 biglycan null membranes while γ-sarcoglycan levels are reduced in P21 biglycan null membranes. Equivalent levels of δ-sarcoglycan and actin expression are seen at both ages. Similar results were observed in muscles from two other sets of mice.
Discussion
In this work we present evidence that biglycan is associated with the native DAPC and has distinct binding sites for α-sarcoglycan, γ-sarcoglycan and α-dystroglycan. To our knowledge, biglycan is the first extracellular ligand reported to associate with any of the sarcoglycans. We used a range of biochemical methods to demonstrate the association of biglycan with two of the sarcoglycans. Blot overlay assays showed that recombinant α- and γ- sarcoglycan bind to both endogenous and recombinant biglycan proteoglycan. The results from this solid-phase assay were confirmed by co-immunoprecipitation (Fig. 2b). Several lines of evidence indicate that this binding is specific. 1) Biglycan is the only binding partner of α- and γ- sarcoglycan detected in Torpedo postsynaptic membranes. 2) Neither β- nor δ- sarcoglycan bind to biglycan as assessed by any of the assays. 3) None of the sarcoglycans bound to decorin, a small leucine-rich repeat proteoglycan that has the same overall structure as biglycan and is ∼55% identical to it. 4) α-Dystroglycan does not bind to the biglycan (or decorin) core polypeptide. 5) Biglycan is associated with the native DAPC isolated from skeletal muscle. 6) The expression of α- and γ-, but neither β- nor δ-, sarcoglycan expression are reduced in developing biglycan null muscle.
Our results indicate that biglycan harbors distinct binding sites for α-dystroglycan, α-sarcoglycan and γ-sarcoglycan. The chondroitin sulfate GAG chains of Torpedo biglycan are necessary for its binding to α-dystroglycan (Bowe et al., 2000). Here, we extended these studies by showing that α-dystroglycan binds to purified recombinant human biglycan proteoglycan, but not to the core polypeptide produced in the same cells (Fig. 1 and 2). It is unknown whether the GAG chains of biglycan are sufficient for this interaction. It is possible that the binding site for α-dystroglycan encompasses a domain on biglycan's polypeptide core as well as its GAG side chains.
Unlike α-dystroglycan, α- and γ- sarcoglycan bind to both the biglycan proteoglycan and core polypeptide (Figs. 1 and 2). Thus, biglycan's GAG side chains are dispensable for sarcoglycan binding. The use of a biglycan-decorin chimera and an MBP fusion protein containing the first 30 amino acids of the mature biglycan polypeptide (MBP-Bgn38-77) revealed that the N-terminal domain is sufficient for α-sarcoglycan binding (Fig. 3). In contrast, γ- sarcoglycan did not bind the biglycan-decorin chimera, nor was its binding to intact biglycan inhibited by MBP-Bgn38-77. Therefore, the biglycan core polypeptide harbors distinct binding sites for α- and γ- sarcoglycan. The presence of two binding sites on biglycan is not unexpected in view of the topologies of α- and γ- sarcoglycan, which are type I and type II membrane proteins, respectively (Ozawa et al., 2005).
α-Sarcoglycan is the first protein demonstrated to bind the N-terminal cysteine-rich domain of biglycan. Other molecules reported to bind biglycan, such as collagens I and VI, as well as TGF-ß and BMP-4, interact via its LRR domains. Moreover, these three proteins also bind decorin (Ameye and Young, 2002; Hocking et al., 1998; Moreno et al., 2005; Wiberg et al., 2001). These observations are consistent with the structural relationship of decorin and biglycan, which are most divergent in their N-terminal cysteine-rich domains. Finally, it is noteworthy that the primary structure of biglycan is highly conserved. Mouse and human biglycans are >95% identical at the amino acid level (mature polypeptides) and the partial sequence of Torpedo biglycan is 76% identical to the human homolog (28 of 37 amino acids) (Bowe et al., 2000). This strong conservation supports the idea that biglycan's core polypeptide is particularly important for its function. (At present, we do not know whether the non-glycanated form of biglycan is present in Torpedo membranes, since our antibodies do not cross react with any biglycan in this organism.) The presence of both a non-glycanated and proteoglycan form of biglycan in mammalian muscle also raises the possibility that these two biglycan species may have distinct functions.
Our results provide in vivo evidence for the selective regulation of different sarcoglycans in development. In mature muscle there is general agreement that the major sarcoglycan complex consists of a tetramer of α,β,γ, and δ subunits (Holt and Campbell, 1998; Ozawa et al., 2005; Wheeler et al., 2002). However, genetic and cell culture studies indicate that other complexes can also occur. For example, α–,β–, and δ– sarcoglycan are expressed, albeit at lower levels, in the absence of γ–sarcoglycan (Hack et al., 2000a). β– and δ– Sarcoglycan appear to be particularly important for assembly in some systems (Adams et al., 2004; Chan et al., 1998), and mutations in them leads to complete loss of the complex in vivo. Mutations α–sarcoglycan can be at least partially compensated for by ε–sarcoglycan (Imamura et al., 2005; Liu and Engvall, 1999; Straub et al., 1999). Our results showing selective regulation of α– and γ- sarcoglycan are in accord with studies showing a stepwise assembly of sarcoglycans in cultured cells. For example, Liu and Engvall (1999) showed that levels of δ–sarcoglycan are unchanged during myotube formation, while α– and γ- increase coordinately at a later stage. Further, Noguchi et al. (Noguchi et al., 2000) detected a complex consisting of α– and γ–, but without β– and δ– sarcoglycan, in association with dystroglycan. Although it is not currently possible to fully reconcile data obtained from genetic and various cell culture systems, it is clear that multiple sarcoglycan complexes are likely to be present in muscle, particularly during development.
The identification of biglycan as an extracellular ligand for three components of the DAPC could also have implications for muscular dystrophy therapy. As noted above, disruption of the sarcoglycan complex is thought to be the cause of muscle cell death in many muscular dystrophies. Our finding that biglycan plays a particularly important role in regulating DAPC assembly in early postnatal mice suggests a novel avenue for rescuing DAPC function, even in the absence of dystrophin.
Materials and Methods
In Vitro Transcription/Translation
The in vitro expression plasmids encoding full length human α-, β-, γ- and δ-sarcoglycan in the vector pMGT, developed by A. Ahn, were generously provided by Louis Kunkel (Ahn et al., 1996). The sarcoglycan polypeptides and α-dystroglycan were generated by in vitro transcription/translation using the Promega TNT T7 coupled reticulocyte system as per the manufacturer's instructions as described previously (Bowe et al., 2000). Recombinant proteins produced from these vectors often show multiple bands (e.g. Fig. 2), presumably due to alternative translation start sites in the plasmid (Ahn et al., 1996). For protein to be used in radioactive ligand blot overlay assay, the reaction mixture contained methionine (with no unlabeled methionine). For protein to be used in non-radioactive ligand blot overlay assay, the reaction mixture contained biotin-lysyl-tRNA with unlabeled methionine.
Preparation of recombinant biglycan and biglycan-decorin chimeras
Recombinant human decorin and biglycan proteoglycan and core protein glycoforms were expressed and purified using the vaccinia virus/T7 bacteriophage expression system (Elroy-Stein et al., 1989; Fuerst et al., 1986), as previously described (Hocking et al., 1996; Ramamurthy et al., 1996). Briefly, conditioned media was applied to Sephadex G-50 columns equilibrated and eluted with 5 mM imidazole, 0.5 M NaCl, 20 mM Tris-HCl, pH 8.0, 0.2% CHAPS to separate macromolecules from unincorporated radioactive precursors. The eluted macromolecular fraction was applied to iminodiacetic acid immobilized on Sepharose 6B that had been equilibrated with nickel chloride. After sample application, the column was washed with 5 column volumes of 30 mM imidazole, 0.5 M NaCl, 20 mM Tris-HCl, pH 8.0, 0.2% CHAPS and bound material eluted with 150 mM imidazole, 0.5 M NaCl, 20 mM Tris-HCl, pH 8.0, 0.2% CHAPS. Pooled fractions were dialyzed against phosphate-buffered saline, pH 7.4 and concentrated on Ultrafree-15 centrifugal filter devices. Protein concentrations were determined by the molar extinction coefficient (Pace et al., 1995).
Domain substitution between cDNAs encoding human biglycan (pBGN4) and human decorin (pDCN1) was done by a strategy of strand-overlap extension (SOE) PCR utilizing specific overlapping oligonucleotide primers (Ho et al., 1989; Horton et al., 1989). Each cDNA construct was created in the plasmid backbone of pTcam1 (McQuillan et al., 2001) downstream of a sequence encoding the human insulin signal sequence and a poly-histidine tag. The resulting pBD2 chimera contains sequence encoding biglycan protein from amino acid 38 to 78 (amino-terminal domain of the mature core protein) and decorin protein from amino acid 70 to 359 (leucine-rich repeats 1-10 and the C-terminal domain), BGN38-78DCN70-359). All constructs were verified by DNA sequencing and by in vitro transcription and translation directly from the plasmid vector (TNT Transcription/Translation System, Promega) in the presence of 40 μCi of Trans[35S]-label and reaction products were analyzed by SDS-PAGE. All recombinant vaccinia viruses expressing wild type and chimeric proteins were generated by homologous recombination between plasmid constructs and wild type vaccinia virus (vTF7-3), as described previously (Hocking et al., 1996; McQuillan et al., 2001).
Preparation of maltose binding protein or maltose binding protein fused to amino acids
Maltose binding protein or maltose binding protein fused to amino acids 38 to 77 of human biglycan (38-DEEASGSDTTSGVPDLDSVTPTFSAMCPFGCHCHLRVVQC-77) or amino acids 31 to 71 of human decorin (31-DEASGIIPYDPDNPLISMCPYRCQCHLRVVQCSDLGLDKVP-71) were expressed from a plasmid derived from pMALp2 (New England Biolabs, Beverly, MA) as previously described (Joh et al., 1998).
Antibodies
Antibodies to the sarcoglycans and other DAPC components were obtained from NovoCastra (Newcastle upon Tyne, UK). Rabbit anti-ζ-sarcoglycan was generously provided by Elizabeth McNally (The University of Chicago, Chicago, IL) and rabbit anti-dystrophin (anti 6-10) was generously provided by Louis Kunkel (Lidov et al., 1990). The anti-6-His was from Sigma (St. Louis, MO). Rabbit anti-decorin was generously provided by Larry Fisher (Bianco et al., 1990). The antiserum to biglycan was produced in rabbit using a full-length human biglycan bacterial fusion protein produced as described previously (Bowe et al., 2000). This antiserum recognizes mouse and human biglycan (see Results) but does not recognize decorin (not shown).
Skeletal muscle membrane preparations and digitonin solubilization
Adult muscle (0.25 gm) was homogenized in 1ml dissection buffer containing: 0.3 M sucrose, 35 mM Tris (pH 7.4), 10mM EDTA, 10mM EGTA, PMSF, leupeptin, aprotinin, benzamidine, NEM, pepstatin A and sodium azide. Samples were sonicated on ice for 3 × 10 s and centrifuged at 7000 × g at 4°C for 20 min. The supernatant was filtered over two layers of gauze, solid KCl was added to a final concentration of 0.6 M and samples were centrifuged as above. The membranes were then collected by centrifugation at 140,000 × g for 60 min at 4°C. Digitonin solubilization was performed as described by Campbell and Kahl (1989). Protein levels were determined using the BCA protein assay kit (Pierce, Rockford, IL).
Ligand Blot Overlay
Purified recombinant biglycan and decorin proteins were separated by SDS-PAGE (5-15% gradient gels) and transferred to nitrocellulose. The blots were rinsed and blocked for 3 h in HBSS containing 1mM CaCl2, 1mM MgCl2, 1% BSA, 1% nonfat dry milk, 1mM DTT, 10mM HEPES pH 7.4, and then incubated 16 h in the same buffer containing either [35S]methionine-labeled or biotinylated sarcoglycans +/- 1.6 μM MBP-Bgn38-77 or MBP-Dcn31-71. For [35S]-methionine-labeled sarcoglycans, blots were rinsed and dried, and bound sarcoglycan was visualized by autoradiography. For biotin-lysyl-tRNA-labeled sarcoglycans, blots of bacterially-expressed biglycan (Bowe et al., 2000) were rinsed and incubated for 1 hr with streptavidin-HRP (1:1500) in TBS + 0.5% Tween® 20. Bound sarcoglycan was detected by enhanced chemiluminescence (Amersham, Little Chalfon, UK).
Co-Immunoprecipitation
His-tagged biglycan core polypeptide was incubated with the indicated 35S-methionine labeled, in vitro-translated sarcoglycans in solution for 1 hr. The mixtures were then incubated with a rabbit anti-biglycan antiserum, an anti-6XHis antibody, or normal rabbit IgG as indicated and immune complexes were precipitated with protein G beads. Immunoprecipitation of digitonin solubilized skeletal muscle membrane fractions with anti-dystrophin (anti 6-10) or anti-sarcoglycan antibodies (NovoCastra) were performed using the Seize × Immunoprecipitation kit (Pierce, Rockford, IL). The solubilized fractions were incubated with 50μg of antibodies or normal IgG (rabbit or mouse). The precipitated products were eluted and collected according to manufacturer's protocols.
Western blot analysis
Proteins were transferred from SDS-PAGE gels to nitrocellulose membranes and probed as previously described (Bowe et al., 2000). Bound antibodies were detected with enhanced chemiluminescence according to manufacturer's protocols (Amersham).
Biglycan null mice
Biglycan null mice (Xu et al., 1998) on either a C57Bl/6 or a C3H (congenic; Jackson Laboratories) background were housed at the Brown University animal care facility and maintained according to IACUC regulations. For experiments using the C57/Bl6, mutant male mice were generated by breeding heterozygous females to wild-type males. PCR genotyping was performed on genomic DNA using primer pairs specific for mutant and wild type biglycan alleles. In these experiments material from the mutant male mice was compared with that of their wild type male littermates. For experiments with the (congenic) C3H mice, age-matched wild type mice (Jackson Laboratories) were used. Equivalent results were observed with both strains.
Immunohistochemistry
Quadriceps femoris muscles from P14 or P21 wild type or biglycan null mice were harvested and flash frozen in isopentane. 10 μM sections were mounted onto slides and immunostaining was performed using the M.O.M. Basic Kit (Vector Laboratories, Burlingame, CA) as per the manufacturer's instructions. Sections were mounted in Permafluor (Thermo Electron Corporation, Pittsburgh, PA) and observed using a Nikon (Melville, NY) Eclipse E800 microscope. Images were acquired with Scanalytics (Fairfax, VA) IP Lab Spectrum software. Additionally, sections were analyzed using confocal laser scanning microscopy (Leica TCS SP2 Acousto-Optical Beam Splitter (AOBS)). Images were acquired using Leica LCS acquisition software and imported into Adobe Photoshop.
Acknowledgments
We thank Beth McKechnie for providing superb technical assistance for this work, Louis Kunkel and Andrew Ahn for generously providing sarcoglycan clones, Larry Fisher for the gift of anti-decorin antisera and Hilliary Creely for producing the bacterially-expressed biglycan. We also thank Mark Bowe for participation in early experiments and Beatrice Lechner, Alison Amenta and H. Creely for comments on the manuscript. This work was supported by grants from the NIH (HD23924 and RR15578).
Abbreviations
- DGC
dystrophin-glycoprotein complex
- DMD
Duchenne muscular dystrophy
- LGMD
Limb-girdle muscular dystrophy
- CK
creatine kinase
- PRELP
proline-arginine rich and leucine-rich repeat protein
- LRR
leucine-rich repeat
- CSPG
chondroitin sulfate proteoglycan
- GAG
glycosaminoglycan
References
- Adams ME, Butler MH, Dwyer TM, Peters MF, Murnane AA, Froehner SC. Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron. 1993;11(3):531–540. doi: 10.1016/0896-6273(93)90157-m. [DOI] [PubMed] [Google Scholar]
- Adams ME, Kramarcy N, Fukuda T, Engel AG, Sealock R, Froehner SC. Structural abnormalities at neuromuscular synapses lacking multiple syntrophin isoforms. J Neurosci. 2004;24(46):10302–10309. doi: 10.1523/JNEUROSCI.3408-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn AH, Freener CA, Gussoni E, Yoshida M, Ozawa E, Kunkel LM. The three human syntrophin genes are expressed in diverse tissues, have distinct chromosomal locations, and each bind to dystrophin and its relatives. J Biol Chem. 1996;271(5):2724–2730. doi: 10.1074/jbc.271.5.2724. [DOI] [PubMed] [Google Scholar]
- Ameye L, Young MF. Mice deficient in small leucine-rich proteoglycans: novel in vivo models for osteoporosis, osteoarthritis, Ehlers-Danlos syndrome, muscular dystrophy, and corneal diseases. Glycobiology. 2002;12(9):107R–116R. doi: 10.1093/glycob/cwf065. [DOI] [PubMed] [Google Scholar]
- Bianco P, Fisher LW, Young MF, Termine JD, Robey PG. Expression and localization of the two small proteoglycans biglycan and decorin in developing human skeletal and non-skeletal tissues. J Histochem Cytochem. 1990;38(11):1549–1563. doi: 10.1177/38.11.2212616. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82(2):291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- Bowe MA, Deyst KA, Leszyk JD, Fallon JR. Identification and purification of an agrin receptor from Torpedo postsynaptic membranes: A heteromeric complex related to the dystroglycans. Neuron. 1994;12:1173–1180. doi: 10.1016/0896-6273(94)90324-7. [DOI] [PubMed] [Google Scholar]
- Bowe MA, Mendis DB, Fallon JR. The small leucine-rich repeat proteoglycan biglycan binds to alpha- dystroglycan and is upregulated in dystrophic muscle. J Cell Biol. 2000;148(4):801–810. doi: 10.1083/jcb.148.4.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandan E, Fuentes ME, Andrade W. Decorin, a chondroitin/dermatan sulfate proteoglycan is under neural control in rat skeletal muscle. J Neurosci Res. 1992;32(1):51–59. doi: 10.1002/jnr.490320107. [DOI] [PubMed] [Google Scholar]
- Campbell KP, Stull JT. Skeletal muscle basement membrane-sarcolemma-cytoskeleton interaction minireview series. J Biol Chem. 2003;278(15):12599–12600. doi: 10.1074/jbc.R300005200. [DOI] [PubMed] [Google Scholar]
- Casar JC, McKechnie BA, Fallon JR, Young MF, Brandan E. Transient up-regulation of biglycan during skeletal muscle regeneration: delayed fiber growth along with decorin increase in biglycan-deficient mice. Dev Biol. 2004;268(2):358–371. doi: 10.1016/j.ydbio.2003.12.025. [DOI] [PubMed] [Google Scholar]
- Chan YM, Bonnemann CG, Lidov HGW, Kunkel LM. Molecular organization of sarcoglycan complex in mouse myotubes in culture. J Cell Biol. 1998;143(7):2033–2044. doi: 10.1083/jcb.143.7.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalkilic I, Kunkel LM. Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev. 2003;13(3):231–238. doi: 10.1016/s0959-437x(03)00048-0. [DOI] [PubMed] [Google Scholar]
- Durbeej M, Sawatzki SM, Barresi R, Schmainda KM, Allamand V, Michele DE, Campbell KP. Gene transfer establishes primacy of striated vs. smooth muscle sarcoglycan complex in limb-girdle muscular dystrophy. Proc Natl Acad Sci U S A. 2003;100(15):8910–8915. doi: 10.1073/pnas.1537554100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elroy-Stein O, Fuerst TR, Moss B. Cap-independent translation of mRNA conferred by encephalomyocarditis virus 5′ sequence improves the performance of the vaccinia virus/bacteriophage T7 hybrid expression system. Proc Natl Acad Sci U S A. 1989;86(16):6126–6130. doi: 10.1073/pnas.86.16.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher LW, Termine JD, Young MF. Deduced protein sequence of bone small proteoglycan I (biglycan) shows homology with proteoglycan II (decorin) and several nonconnective tissue proteins in a variety of species. J Biol Chem. 1989;264(8):4571–4576. [PubMed] [Google Scholar]
- Fuerst TR, Niles EG, Studier FW, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci U S A. 1986;83(21):8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan-alpha, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77(5):675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Hack AA, Groh ME, McNally EM. Sarcoglycans in muscular dystrophy. Microsc Res Tech. 2000a;48(3-4):167–180. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<167::AID-JEMT5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Hack AA, Lam MY, Cordier L, Shoturma DI, Ly CT, Hadhazy MA, Hadhazy MR, Sweeney HL, McNally EM. Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J Cell Sci. 2000b;113(Pt 14):2535–2544. doi: 10.1242/jcs.113.14.2535. [DOI] [PubMed] [Google Scholar]
- Haliloglu G, Topaloglu H. Glycosylation defects in muscular dystrophies. Curr Opin Neurol. 2004;17(5):521–527. doi: 10.1097/00019052-200410000-00002. [DOI] [PubMed] [Google Scholar]
- Hemler ME. Dystroglycan versatility. Cell. 1999;97(5):543–546. doi: 10.1016/s0092-8674(00)80764-3. [DOI] [PubMed] [Google Scholar]
- Henry MD, Satz JS, Brakebusch C, Costell M, Gustafsson E, Fassler R, Campbell KP. Distinct roles for dystroglycan, beta1 integrin and perlecan in cell surface laminin organization. J Cell Sci. 2001;114(Pt 6):1137–1144. doi: 10.1242/jcs.114.6.1137. [DOI] [PubMed] [Google Scholar]
- Hildebrand A, Romaris M, Rasmussen LM, Heinegard D, Twardzik DR, Border WA, Ruoslahti E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem J. 1994;302(Pt 2):527–534. doi: 10.1042/bj3020527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77(1):51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Hocking AM, Shinomura T, McQuillan DJ. Leucine-rich repeat glycoproteins of the extracellular matrix. Matrix Biol. 1998;17(1):1–19. doi: 10.1016/s0945-053x(98)90121-4. [DOI] [PubMed] [Google Scholar]
- Hocking AM, Strugnell RA, Ramamurthy P, McQuillan DJ. Eukaryotic expression of recombinant biglycan. Post-translational processing and the importance of secondary structure for biological activity. J Biol Chem. 1996;271(32):19571–19577. [PubMed] [Google Scholar]
- Hoffman EP, Brown RJ, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Holt KH, Campbell KP. Assembly of the sarcoglycan complex. Insights for muscular dystrophy. J Biol Chem. 1998;273(52):34667–34670. doi: 10.1074/jbc.273.52.34667. [DOI] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77(1):61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Imamura M, Mochizuki Y, Engvall E, Takeda S. Epsilon-sarcoglycan compensates for lack of alpha-sarcoglycan in a mouse model of limb-girdle muscular dystrophy. Hum Mol Genet. 2005;14(6):775–783. doi: 10.1093/hmg/ddi072. [DOI] [PubMed] [Google Scholar]
- Iozzo RV. Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem. 1998;67:609–652. doi: 10.1146/annurev.biochem.67.1.609. [DOI] [PubMed] [Google Scholar]
- Joh D, Speziale P, Gurusiddappa S, Manor J, Hook M. Multiple specificities of the staphylococcal and streptococcal fibronectin-binding microbial surface components recognizing adhesive matrix molecules. Eur J Biochem. 1998;258(2):897–905. doi: 10.1046/j.1432-1327.1998.2580897.x. [DOI] [PubMed] [Google Scholar]
- Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP. Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. J Biol Chem. 1995;270(45):27305–27310. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- Krishnan P, Hocking AM, Scholtz JM, Pace CN, Holik KK, McQuillan DJ. Distinct secondary structures of the leucine-rich repeat proteoglycans decorin and biglycan. Glycosylation-dependent conformational stability. J Biol Chem. 1999;274(16):10945–10950. doi: 10.1074/jbc.274.16.10945. [DOI] [PubMed] [Google Scholar]
- Lechner BE, Lim JH, Mercado ML, Fallon JR. Developmental regulation of biglycan expression in muscle and tendon. Muscle and Nerve. 2006 doi: 10.1002/mus.20596. In Press. [DOI] [PubMed] [Google Scholar]
- Liu LA, Engvall E. Sarcoglycan isoforms in skeletal muscle. J Biol Chem. 1999;274(53):38171–38176. doi: 10.1074/jbc.274.53.38171. [DOI] [PubMed] [Google Scholar]
- Lorenzo P, Aspberg A, Onnerfjord P, Bayliss MT, Neame PJ, Heinegard D. Identification and characterization of asporin. a novel member of the leucine-rich repeat protein family closely related to decorin and biglycan. J Biol Chem. 2001;276(15):12201–12211. doi: 10.1074/jbc.M010932200. [DOI] [PubMed] [Google Scholar]
- McQuillan DJ, Seo NS, Hocking AM, McQuillan CI. Recombinant expression of proteoglycans in mammalian cells. Utility and advantages of the vaccinia virus/T7 bacteriophage hybrid expression system. Methods Mol Biol. 2001;171:201–219. doi: 10.1385/1-59259-209-0:201. [DOI] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418(6896):422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- Moreno M, Munoz R, Aroca F, Labarca M, Brandan E, Larrain J. Biglycan is a new extracellular component of the Chordin-BMP4 signaling pathway. Embo J. 2005;24(7):1397–1405. doi: 10.1038/sj.emboj.7600615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntoni F, Brockington M, Blake DJ, Torelli S, Brown SC. Defective glycosylation in muscular dystrophy. Lancet. 2002;360(9343):1419–1421. doi: 10.1016/S0140-6736(02)11397-3. [DOI] [PubMed] [Google Scholar]
- Nawrotzki R, Loh NY, Ruegg MA, Davies KE, Blake DJ. Characterisation of alpha-dystrobrevin in muscle. J Cell Sci. 1998;111(Pt 17):2595–2605. doi: 10.1242/jcs.111.17.2595. [DOI] [PubMed] [Google Scholar]
- Noguchi S, Wakabayashi E, Imamura M, Yoshida M, Ozawa E. Formation of sarcoglycan complex with differentiation in cultured myocytes. Eur J Biochem. 2000;267(3):640–648. doi: 10.1046/j.1432-1327.2000.00998.x. [DOI] [PubMed] [Google Scholar]
- Ozawa E, Mizuno Y, Hagiwara Y, Sasaoka T, Yoshida M. Molecular and cell biology of the sarcoglycan complex. Muscle Nerve. 2005 doi: 10.1002/mus.20349. [DOI] [PubMed] [Google Scholar]
- Ozawa E, Nishino I, Nonaka I. Sarcolemmopathy: muscular dystrophies with cell membrane defects. Brain Pathol. 2001;11(2):218–230. doi: 10.1111/j.1750-3639.2001.tb00394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4(11):2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters MF, Sadoulet-Puccio HM, Grady MR, Kramarcy NR, Kunkel LM, Sanes JR, Sealock R, Froehner SC. Differential membrane localization and intermolecular associations of alpha-dystrobrevin isoforms in skeletal muscle. J Cell Biol. 1998;142(5):1269–1278. doi: 10.1083/jcb.142.5.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthy P, Hocking AM, McQuillan DJ. Recombinant decorin glycoforms. Purification and structure. J Biol Chem. 1996;271(32):19578–19584. doi: 10.1074/jbc.271.32.19578. [DOI] [PubMed] [Google Scholar]
- Rybakova IN, Amann KJ, Ervasti JM. A new model for the interaction of dystrophin with F-actin. J Cell Biol. 1996;135(3):661–672. doi: 10.1083/jcb.135.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub V, Campbell KP. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr Opin Neurol. 1997;10(2):168–175. doi: 10.1097/00019052-199704000-00016. [DOI] [PubMed] [Google Scholar]
- Straub V, Ettinger AJ, Durbeej M, Venzke DP, Cutshall S, Sanes JR, Campbell KP. epsilon-sarcoglycan replaces alpha-sarcoglycan in smooth muscle to form a unique dystrophin-glycoprotein complex. J Biol Chem. 1999;274(39):27989–27996. doi: 10.1074/jbc.274.39.27989. [DOI] [PubMed] [Google Scholar]
- Tufvesson E, Westergren-Thorsson G. Biglycan and decorin induce morphological and cytoskeletal changes involving signalling by the small GTPases RhoA and Rac1 resulting in lung fibroblast migration. J Cell Sci. 2003;116(Pt 23):4857–4864. doi: 10.1242/jcs.00808. [DOI] [PubMed] [Google Scholar]
- Wheeler MT, Allikian MJ, Heydemann A, McNally EM. The sarcoglycan complex in striated and vascular smooth muscle. Cold Spring Harb Symp Quant Biol. 2002;67:389–397. doi: 10.1101/sqb.2002.67.389. [DOI] [PubMed] [Google Scholar]
- Wiberg C, Hedbom E, Khairullina A, Lamande SR, Oldberg A, Timpl R, Morgelin M, Heinegard D. Biglycan and decorin bind close to the n-terminal region of the collagen VI triple helix. J Biol Chem. 2001;276(22):18947–18952. doi: 10.1074/jbc.M100625200. [DOI] [PubMed] [Google Scholar]
- Xu T, Bianco P, Fisher LW, Longenecker G, Smith E, Goldstein S, Bonadio J, Boskey A, Heegaard AM, Sommer B, Satomura K, Dominguez P, Zhao C, Kulkarni AB, Robey PG, Young MF. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat Genet. 1998;20(1):78–82. doi: 10.1038/1746. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Suzuki A, Yamamoto H, Noguchi S, Mizuno Y, Ozawa E. Dissociation of the complex of dystrophin and its associated proteins into several unique groups by n-octyl beta-D-glucoside. Eur J Biochem. 1994;222(3):1055–1061. doi: 10.1111/j.1432-1033.1994.tb18958.x. [DOI] [PubMed] [Google Scholar]