

Abstract
We used two virtual screening programs, ICM and GOLD, to dock nearly 50,000 compounds into each of two conformations of the target protein ricin A chain (RTA). A limited control set suggests that candidates scored highly by two programs may have a higher probability of being ligands than those in a list from a single program. Based on the virtual screens, we purchased 306 compounds that were subjected to a kinetic assay. Six compounds were found to give modest, but significant, inhibition of RTA. They also tended to inhibit shiga toxin A chain, with roughly the same IC50. The compounds generally represent novel chemical platforms that do not resemble RTA substrates, as currently known inhibitors do. These six were also tested in a cell-based assay for their ability to protect cells from intact ricin. Two compounds were effective in this regard, showing modest to strong ricin inhibition, but also showing some cytotoxicity. RTA, with its large, polar active site is a difficult drug design target which is expected to bind small molecules only weakly. The ability of the method to find these novel platforms is encouraging and suggests virtual screening can contribute to the search for ricin and shiga toxin inhibitors.
Keywords: Ricin, shiga toxin, virtual screen, drug design
1. INTRODUCTION
Ricin is the archetypal example of the highly cytotoxic family of ribosome-inhibiting proteins, or RIPs (Olsnes and Pihl, 1982; Lord et al., 1994; Robertus, 1996). It is a class II RIP; that is, it has an A chain and a B chain. Class I RIPs have only the A chain. The A chain, called RTA for ricin, is an N-glycosidase that specifically depurinates a key adenosine in a conserved stem-loop structure, called the sarcin/ricin loop, within the 28S rRNA (Endo and Tsurugi, 1987). This depurination inactivates protein synthesis and leads to cell death. The B chain, RTB, is a lectin that binds to cell surface galactosides and facilitates toxin uptake. Ricin has an intravenous LD50 of about 3 µg/KG for mice (Fodstad et al., 1976). It is less toxic when ingested, but can be quite potent when dispersed as an aerosol; the LD50 for humans by injection or aerosol is estimated to be 3 – 5 µg/Kg with a minimal adult lethal dose of 500 µg (Franz, 1997). Ricin is not infectious and therefore is classed by the CDC as a Class B biohazard. Because of its toxicity and ease of preparation, ricin has been used by terrorist groups (Loyd, 2001).
The X-ray structure of ricin has been determined (Montfort et al., 1987; Rutenber et al., 1991). Complexes with substrate analogs like FMP (Monzingo and Robertus, 1992) permitted an understanding of substrate binding and recognition. This information, together with site directed mutagenesis of key active site residues, (Frankel et al., 1990; Ready et al., 1991; Kim and Robertus, 1992) allowed a plausible mechanism of action to be proposed for the N-glycosidase family. This included the notion that cleavage of the key adenine base from the ribose involved stabilization of an oxocarbenium ion on the sugar by Glu 177, and partial protonation of the leaving adenine by Arg 180. The mechanism has subsequently been refined using kinetic isotope data to show that it proceeds as a two step SN1 reaction. The Schramm group showed that the adenine leaves, creating cation character on the ribose, and this is followed by an attack on the sugar by water (Chen et al., 2000).. Although ricin is the most famous RIP, there are other important members of this large family of enzymes; the structural relationships within the family have been reviewed recently (Robertus and Monzingo, 2004). The amino acid sequence of ricin A chain is about 38% identical to that of the plant RIP called abrin, and about 20% identical to that of the bacterial shiga toxin. One of the most important is the Shiga toxin (Stx) family, which includes the shiga-like toxins found in pathogenic strains of Escherichia coli (O'Brien and Holmes, 1987). These toxins are class II RIPs; but in place of a single B chain as in the plant-derived toxins, they have a pentamer of cell surface-binding proteins. The A chain of the toxin is activated by cleavage into the A1 enzyme (StxA1) and an A2 fragment that can bind and block the active site until reduction of a disulfide bond allows it to diffuse away (Olsnes et al., 1981). It has been shown that the isolated StxA1 chain, unlike RTA, can attack bacterial ribosomes as well as eucaryotic ones (Suh et al., 1998). The X-ray structure of Shiga toxin has been solved and shows StxA is a structural homolog of RTA (Fraser et al., 1994).
There is interest among the biodefense and public health communities in identifying inhibitors of RIP enzymes to act as antidotes to ricin and Shiga (Stx) intoxication. One strategy is to identify ligands that bind strongly to the A chain and retard the depurination reaction.
Historically, the search for inhibitors of appropriate drug targets has depended on high throughput (HTP) screening assays, testing large chemical libraries against the target protein (Kenny et al., 1998; Persidis, 1998; Pereira and Williams, 2007) We have recently completed the first stage of a HTP, cell-based, screen for ricin inhibitors (Wahome et al., 2010 submitted).
In addition to physical HTP screening, there have been recent efforts to reduce the screening burden by using computer programs to carry out “virtual”, or in silico screens; the hope is that this might eliminate many chemical candidates and enrich the percentage of inhibitors in the list of physically screened molecules (Taylor et al., 2002; Shoichet, 2004; Chen et al., 2006).
The first small molecule inhibitor of RTA was identified by virtual screening. Pteroic acid (PTA) was predicted to bind to the RTA specificity pocket; it was shown by X-ray crystallography to bind as predicted, and kinetically shown to inhibit RTA with an IC50 value of about 600 µM (Yan et al., 1997). Subsequent work showed that guanine platforms could also be useful for RTA inhibitor design (Yan et al., 1998; Miller et al., 2002). Recently, virtual screening identified dihydroxy amino pyrimidine as a useful platform (Bai et al., 2009). In particular 4-[3-(2-amino-1,4-dihydro-6-hydroxy-4-oxo-5-pyrimidinyl)propyl]-benzoic acid (PBA) was shown to have an IC50 value of 270 µM. X-ray analysis revealed that PBA occupied the adenine binding pocket of RTA and made the same kind of specific hydrogen bonds as did the pterin- and guanine-based inhibitors. However, this new platform is more soluble and offers some potential advantages in inhibitor design. In this paper we report the results of a large virtual screen aimed at identifying additional novel inhibitor platforms; 306 high ranking candidates from a virtual screen were purchased and tested for RTA inhibition based on their computer docking.
2. MATERIALS AND METHODS
2.1 Protein Expression
Recombinant RTA was expressed and purified as described previously (Bai et al., 2009). Recombinant StxA1 was originally engineered as a His tagged protein (Suh et al., 1998). Poor expressions levels led to a re-engineering as an intein fusion (Miller et al., 2002). The gene coding for StxA1 was cloned into a pTYB2 plasmid from the Impact-CN system (New England Biolabs, Ipswich, MA), and is referred to as pTYB2SLT.
One colony of BL21AI cells containing the pTYB2SLT plasmid was added to 50 mL LB media containing 0.1 mg/L ampicillin. The culture was grown at 37 °C overnight while shaking and was added to 500 mL of LB media containing ampicillin and 0.1%glucose to obtain a starting OD600 of 0.1. The cells were grown for approximately 1.5 hours at 37 °C while shaking, until the OD600 reached 0.5 – 1.0. Protein expression was induced in the cells with the addition of 1 mM IPTG and 0.2% L-arabinose. The induced culture continued to grow at 30 °C for 4 hours. The cells were harvested by centrifugation for 20 minutes at 4 °C in a Beckman JA10 rotor at 3000×g. The cell pellets were resuspended in column buffer (20 mM Tris-HCl, pH 8.0, 0.5 M NaCl, 0.1 mM EDTA) and lysed using the French Press at 1000 psi three times. The lysed cells were ultracentrifuged in a Beckman type60Ti rotor for 1 hour at 4 °C at 80,000×g. The supernatant was filter sterilized with a 0.22 µm syringe filter before being applied to the chitin column for purification.
StxA1 protein was purified on a 20 mL column bed volume of chitin beads (New England Biolabs, Ipswich, MA). The column was equilibrated with column buffer, and the supernatant was applied to the column. The column was washed with column buffer and cross reacted for at least 20 hours with 39 mM DTT in column buffer. Elution buffer (10mM Tris-HCl, pH 7.6,75 mM NaCl) was added to the column, and fractions were collected until Bradford Reagent testing no longer detected the presence of protein. Fractions found to contain StxA1 protein were pooled and dialyzed in elution buffer at 4 °C. A typical yield is 5 mg for 1 liter of cell culture.
2.2 Translation Assay for A Chain Activity
RTA and StxA1 activity were assessed by monitoring their ability to inhibit luciferase expression in a reticulocyte lysate system. The method has been described recently (Bai et al., 2009). Candidate inhibitors were dissolved in DMSO to a nominal concentration of 10 mM; these were diluted 100 fold in the translation assay to a nominal concentration of 100 µM, making the assay solution 1% DMSO. In addition, the assays contained 1 mg/ml of BSA which retards false positives arising from small molecule aggregation that can adsorb and inhibit RTA (Seidler et al., 2003; Feng et al., 2007).
2.3 Virtual Screens
For this experiment we screened the ChemBridge Diversity Library. It contained three-dimensional coordinates for 49,797 compounds in a concatenated sdf file format. The three dimensional structures and bond patterns were prepared by the distributor (ChemBridge, San Diego CA). The ligands all have explicit hydrogen atoms, required by the search programs. We screened the library using two commercial programs, ICM (Abagyan et al., 1994) and GOLD (Jones et al., 1997). ICM uses a docking algorithm based around a global energy optimization of the entire ligand described as an internal coordinate system (Abagyan and Totrov, 2001) within the protein binding site. GOLD uses a genetic algorithm to randomly sample conformational space and evolve an optimum configuration for the ligand.
The target protein, RTA, was screened in two forms, open and closed. The wild type protein has the adenine-binding specificity pocket blocked by the side chain of Tyr 80; this is the closed configuration (Mlsna et al., 1993); the RTA coordinates from this form are from PDB file 1RTC. Specific ligands, like PTA, guanines, and PBA, will displace this side chain by rotation and bind in the specificity pocket, as described previously (Yan et al., 1997; Miller et al., 2002; Bai et al., 2009). The open configuration used for this screen PDB file 1BR5, with the neopterin ligand removed. In addition to the specificity pocket, RTA contains a second pocket, thought to a bind guanine in natural substrates (Monzingo and Robertus, 1992). Docking in the open configuration centered on the specificity pocket defined by previous X-ray structures, but extended to include the second pocket. In the closed form, docking centered on the second pocket but extended to include the side chain of Tyr 80.
ICM was run using the default thoroughness parameter of 1. The docking site was identified automatically by ICM’s cavity search, but the search box size was manually adjusted to allow ligands to also be docked into the second pocket region of RTA. We altered the default ICM score filter cutoff to allow us to analyze all compounds in the search. GOLD was run using the default parameter file in auto select mode, set to 100% thoroughness. Auto mode sets a minimum of 10,000 starting conformations and a maximum of 125,00 per compound, but optimizes the search based on the compound. Our tests showed this mode was faster than presetting operation numbers, which wastes search time for ligands with few rotatable bonds. GOLD was set to stop searching a given ligand if the top three conformations deviated by less than 1.5 Å rms per atom. The GOLD binding site was identified using a docked ligand; this was PTA for the open configuration and 2,5-diamino-4,6-dihydroxypyrimidine for the closed configuration. Scoring was based on the default GOLD score algorithm. Both programs were run with RTA as a fixed target; the computational cost to run flexible docking on such a large data base is prohibitive.
Docking was carried out using parallel processing on the TI3D computer cluster. The cluster has 16 HP Proliant BL35P blades, each having two dual core AMD Opteron 2.4GHz processors and 8 GB of memory. Docking poses were ranked by scores, and filtered using preferred chemical criteria; the common Lipinski rule of 5 (Lipinski et al., 2001) was modified to include compounds with logP values < 4. The docking poses for top scoring compounds were also examined by eye, using PyMol (DeLano, 2002), to see if reasonable polar interactions were made between the candidate and target protein. Certain chemical families were strongly represented in the scoring lists, and these were pared so that a larger number of chemical platforms could be tested. In this fashion, we selected 306 candidates that strongly targeted either the open or closed form of RTA.
The 306 candidate inhibitors were purchased from ChemBridge (San Diego, CA). Approximately 1 mg of each compound was dissolved in 200 µl of DMSO in a 384 well plate, for a nominal concentration of 10 mM. Subsequently, discovered hit compounds were reordered as dry powders from the same source.
2.4 Cell Cytotoxicity Assay
Briefly, Vero cells (5000 cells/well) grown in Dulbecco’s minimal essential medium DMEM (+ 10% fetal bovine serum (FBS) were seeded in a 96 well plate and incubated at 37°C (5% CO2 and ~ 95% humidity) for ~ 15 hr. 1.0 µl of a serially diluted compound was added to Vero cells in triplicate while an equal volume of DMSO (100%) was added to Vero cells in positive and negative control wells. The plate was incubated (5% CO2 and ~ 95% humidity) at 37 °C for 1 hr.
7 µl of ricin (5 ng/ml) were added to the assay and to the negative control wells while an equal volume of growth medium was added to the positive control wells. The plate was incubated at 37 °C for 24 hr or 48 hr before cell viability was determined using an luciferase-based cell viability assay Celltiter-Glo, Promega, Madison, WI) (Neal et al., 2010) The plate was incubated at room temperature for ~ 5 min before reading luminescence in a SpectraMax L luminometer.
3. RESULTS
3.1 Protein targets
We carried out a virtual screening of nearly 50,000 compounds from the ChemBridge Diversity Library, using ICM and GOLD. We used both the open and closed active site forms of RTA as targets. Figure 1a shows the active site of RTA in the open conformation; a PTA ligand is bound in the specificity site of the larger active site (Yan et al., 1997). The side chain of Tyr 80 closes over this pocket in the empty enzyme, but is displaced by adenine and related compounds, as shown. The search box for this form centers on the specificity pocket but extends toward the large “second pocket” at the upper right, which we predicted would bind the guanine of a natural substrate (Monzingo and Robertus, 1992). Figure 1b shows the active site region of RTA in the closed form, with the inhibitor 2,5-diamino-4,6-dihydroxypyrimidine (DDP) bound. In this case the ligand pyrimidine ring stacks on the side chain of Tyr 80, which remains closed over the specificity pocket. The search pocket for this form uses the region between Tyr 80, marked by DDP, and the second pocket.
Figure 1.


Solvent-accessible surface structure of RTA. On the surface, oxygens are red, nitrogens blue, and carbons green. A. RTA in the open form with the inhibitor PTA (PDB:1BR6) occupying the adenine specificity pocket, and shown as a stick molecule. B. RTA in the closed form with the inhibitor 2,5-diamino-4,6-dihydroxypyrimidine (PDB:1IL5) shown in stick bonds.
3.2 Docking scores
Virtual screening has had some successes (Cournia et al., 2009), but computer docking has strong limitations, particularly in ranking compound affinities (Warren et al., 2006). Figure 2a shows a correlation between ~48,000 docking scores for two independent screens of the RTA open configuration using ICM. Each operator has some selection over the screening site, and each run has a different random starting seed for each compound. The correlation coefficient for these two screens is 0.56. We also correlated the docking scores for ICM and GOLD; strong binding is reflected in large negative scores for ICM and large positive scores for GOLD. Figure 2b shows there is essentially no correlation between the ~48,000 scores which are shown as small black dots; the solid line is the best fit to the data but the correlation is near zero. This suggests the two programs show very little agreement about the binding affinity of most compounds, which are in fact not really ligands at all. We also docked seven known RTA inhibitors for which the X-ray structures in complex with RTA have been determined; these data are shown as the large gray circles in Figure 2b. All of these inhibitors bind RTA in the open conformation. The broken line is a linear fit of that data; correlation for docking these known inhibitors is 0.71. This suggests that the two programs do agree modestly well when scoring compounds that do bind the target. Furthermore, the strongest binding of these known inhibitors do occupy the upper left corner of the plot where stronger binding is indicated. Although there is not a perfect ranking of predicted scores with observed IC50 values, both programs rank strong ligands like PTA (IC50=0.6 mM) higher than weak ones, like 9-deazaguanine (IC50=1.4 mM) (Miller et al., 2002). In addition to the scoring values, the docking poses by both ICM and GOLD for these known ligands are quite accurate when compared with the observed X-ray structures, as has been described earlier (Bai et al., 2009).
Figure 2.


Correlation of virtual screening scores against the ChemBridge diversity library. A. Correlation between docking scores for two independent screens using the program ICM; the correlation coefficient is 0.56. B. Correlation scores between programs ICM and GOLD; the correlation coefficient is 0.05(Minetti et al., 2003). The large circles show scores for a positive control of known RTA open form inhibitors.
For the open RTA target, ICM ranked the known inhibitors higher than GOLD. ICM ranked the known inhibitors with scores corresponding to the top 3% of the large ChemBridge screen, whereas the GOLD scores corresponded to the top two thirds. If we confine the scores to the known inhibitors with IC50 values below 0.6 mM (these correspond to the top four in Figure 2b), then the GOLD scores are in the top half. The ability to accurately predict binding scores depends on the nature of the target, and no one program performs best on all target types (Kellenberger et al., 2004; Perola et al., 2004; Chen et al., 2006; Warren et al., 2006). It appears that for the open form of RTA, ICM may outperform GOLD, and we used it in creating our list of inhibitors to screen. This list was then filtered for those compounds that also had strong GOLD scores. Scores for the known RTA inhibitors suggested that candidate compounds of interest might be those that have both an ICM score less than −20 and a GOLD score above 30. Furthermore, our experience with kinetic assays suggests that compounds with a logP value above 4 are too insoluble to test in our kinetic assay, and that those not predicted to have a strong hydrogen bonding component to their docking score tend to be poor candidates (Miller et al., 2002; Bai et al., 2009). Therefore, we also applied these filters to our list and then visually inspected several hundred docked candidates to assess the reasonableness of their predicted chemical interactions. In this manner, we chose 306 compounds, representing a range of chemical platforms, to purchase and assay. About 65% of these had been docked in the open form, and the rest in the closed form of RTA.
3.3 In vitro kinetic assays
The 306 candidate inhibitors, together with positive and negative controls, were assayed in an in vitro protein translation assay. Initially the candidates were present at a nominal concentration of 100 µM; that is, compound solutions were diluted into the assay solution making the latter 1% DMSO. About 13 compounds showed measurable inhibition of RTA activity. These compounds were then retested to eliminate false positives. Eight compounds were then purchased as dry powders, dissolved in DMSO and used in a dose response assays to determine their IC50 values. Of these, six gave weak, but measurable inhibition values. Figure 3 shows the dose response curve for compound CB7543758 (CID 849809). The graph shows an IC50 value of about 250 µM, but the shape of the curve shows the compound has limited solubility. The inhibition at a nominal 500 µM is nearly the same as that at 250 µM, suggesting the compound has saturated at about 250 µM. We used an online solubility computation, log S, (Tetko et al., 2005) that predicted the aqueous solubility for CB7543758 to be about 100 µM, consistent with our observation. A list of estimated IC50 values from the dose response experiments is shown in Table 1. In general, the compounds showed weak to modest IC50 values from about 0.2 – 2.0 mM; these values are similar to those exhibited by other small molecule inhibitors of RTA (Yan et al., 1997; Miller et al., 2002; Mandal et al., 2008; Bai et al., 2009).
Figure 3.

Dose response inhibition of RTA. The data reflect the inhibition of the enzyme by increasing concentrations of CB7543758, dissolved in DMSO.
Table 1.
Inhibitors identified from the virtual screen
| Structure | CB ID CID |
RTA IC50 µM |
StxA1 IC50 µM |
Ricin inhibition |
Cytotoxicity IC50 µM |
|---|---|---|---|---|---|
 |
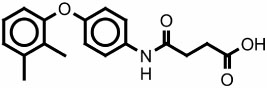
5131604 785308 |
1800 | >1000 | none | >300 |
 |
5225540 609529 |
180 | none | Strong at 25 µM |
>30 |
 |
5282931 766985 |
250 | >125 | None | none |
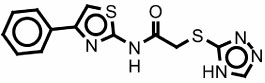 |
5978818 767227 |
550 | >1000 | Modest at 60 µM |
>60 |
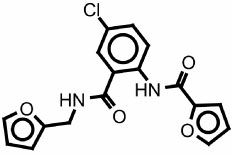 |
7543758 849809 |
250 | 400 | none | >125 |
 |
9062573 933869 |
500 | >1000 | None | >300 |
In addition to measuring the inhibition of RTA, we used the same dose response methods to measure the inhibition of the A chain of shiga toxin 1 (StxA1). Those results are also summarized in Table 1. In general, RTA inhibitors also inhibit StxA1, although the IC50 values vary within a small range, generally with a factor of 2 to 3 as seem previously (Miller et al., 2002). Compound CB5225540 was a strong RTA inhibitor, but failed to inhibit StxA1.
3.4 Cell based assays
The compounds which gave reproducible inhibition of RTA were then measured in a cell based assay for their ability to inhibit intact ricin intoxication. These tests also assess the cytotoxicity of the compounds themselves. Figure 4 shows the results for the only two compounds, CB5225540 and CB5978818 that showed any ability to protect Vero cells against ricin intoxication. CB5225540 shows little cytotoxicity at concentrations below 25 µM and strongly protected against ricin-induced cell death. Indeed, ricin was about 90% inhibited at that concentration and the compound appears to have an 50% effective inhibitory concentration (EC50) value around 10 µM in the cell assay. Unfortunately, CB5225540 becomes strongly cytotoxic at higher concentrations. Figure 4b shows the dose response for CB5978818. It was not cytotoxic at concentrations below about 60 µM, and offered modest protection against ricin, inhibiting about one third of the enzyme activity at that concentration.
Figure 4.


Dose response inhibition of ricin in a cell based assay. The dashed line shows cell viability after ricin intoxication. Open circles show the dose dependent cytotoxicity of the inhibitor without ricin, and closed circles show the viability of cells challenged with ricin and varying doses of inhibitor.
4. DISCUSSION
Ricin A chain, RTA, binds to rRNA and hydrolyzes an adenine base from a conserved stem loop structure. The enzyme has a specificity pocket that binds adenine, making an aromatic stacking with active site Tyr side chains and making hydrogen bonds with specific enzyme groups. We have previously identified a number of inhibitors that bind to this site and exploit the adenine specificity groups as seen crystallographically. These ligands include derivatives of adenine, guanine, pterin, and dihydroxy-aminopyrimidine (Monzingo and Robertus, 1992; Yan et al., 1997; Miller et al., 2002; Bai et al., 2009). Several of these inhibitors were initially discovered by computer-based virtual screening (Yan et al., 1997; Bai et al., 2009). However, the chemical libraries that produced these hits contained a variety of biological molecules that are aromatic compounds substituted with polar hydrogen bonding functionalities that can interact with the RTA specificity site. These kinds of biological compounds are often difficult to synthesize and are not common in the large diversity libraries created by manifold synthesis for high throughput chemical screens. Even so, we wanted to test if virtual screening of a diversity library might elucidate novel chemical platforms, with drug design potential, even if they did not resemble natural RTA substrates.
Our strategy for the virtual screen against RTA was to define two binding modes for the protein (Figure 1). One emphasized docking into the specificity pocket, which has evolved to bind adenine. This open conformation has been seen to accommodate a range of inhibitor platforms, like pterins, and guanines, that stack between the side chains of tyrosines 80 and 123, and make specific hydrogen bonds with groups in the specificity site (Yan et al., 1997; Miller et al., 2002; Bai et al., 2009). The second, closed, mode has only been seen to bind one pyrimidine platform (Miller et al., 2002) but could bind many candidate compounds that cannot make the specific hydrogen bonds required for optimum binding in the specificity pocket.
Our screening strategy also involved using two programs for docking. This is based on our observation (see Fig 2b) that virtual screening programs show much better agreement in docking poses and scores for real inhibitors than for random compounds that are not, in fact, target ligands. As a general strategy, using only candidates predicted to bind strongly by two, or more, programs may enrich the selection pool for true ligands.
We chose high ranking candidates from both open and closed conformation screens. Many chemical platforms exhibited redundancy. For example, a number of compounds with alkyl amides docked that amide into the RTA specificity site, making hydrogen bonds to the natural adenine receptors. We restricted their representation to three of the top scoring compounds, but rejected dozens to allow other platforms to be tested. We also observed that some dockings appeared by eye to be chemically unreasonable; for example some dockings placed a carboxylate group within the specificity pocket which is known to bind aromatic groups. After filtering the lists based on solubility, redundancy, and stereochemical reasonableness, we purchased 306 compounds dissolved in DMSO and plated in a 384 well format. These compounds were screened in a kinetic assay system where we identified seven RTA and/or StxA1 inhibitors. We feel that the poor solubility of many candidates prevented them from being detected in the translation assay and that a robust and sensitive screen might have shown more ligands.
Of the new inhibitors, only CB5131604 (see Table 1), resembles an adenine substrate; it probably binds in the specificity site of RTA. It was a poor inhibitor, however, with an IC50 value much higher than known inhibitors from the adenine, guanine, or pterin groups. Compound CB5225540 resembles the known DDP inhibitor and may bind to the closed form. Its IC50 of ~200 µM makes it among the stronger small molecule RTA inhibitors identified to date. The other five inhibitors cover a range of chemical platforms and bear no resemblance to currently known RTA inhibitors. Until their X-ray structures are known, we cannot be sure how they interact with RTA and how they might be diversified to improve their binding affinity and specificity.
We tested the new inhibitors of RTA in a cell based assay using intact ricin. This assay follows their ability to protect Vero cells against a ricin challenge. The assay also assesses the cytotoxicity of the compounds and their ability to enter cells and function effectively. Two compounds, CB5225540 and CB5978818, were the only compounds from the virtual screen that confer ricin resistance to cells, with CB5225540 essentially eliminating ricin action when incubated at 25 µM. Both compounds exhibited cytotoxicity, however, at increasing dosage. In spite of this problem, both compounds appeared to be useful lead candidates for further inhibitor development. We assume the failure of the other RTA inhibitors to protect against ricin is due to their poor bioavailability. They may be unable to reach the cytoplasm in sufficient concentration to inhibit ricin A chain.
Failure to protect cells, or to exhibit some cytotoxicity, does not preclude further development of chemical platforms as potential inhibitors, although that development would be more complex. It may be, for example, that modification of an inhibitor compound would not only make it bind more strongly to the RTA target, but would prevent it from deleteriously binding to some other cellular target and thereby reduce its cytotoxicity. We intend to explore both CB5225540 and CB5978818 as platforms for improved anti-ricin compounds. Both are novel platforms that are chemically unrelated to other known small molecule inhibitors of RTA, and represent successes for the virtual screening method.
The method of virtual screening has had mixed success in drug design. It has proved useful in some projects (Cournia et al., 2009), but controlled experiments have called the general utility of the method into question (Chen et al., 2006; Warren et al., 2006). In assessing the success of this experiment it is important to note that RTA is a very difficult drug screening or drug design target; the methods employed here might be even more successful on other proteins. As seen in Figure 1, RTA has a very large, open, polar active site. It is generally considered that proteins with large polar active sites are difficult targets for drug development. Such proteins may depend on numerous weak polar interactions with their ligands that summed together create a favorable, specific interaction. Such large active sites, in the absence of substrates, bind water and a ligand must displace the solvent and make a compensating hydrogen bond, which may not be any more favorable. To date, small molecule RTA inhibitors have only modest IC50 values, often in the millimolar range. Further affinity must be engineered into candidates, for example by derivatizing a ligand-like PTA (Figure 1a) to reach into the second pocket, thereby making additional favorable interactions. Of course a large drug-like ligand interacting over a large area will pay a high entropic cost upon binding. Our virtual screen used small ligands, generally less than 500 MW, in keeping with drug design rules (Lipinski et al., 2001). Such compounds would be expected to bind weakly to this target.
5. CONCLUSION
Computer based virtual screening identified a number of compounds as potential ligand/inhibitors of ricin A chain. 306 top compounds were assayed and 6 were inhibitors. Two of these were able to protect Vero cells from ricin intoxication. The identification of these new platforms, with IC50 values similar to those of substrate-based ligands, is quite encouraging and provides new avenues for the design of ricin antidotes.
ACKNOWLEDGMENTS
This work was supported by NIH grant AI 075509, by Robert A. Welch Foundation grant F1225, by the College of Natural Sciences support to the Center for Structural Biology, and by a grant from the Texas Institute for Drug Discovery and Development (TI3D).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abagyan R, Totrov M. High-throughput docking for lead generation. Curr Opin Chem Biol. 2001;5(4):375–382. doi: 10.1016/s1367-5931(00)00217-9. [DOI] [PubMed] [Google Scholar]
- Abagyan RA, Totrov M, Kuznetsov D. ICM—a new method for protein modeling and design: Applications to docking and structure prediction from the disordered native conformation. J. Comput. Chem. 1994;15:488–506. [Google Scholar]
- Bai Y, Monzingo AF, Robertus JD. The X-ray structure of ricin A chain with a novel inhibitor. Arch Biochem Biophys. 2009;483:23–28. doi: 10.1016/j.abb.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Chen H, Lyne PD, Giordanetto F, Lovell T, Li J. On evaluating molecular-docking methods for pose prediction and enrichment factors. J Chem Inf Model. 2006;46(1):401–415. doi: 10.1021/ci0503255. [DOI] [PubMed] [Google Scholar]
- Chen X-Y, Berti PJ, Schramm VL. Ricin A-Chain: Kinetic Isotope Effects and Transition State Structure with Stem-Loop RNA. J. Am. Chem. Soc. 2000;122(8):1609–1617. [Google Scholar]
- Cournia Z, Leng L, Gandavadi S, Du X, Bucala R, Jorgensen WL. Discovery of human macrophage migration inhibitory factor (MIF)-CD74 antagonists via virtual screening. J Med Chem. 2009;52(2):416–424. doi: 10.1021/jm801100v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- Endo Y, Tsurugi K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J Biol Chem. 1987;262(17):8128–8130. [PubMed] [Google Scholar]
- Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J Med Chem. 2007;50(10):2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- Fodstad O, Olsnes S, Pihl A. Toxicity, distribution and elimination of the cancerostatic lectins abrin and ricin after parenteral injection into mice. Br J Cancer. 1976;34(4):418–425. doi: 10.1038/bjc.1976.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel A, Welsh P, Richardson J, Robertus JD. Role of arginine 180 and glutamic acid 177 of ricin toxin A chain in enzymatic inactivation of ribosomes. Mol Cell Biol. 1990;10(12):6257–6263. doi: 10.1128/mcb.10.12.6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz DR, J NK. Ricin Toxin. United States of America: Office of the Surgeon General, Department of the Army; Textbook of Military Medicine: Medical Aspects of Chemical and Biological Warfare. 1997
- Fraser ME, Chernaia MM, Kozlov YV, James MN. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 A resolution. Nat Struct Biol. 1994;1(1):59–64. doi: 10.1038/nsb0194-59. [DOI] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267(3):727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins. 2004;57(2):225–242. doi: 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- Kenny BA, Bushfield M, Parry-Smith DJ, Fogarty S, Treherne JM. The application of high-throughput screening to novel lead discovery. Prog Drug Res. 1998;51:245–269. doi: 10.1007/978-3-0348-8845-5_7. [DOI] [PubMed] [Google Scholar]
- Kim Y, Robertus JD. Analysis of several key active site residues of ricin A chain by mutagenesis and X-ray crystallography. Protein Eng. 1992;5(8):775–779. doi: 10.1093/protein/5.8.775. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Lord JM, Roberts LM, Robertus JD. Ricin: structure, mode of action, and some current applications. Faseb J. 1994;8(2):201–208. [PubMed] [Google Scholar]
- Loyd A, F M. Bin Laden's poison manual. London, England: London Times; 2001. [Google Scholar]
- Mandal S, Li WT, Bai Y, Robertus JD, Kerwin SM. Synthesis of 2-substituted 9-oxa-guanines {5-aminooxazolo[5,4-d]pyrimidin-7(6H)-ones} and 9-oxa-2-thio-xanthines {5-mercaptooxazolo[5,4-d]pyrimidin-7(6H)-ones} Beilstein J Org Chem. 2008;4:26. doi: 10.3762/bjoc.4.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DJ, Ravikumar K, Shen H, Suh JK, Kerwin SM, Robertus JD. Structure-based design and characterization of novel platforms for ricin and shiga toxin inhibition. J Med Chem. 2002;45(1):90–98. doi: 10.1021/jm010186s. [DOI] [PubMed] [Google Scholar]
- Minetti CA, Remeta DP, Zharkov DO, Plum GE, Johnson F, Grollman AP, Breslauer KJ. Energetics of lesion recognition by a DNA repair protein: thermodynamic characterization of formamidopyrimidine-glycosylase (Fpg) interactions with damaged DNA duplexes. J Mol Biol. 2003;328(5):1047–1060. doi: 10.1016/s0022-2836(03)00365-6. [DOI] [PubMed] [Google Scholar]
- Mlsna D, Monzingo AF, Katzin BJ, Ernst S, Robertus JD. Structure of recombinant ricin A chain at 2.3 A. Protein Sci. 1993;2(3):429–435. doi: 10.1002/pro.5560020315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montfort W, Villafranca JE, Monzingo AF, Ernst SR, Katzin B, Rutenber E, Xuong NH, Hamlin R, Robertus JD. The three-dimensional structure of ricin at 2.8 A. J Biol Chem. 1987;262(11):5398–5403. [PubMed] [Google Scholar]
- Monzingo AF, Robertus JD. X-ray analysis of substrate analogs in the ricin A-chain active site. J Mol Biol. 1992;227(4):1136–1145. doi: 10.1016/0022-2836(92)90526-p. [DOI] [PubMed] [Google Scholar]
- Neal LM, O'Hara J, Brey RN, 3rd, Mantis NJ. A monoclonal immunoglobulin G antibody directed against an immunodominant linear epitope on the ricin A chain confers systemic and mucosal immunity to ricin. Infect Immun. 2010;78(1):552–561. doi: 10.1128/IAI.00796-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien AD, Holmes RK. Shiga and Shiga-like toxins. Microbiol Rev. 1987;51(2):206–220. doi: 10.1128/mr.51.2.206-220.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsnes S, Pihl A. Toxic Lectins and Related Proteins. In: Coen P, van Heynigen S, editors. The Molecular Action of Toxins and Viruses. New York: Elsevier Biomedical Press; 1982. [Google Scholar]
- Olsnes S, Reisbig R, Eiklid K. Subunit structure of Shigella cytotoxin. J Biol Chem. 1981;256(16):8732–8738. [PubMed] [Google Scholar]
- Pereira DA, Williams JA. Origin and evolution of high throughput screening. Br J Pharmacol. 2007;152(1):53–61. doi: 10.1038/sj.bjp.0707373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perola E, Walters WP, Charifson PS. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins. 2004;56(2):235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- Persidis A. High-throughput screening. Advances in robotics and miniturization continue to accelerate drug lead identification. Nat Biotechnol. 1998;16(5):488–489. doi: 10.1038/nbt0598-488. [DOI] [PubMed] [Google Scholar]
- Ready MP, Kim Y, Robertus JD. Site-directed mutagenesis of ricin A-chain and implications for the mechanism of action. Proteins. 1991;10(3):270–278. doi: 10.1002/prot.340100311. [DOI] [PubMed] [Google Scholar]
- Robertus JD, Monzingo AF. The structure of ribosome inactivating proteins. Mini Rev Med Chem. 2004;4(5):477–486. doi: 10.2174/1389557043403837. [DOI] [PubMed] [Google Scholar]
- Robertus JD, M AF. The structure of ribosome inactivating proteins from plants. In: Parker MW, Austin RG, editors. Protein toxin structure. Landes Company; 1996. pp. 253–270. [Google Scholar]
- Rutenber E, Katzin BJ, Ernst S, Collins EJ, Mlsna D, Ready MP, Robertus JD. Crystallographic refinement of ricin to 2.5 A. Proteins. 1991;10(3):240–250. doi: 10.1002/prot.340100308. [DOI] [PubMed] [Google Scholar]
- Seidler J, McGovern SL, Doman TN, Shoichet BK. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J Med Chem. 2003;46(21):4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432(7019):862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh JK, Hovde CJ, Robertus JD. Shiga toxin attacks bacterial ribosomes as effectively as eucaryotic ribosomes. Biochemistry. 1998;37(26):9394–9398. doi: 10.1021/bi980424u. [DOI] [PubMed] [Google Scholar]
- Taylor RD, Jewsbury PJ, Essex JW. A review of protein-small molecule docking methods. J Comput Aided Mol Des. 2002;16(3):151–166. doi: 10.1023/a:1020155510718. [DOI] [PubMed] [Google Scholar]
- Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, Palyulin VA, Radchenko EV, Zefirov NS, Makarenko AS, Tanchuk VY, Prokopenko VV. Virtual computational chemistry laboratory--design and description. J Comput Aided Mol Des. 2005;19(6):453–463. doi: 10.1007/s10822-005-8694-y. [DOI] [PubMed] [Google Scholar]
- Warren GL, Andrews CW, Capelli AM, Clarke B, Lalonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A Critical Assessment of Docking Programs and Scoring Functions. J Med Chem. 2006;49(20):5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- Yan X, Day P, Hollis T, Monzingo AF, Schelp E, Robertus JD, Milne GW, Wang S. Recognition and interaction of small rings with the ricin A-chain binding site. Proteins. 1998;31(1):33–41. doi: 10.1002/(sici)1097-0134(19980401)31:1<33::aid-prot4>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Yan X, Hollis T, Svinth M, Day P, Monzingo AF, Milne GW, Robertus JD. Structure-based identification of a ricin inhibitor. J Mol Biol. 1997;266(5):1043–1049. doi: 10.1006/jmbi.1996.0865. [DOI] [PubMed] [Google Scholar]