

Certain nucleic-acid-binding proteins can recognize varied substrate structures by making use of multiple binding domains or by undergoing conformational changes in a specific binding domain. For example, the Xenopus zinc-finger protein TFIIIA recognizes a specific sequence in duplex DNA and a significantly different sequence in ribosomal RNA;[1–5] this switching of binding site is implicated in feedback-type control of gene expression. We have recently begun to examine various approaches for the design of multifunctional DNA-recognizing molecules which can mimic this kind of multisite-binding behavior.[6] The ability to control binding in this way not only serves to mimic a complex biological function, but also may have some practical applications as well. For example, recent studies in the use of oligonucleotides as sequence-specific inhibitors of gene expression have shown that the combination of agents targeted to multiple sites can be more effective in vitro than single-site binding alone.[7–9]
We recently described the design and synthesis of a cyclic oligodeoxynucleotide molecule which can bind strongly to two separate DNA sequences by switching conformation.[6] In that earlier prototypical structure, a cyclic DNA molecule which contained separate pairs of nine-base binding domains (36 nucleotides altogether) was used to recognize two nine-base sequences by interchanging binding and bridging domains (totalling 18 nucleotides of sequence recognition by the 36 nucleotides in the cyclic compound). In order to explore the limits of this kind of multifunctional binding, we now report the synthesis and properties of a new cyclic DNA which is designed to recognize six different sequences by undergoing six conformational changes. At the same time, careful design allows the elimination of duplicated nucleotides, leading to very high economy of function: the molecule selectively recognizes 48 nucleotides of sequence although the molecule itself contains only 35 nucleotides. Several novel design principles are discussed.
The design of the DNA macrocycle 1 involves the combination of several different principles (Fig. 1). First, binding would occur by triplex formation between opposing pyrimidine domains (the Watson–Crick and Hoogsteen domains) in the circle and a complementary sequence of purines in the target molecule. This would result in stronger binding and higher sequence selectivity than is possible for simple Watson–Crick binding.[10, 11] Second, because the two pyrimidine binding domain sequences in such a circle are related by a pseudo-mirror-plane of symmetry, a pair of binding domains might bind the forward (5′ → 3′ direction) or the reverse (3′ → 5′ direction) of a given sequence by switching the roles of the Watson–Crick and Hoogsteen domains. Third, by contiguous alternating placement of three binding domain pairs, this might allow recognition of six different sequences (three in one orientation and three in the reverse). Recognition of a sequence with a given binding domain pair would then make use of the other domains as bridging loops in the complex.[6] Finally, one might achieve this multiple recognition with fewer nucleotides by eliminating those whose function is duplicated in a neighboring domain, thus overlapping the boundaries of the domains. With this approach, many of the individual nucleotides have more than one function.
Fig. 1.
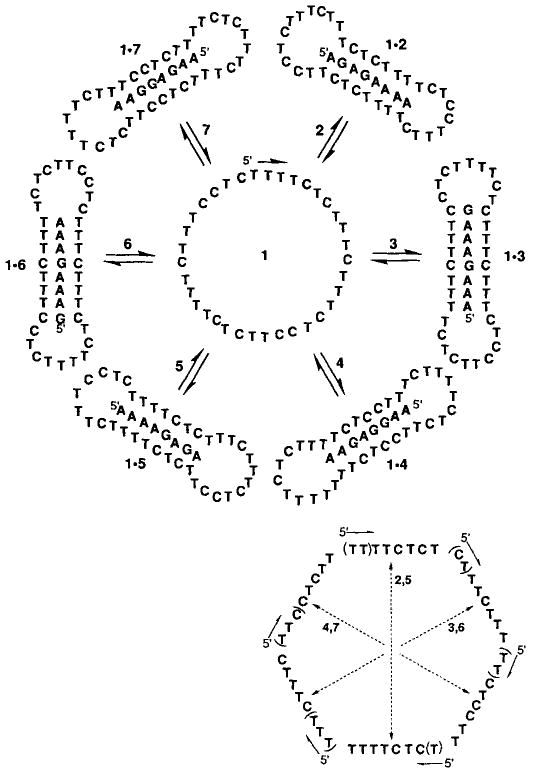
Top: The sequence of circular oligodeoxynucleotide 1 (in center) and the adducts of 1 with complementary sequences 2–7. Bottom: Strategies for construction of 1. The three pairs of domains necessary for recognition by triplex formation were placed in alternating configuration such that two halves of a pair oppose one another. (The two domains in each pair are connected by an arrow.) Parentheses indicate nucleotides which are eliminated because they are repeated from the previous domain.
Specifically, we designed and synthesized the macrocycle 1 with the ability to recognize the six eight-base sequences 2–7 (Fig. 1). The total sequence recognition thus encompasses 48 bases of DNA. If the pyrimidine binding domains of the circle were kept separate and incorporated contiguously into a circle, this would require a circle 48 nucleotides in size. By overlapping the ends of the required domains (Fig. 1 bottom), we eliminate the need for 13 of the nucleotides, and so the circle 1 contains only 35 nucleotides.
The structure of 1 should, in principle, retain the high sequence selectivity previously seen for this kind of circular ligand.[10] To test this selectivity, we also synthesized two sequences, 8 and 9 (sequences shown in Fig. 2), which are the same as two of the recognition sequences but with two nucleotides interchanged.
Fig. 2.
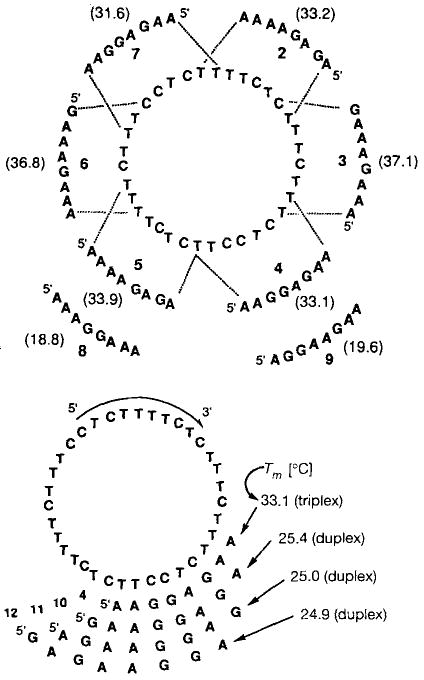
Top: Melting temperatures Tm [°C] for complexes of 1 with complementary sequences 2–7 and mismatched sequences 8 and 9. Bottom: The relative affinity of 1 for a triple helical complement (4) as compared to other Watson–Crick complements (10–12), as determined by thermal denaturation. The eight-base complements 4 and 10–12 are overlapping sequences related by single-nucleotide frameshifts.
Binding studies of 1 with these eight purine sequences were carried out by optical monitoring of thermal denaturation of the 1:1 mixtures at pH 7.0 (100 mm NaCl, 10 mm MgCl2). Melting curves reveal sharp, apparently two-state transitions at 260 nm for the complexes of 1 with 2–7. As expected for such triple helical complexes,[11] the melting temperatures are considerably higher than those for simple Watson–Crick complements containing the same base pairs (see below). The melting transitions of 1 with 2–7 are all within the range 33–37 °C (Fig. 2 top); the hyperchromicities are 18–25%. Also consistent with this mode of binding is the finding that the stability of all six complexes are pH-dependent (data not shown), as expected for triplexes involving C · G · C triads and not duplexes.[12]
By contrast, mismatched sequences 8 and 9 are bound much more weakly than 1, with Tm values of 18.8 and 19.6 °C, respectively, and are essentially fully dissociated at room temperature. Curve-fitting algorithms[13] allow estimates of free energy for the complexes of 1 with 2–9. Under these conditions, the complexes 1 · 2–1 · 7 are bound with free energies of 10.3–12.2 kcal mol−1. By comparison, the complexes of the two mismatched purine sequences (8, 9) are bound much weaker to 1, with free energies of 6.5 and 6.6 kcal mol−1. Thus, 1 is selective for the complementary sequences over these two mismatched sequences by three to four orders of magnitude in association constant.
A different issue of complementarity and selectivity arises from the structure of 1. While it is designed to recognize sequences 2–7 by triplex formation, it is also complementary in Watson–Crick fashion to 29 additional eight-nucleotide sequences. To test whether the circle 1 binds selectively to the six intended sequences (2–7) relative to the 29 other sequences, we synthesized three purine octamers, 10–12, which are fully complementary to the circle only in Watson–Crick fashion. The complementary sites for the sequences occur adjacent to the binding domain for the triplex-bound oligomer 4, and is frameshifted by one, two, or three nucleotides, respectively (Fig. 2 bottom).
Results of these additional binding studies show that while the circle 1 is indeed complementary to the Watson–Crick complements 10–12, it shows considerable selectivity instead for the intended triplex complements 2–7. Thus, 10–12 are bound with Tm values of 24.9–25.4 °C and hypochromicity values of 0.10–0.16, while the triplex complements 2–7 are bound with Tm values of 33.1 – 37.1 °C and hypochromicity values of 0.18–0.25. The free energy of formation for these types of complex differs by 2.1 –4.3 kcal mol−1. The association constant for 1 · 2–1 · 7 is 34- to 1400-fold larger than that for 1 · 10–1 · 12. The sequences which are only complementary in Watson–Crick fashion may thus be viewed as being mismatched sequences, since they are not able to form the full number of stabilizing Hoogsteen bonds that the intended six sequences can. The lower hypochromicitiy values with 1 · 10–1 · 12 are consistent with the Hoogsteen strand not being involved in these complexes, although we cannot rule out partial Hoogsteen-type interaction. The selectivity shown by 1 against Watson–Crick binding alone is, in fact, similar in magnitude to the amount of discrimination which a linear oligodeoxynucleotide shows against single-base mismatches.[10] It is clear, therefore, that compound 1 is readily able to distinguish between its six target sequenes and other complementary and partially complementary DNA sequences.
In summary, the results indicate that reasonably complex multifunctional behavior can be successfully designed into synthetic DNA-recognizing molecules. The design of the oligonucleotide macrocycle 1 illustrates ways to control binding properties in DNA molecules by varying more than just the simple primary sequence of nucleotide bases. In this case, the circularity, the use of opposite domains for binding, and the pseudo-symmetry of the domains all allow for greater control of secondary and tertiary interactions in the final complexes than is commonly used in Watson–Crick binding. It is possible that this multisite binding might be applied to practical problems of nucleic acid recognition; for example, inhibition of gene expression by targeting more than one site in a gene, or more than one gene in an organism. Further study will be necessary to investigate these possibilities.
Experimental Procedure
Oligodeoxynucleotides 2–10 were synthesized by automated methods using β-cyanoethylphosphoramidite chemistry [14] on a Pharmacia LKB instrument, and were purified by preparative denaturing polyacrylamide gel electrophoresis. Quantification was done by measuring absorbance at 260 nm and calculating concentration based on extinction coefficients derived from nearest neighbor analysis [15]. For construction of compound 1, the linear precursor 5′-dTTTTCTTTCCTCTTTTCTCTTTCTTTCTCCTTCTCp was synthesized, along with a template for cyclization having the sequence 5′-dAGAAAAGAGAA. Cyclization was carried out with an aqueous nonenzymatic ligation reaction, with 50 μm each of linear precursor and template. 200 mm imidazole · HCl (titrated to pH 7.0 as a stock solution prior to addition to the reaction mixture). 100 mm NiCl2, and 125 mm BrCN for 12 h at 23°C, as described previously [11, 16]. Workup and isolation were as described previously [11, 17]. The conversion to circular product was about 70% (by gel analysis), and the circular product was isolated in 43% yield by excising, crushing, and elution from the peparative get followed by dialysis to remove salts. The circular structure was confirmed by complete resistance to cleavage by a combination of T4 DNA polymerase (3′ exonuclease activity) and T4 polynucleotide kinase (3′ phosphatase activity in the absence of ATP) under conditions which cleave the linear precursor to mononucleotides. The cleavage was carried out at 37°C (1–40 minutes) with 23 μm DNA, 58 mm tris(hydroxymethyl)aminomethane (Tris) · HCl (pH 7.6), 12 mm MgCl2, 6 mm dithiothreitol, 100 μm spermidine, 100 μm EDTA, 0.5 units/μL kinase, and 0.1–0.2 units/μL polymerase. Enzymes were purchased from New England Biolabs (T4 kinase) and United States Biochemical (T4 polymerase).
Thermal denaturation studies on the 1:1 complexes were carried out at 6 μm total oligonucleotide concentration in a buffer containing 100 mm NaCl, 10 mm MgCl2, and 10 mm Na-PIPES (HPIPES = l,4-piperazinediethanesulfonic acid) at pH 7.0 in stoppered 1 cm pathlength cells. Prior to the denaturation experiment, samples were heated to 90°C for 2 min and then allowed to cool slowly to room temperature. For the denaturation, a thermocontrolled Varian Cary 1 UV/VIS instrument was used, and temperature was increased at a rate of 0.5 K per minute under nitrogen atmosphere. The reference samples contained the identical buffer without DNA. Curve fitting was carried out as described previously [8, 10]. Tm values are obtained from the first derivative of absorbance with respect to T−1, and are estimated to be accurate to within ± 0.5°C. Precision in free energy measurements is estimated to be ± 10–15%.
Footnotes
This work was supported in part by a grant from the National Institutes of Health (RO1-GM46625). E. T. K. acknowledges awards from the Young Investigator Programs of the Office of Naval Research (1992–1995), the Beckman Foundation (1992–1994), and the Army Research Office (1993–1996), and a Camille and Henry Dreyfus Teacher-Scholar Award (1993).
References
- 1.Miller J, McLachan AD, Klug A. EMBO J. 1985;4:1609–1614. doi: 10.1002/j.1460-2075.1985.tb03825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pavletich NP, Pabo CO. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- 3.Pelham HRB, Brown DD. Proc Natl Acad Sci USA. 1980;77:4170–4174. doi: 10.1073/pnas.77.7.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Honda BM, Roeder RG. Cell. 1980;22:119–126. doi: 10.1016/0092-8674(80)90160-9. [DOI] [PubMed] [Google Scholar]
- 5.Sands MS, Bogenhagen DF. Nucleic Acids Res. 1991;19:1791–1796. doi: 10.1093/nar/19.8.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubin E, Kool E. J Am Chem Soc. 1993;115:301–302. doi: 10.1021/ja00054a060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maher LJ, III, Dolnick BJ. Arch Biochem Biophys. 1987;253:214–220. doi: 10.1016/0003-9861(87)90654-0. [DOI] [PubMed] [Google Scholar]
- 8.Lisziewicz J, Sun D, Klotman M, Agrawal S, Zamecnik P, Gallo R. Proc Natl Acad Sci USA. 1992;89:11209–11213. doi: 10.1073/pnas.89.23.11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan R, Edge M, Colman A. Nucleic Acids Res. 1993;21:4615–4620. doi: 10.1093/nar/21.19.4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kool ET. J Am Chem Soc. 1991;113:6265–6266. [Google Scholar]
- 11.Prakash G, Kool ET. J Am Chem Soc. 1992;114:3523–3527. doi: 10.1021/ja00035a056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pilch DS, Levenson C, Shafer RH. Proc Natl Acad Sci USA. 1990;87:1942. doi: 10.1073/pnas.87.5.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersheim M, Turner DH. Biochemistry. 1983;22:256–263. doi: 10.1021/bi00271a004. [DOI] [PubMed] [Google Scholar]
- 14.Beaucage SL, Caruthers MH. Tetrahedron Lett. 1981;22:1859–1862. [Google Scholar]
- 15.Borer PN. In: Handbook of Biochemistry and Molecular Biology. 3rd. Fasman GD, editor. I. CRC; Cleveland, OH, USA: 1985. p. 589. [Google Scholar]
- 16.Rumney S, Kool ET. Angew Chem. 1992;104:1686–1689. [Google Scholar]; Angew Chem Int Ed Engl. 1992;31:1617–1619. doi: 10.1002/anie.199216171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prakash G, Rubin E, Kool ET. unpublished. [Google Scholar]