

Abstract
Inclusion body myositis is a progressive disease of the skeletal muscle. Here, specific theories of its pathogenesis are reviewed and general considerations pertaining to modeling of this disease discussed. Understanding of inclusion body myositis disease mechanism remains extremely poor. Current published animal models do not represent the disease. Future studies need to consider the critical role of biomarkers and methodologic issues in their discovery.
Keywords: Inclusion body myositis
Introduction
Inclusion body myositis (IBM) is a poorly understood disease affecting skeletal muscle. Its distinctive clinical features (onset at age 40 years or older, preference for asymmetric finger and wrist flexors and quadriceps muscles) and pathology, characterized by impressive myofiber abnormalities (termed degeneration), and immune system cell infiltration have yielded many ideas regarding its disease mechanisms. Enigmatic of the confusion in this field, even the name IBM derives from a case report of a patient who did not have the disease (a 26-year-old with quadriceps sparing and a severe limb-girdle pattern of weakness since age 18 years) [1]. Several recent reviews have addressed its potential disease mechanisms [2•, 3–6•, 7•].
Specific Theories
Myofiber Injury by β-Amyloid
The belief that the protein β-amyloid (Aβ) is produced by IBM myofibers and injures them is widely believed to be fact, having been stated as such in at least 200 PubMed-indexed papers as of October 2007. According to this theory, unknown factors result in the overproduction by myofibers of the mRNA of the Aβ precursor protein (βAPP), followed by translation of this transcript to the βAPP protein itself, and the cleavage of this protein into Aβ peptide. The overabundance of Aβ inside of myofibers is then theorized to cause myofiber degeneration, atrophy, and death through a variety of mechanisms of “toxicity” [8]. Congo red staining material (“amyloid”) in IBM myofibers is an entirely distinct issue, and this material, whose nature is likely diverse but certainly unknown, should not be confused with the protein Aβ simply because both substances share a common word in their names.
Several concerns about the scientific foundation for the Aβ toxicity IBM theory and the nature of the widespread published belief in it have been reported [4, 9••]. For example, no Western blot study demonstrating the claimed presence of Aβ in IBM muscle (let alone blots comparing IBM with other muscle diseases) has ever been published, a remarkable fact given the claim’s wide acceptance. Claims regarding Aβ presence in IBM muscle continue to be made using immunohistochemical reagents (eg, the 6E10 antibody) that react with βAPP [10] despite knowledge of this technical pitfall [9••, 11], a critical issue because the protein βAPP has been reported previously as being nonspecifically present in regenerating myofibers in all muscle diseases [12, 13]. However, it is examination of the central claim of this theory, that βAPP transcript is overproduced by IBM myofibers, that most succinctly outlines its critical problems.
The central problem with the claim that βAPP transcript is specifically overproduced by IBM myofibers is that it has no supporting evidence and was contradicted by even the most vigorous current proponents of this theory. The same laboratory reporting that βAPP transcript was overproduced in IBM myofibers in a 1993 publication [14] reported in a 1994 publication [13] that it was similarly overproduced in regenerating muscle fibers in all muscle diseases studied, including polymyositis, dermatomyositis, amyotrophic lateral sclerosis, and Duchenne muscular dystrophy (“the pattern and intensity of βAPP–mRNA expression in regenerating muscle fibers did not differ in relation to the diagnosis or age of a patient”) [13]. However, as of October 2007, the 1993 publication had received 54 citations from other papers supporting claims of this transcript’s overproduction in IBM, whereas the 1994 publication had been cited only four times [9••].
The only independent attempt to confirm the 1993 publication reporting IBM myofiber βAPP transcript overproduction using the same methodology (in situ histochemistry) found the method technically unreliable in IBM because of the presence of one or more sarcoplasmic nucleic acid-binding proteins (“in contrast to the findings of Sarkozi et al., we were unable to show an increase in mRNA for beta-amyloid precursor protein in IBM fibers by using antisense RNA probes”) [15]. This article, whose authors included the late George Karpati, is the most important IBM mechanistic paper published in the 1990s and early-2000s (see the subsequent “Nuclear Abnormalities” discussion), yet it has rarely been cited by the neuromuscular community despite thousands of citations made to other IBM mechanistic papers [9••].
No quantitative studies of βAPP transcript abundance in IBM and appropriate control samples were reported until almost a decade later; these microarray studies showed no difference in βAPP transcript abundance in IBM compared with polymyositis [16]. Not until 2008 were the first polymerase chain reaction-based studies of βAPP transcript in IBM muscle reported [17], and these showed no difference between the transcript’s abundance in IBM and polymyositis, with even greater amounts of βAPP transcript in dermatomyositis than IBM. By this time, nine publications had already reported βAPP transcript-overproducing animals as models of IBM [18–26]. How this theory could have progressed to a point in which transgenic animals overproducing βAPP transcript could be used as models of IBM without the most basic polymerase chain reaction studies examining whether βAPP transcript was even specifically overproduced in IBM muscle is a remarkable story in its own right, a story understood through a study of the citation network around this theory [9••].
Myofiber Injury by Tau
At least 41 publications have stated that accumulation of phosphorylated forms of the microtubule-associated protein tau occurs in and contributes to the pathogenesis of IBM [27•]. IBM has been called a “tauopathy” [28], and the use of lithium in clinical trials for patients with IBM has been recommended based on reductions in tau pathology in βAPP transgenic mice treated with lithium [25].
Evidence that tau is present in IBM myofibers is entirely limited to its immunohistochemical detection by antibodies (“tau” immunoreactivity), with the most commonly used one being SMI-31. What is remarkable about this literature is that immunoreactivity with this antibody has repeatedly been used to claim tau presence even though this antibody’s primary target (to which it was developed) is a different protein, phoshorylated neurofilament H. Recent studies have provided evidence that “tau” immunoreactivity is present in normal muscle myonuclei and that some or all of this immunoreactivity is due to the presence of neurofilament H [27•]. All literature claiming that SMI-31 immunoreactivity demonstrates phosphorylated tau in IBM myofibers is incorrect. There is no evidence that tau plays any role in the mechanism of myofiber injury in IBM.
Myofiber Injury and Other Accumulated Molecules
Many other molecules have been reported to accumulate in IBM myofibers and have been suggested to play a role in IBM myofiber injury. These include, but are not limited to, the following (references not included here because of space limitations): prion protein, ubiquitin (and mutant ubiquitin), cathepsins B and D, α1-antichymotrypsin, αB crystallin, desmin, cdk5, semicarbazide-sensitive amine oxidase, presenilin-1, α-synuclein, ApoE, redox factor-1, BACE1, BACE2, low-density lipoprotein receptor, very low-density lipoprotein receptor, liver-regulating protein, rapsyn, nicotinic acetylcholine receptor, transforming growth factor-β1, fibroblast growth factor, fibroblast growth factor receptor, cystatin C, neuronal nitric oxide synthase, inducible nitric oxide synthase, nitrotyrosine, insulin-like growth factor-1, Hsp27, Hsp40, Hsp65, Hsp70, superoxide dismutase-1, interleukin-1α, interleukin-1β, interleukin-6, calnexin, calreticulin, glucose-regulated protein-78, glucose-regulated protein-94, endoplasmic reticulum protein-72, extracellular signal-regulated kinase, Vaccinia virus complement control protein, clusterin, γ-tubulin, synemin, Bcl-2, Bax, LMP2, LMP7, MECL1, myostatin, glycogen synthase kinase-3β, RNA polymerase II, survival motor neuron protein, NOGO-B, neprilysin, nuclear factor-κB, transglutaminase 1 and 2, γ-tubulin; the 19S, 20S, PSMB8, PSMB9 proteasome subunits, HERP, parkin, RNF5, casein kinase-1a, and sequestosome-1.
The validity and meaning to IBM pathogenesis for many of these biomarkers is uncertain. Many of these studies are incomplete in defining the significance of these biomarkers, lacking disease control studies or quantitation. For example, RNF5 overexpression was interpreted from immunohistochemical studies and formed the basis for a transgenic RNF5 transcript-overproducing murine model claimed to represent IBM [29]. However, RNF5 transcript levels were not studied in IBM, and no studies were conducted in any other inflammatory myopathy samples, such as polymyositis or dermatomyositis. What if RNF5 was overproduced in all inflammatory myopathies, or many other muscle diseases? Many reports, including disease control studies, have stated ambiguously that various accumulated molecules were not present in “immunoreactive inclusions that were characteristic of IBM” [9••], leaving open the possibility that these molecules were present in disease control myofibers in other visible patterns; the statement of the control results is a tautology because the control diseases do not have “inclusions.”
Myofiber Injury by the Immune System
IBM muscle usually contains an impressive adaptive immune system response [30]. Whether the immune system contributes substantially to myofiber injury is uncertain. In considering this area, it is important to recognize implications of cellular constituents of the immune system (generally what is called inflammation based on the microscopic visualization of cells) and soluble secreted molecules of the immune system present, but not visible by microscopy, in IBM muscle.
T Cells
The invasion of apparently viable IBM myofibers by cytotoxic T cells has been emphasized since the mid-1980s [31]. A great deal of evidence indicates that these T cells have been stimulated by antigen and developed through successive generations highly specific antigen-directed T-cell receptors [reviewed in 30]. Although the presence of cytotoxic T-cell myofiber invasion has been widely emphasized, this occurs in a relatively small number of myofibers based on cross-section examination, and many IBM biopsies show far greater numbers of CD4+ T cells (not cytotoxic) surrounding and pushing apart, but not invading, myofibers. Many morphologically abnormal myofibers typically have no nearby T cells visible on cross-sections. Whether these T cells are injuring muscle (eg, through secretion of soluble molecules) or contributing to other immune cell myofiber injury is unknown.
B Cells
Although B cells as defined by the surface markers CD19 and CD20 were long thought to be sparse or absent from IBM muscle, recent studies have shown that differentiated B cells (CD138+ antibody-secreting plasma cells) are not only abundant in IBM muscle but are transcriptionally active, producing and secreting immunoglobulins within muscle, and that these immunoglobulins are from clonally expanded, highly refined antigen-directed plasma cells [32, 33•].
Although the consequences of such antibody production are unknown, the key insight gained from these discoveries is that they open the door to possibly identifying antigens against which both T and B cells may be directed because of the principle of linked recognition (B-cell-aided maturation of T cell requires that both B-cell immunoglobulin and T-cell receptors recognize the same molecular complex). The use of patient-derived antibodies for antigen identification is a technically easier strategy than T-cell approaches. This strategy has been used successfully, identifying an immune response against αB crystallin in several patients with IBM [34]. αB Crystallin previously had been identified as a molecule of interest in IBM because of its distinctive immunohistochemical appearance in IBM compared with other inflammatory myopathies [35].
Soluble Immune Molecules
IBM muscle is likely an environment rich in soluble immune cell-secreted proteins. Certainly the RNA transcripts of such immune molecules are greatly amplified in IBM muscle [16]. Studies of their proteins are hampered by technical challenges: most of these are likely washed away during the preparation of immunohistochemical sections. The accurate measurement of cytokine proteins in IBM muscle by other methods is fraught with difficulties. The mechanistic consequences of this likely cytokine-rich environment, containing particularly abundant interferon-γ and possibly tumor necrosis factor-α based on transcript studies and the abundance of T cells and macrophages present, are unknown.
Nuclear Abnormalities
Nuclear abnormalities and their implications in IBM recently have been reviewed [7•]. The first published reports delineating distinct pathological features of IBM from polymyositis were written by Chou [36, 37] in 1967 and 1968. These emphasized substantial myonuclear abnormalities that were further detailed by Carpenter and colleagues in 1978 [38] and between 1993 and 1996 [15, 39, 40]. These investigators formulated a hypothesis that rimmed vacuoles, a feature that distinguishes IBM from polymyositis on hematoxylin- and eosin- and trichrome-stained muscle sections, derived from the breakdown of myonuclei. Between 1996 and 2007, few published papers mentioned these data. No review papers, typically the most influential type of publication in shaping opinion, including at least 31 written during this period, mentioned the existence of these data or their implications.
Most rimmed vacuoles are lined with nuclear membrane proteins, suggesting they frequently derive from myonu-clear breakdown [41]. Further evidence for this hypothesis is reviewed elsewhere [7•]. Fifteen years ago, experiments attempting (and failing) to confirm claims of specific Aβ precursor protein transcript abundance instead found a nucleic acid-binding protein lining vacuoles of some IBM myofibers [15]. The recent discovery of the nucleic acid-binding protein TDP-43 in IBM non-nuclear sarcoplasm is a major advance in this long dormant theory [42•, 43••, 44, 45]. Abnormalities in the distribution of TDP-43 in IBM myofibers with fluorescent microscopy are the most impressive of all microscopic IBM biomarkers I have seen, present in a mean of 23% of IBM myofibers, most of which appear morphologically normal or only minimally abnormal on parallel hematoxylin and eosion sections [43••]. In these fibers, TDP-43 has redistributed from its normally nuclear location to the sarcoplasm.
The mechanisms and consequences of TDP-43 redistribution from myonuclei to sarcoplasm in a high percentage of IBM myofibers are uncertain. Abnormal accumulation of extranuclear TDP-43 may lead to deleterious interaction with or sequestration of certain particular mRNA or microRNAs, and affect the translation of specific proteins in IBM.
Future Study of IBM Pathogenesis
There are lessons to be learned from several decades of research into IBM disease mechanism that has not disclosed fundamental insights benefiting people. Some of these are outlined here.
Animal Models in IBM: Progress or Circularity?
Thirteen publications have reported animal experiments as actual or potential models of IBM [18–26, 29, 46–48]. It is my opinion that physicians have inordinate reverence for such animal models and need to consider more critically their validities and potential utilities for delivering translational results to patients. Reasonable animal models based on unequivocal temporally and causally defined events (such as DNA mutations, infectious organisms, or mechanical trauma) can potentially be useful in the study of disease mechanism. Because in IBM, no such temporally and causally defined events have been defined from the study of human tissue samples, two approaches have been used to justify animal models: circularity and invention of temporality.
Circularity involves the production of an animal model and then the claim that “it looks like” the human disease to persuade others that the animal model includes the “key” disease-causing molecular events. For example, animals are genetically engineered to overproduce βAPP, their tissues examined to show βAPP overproduced, and they are then confirmed as IBM models because IBM also is reported as having βAPP in muscle. In another example, histochemical studies from animals fed high-cholesterol diets containing abundant artifact have been interpreted as resembling the degenerative changes of IBM myofibers [47].
Invention of temporality has been carefully documented for the claim that Aβ accumulation occurs early and precedes other abnormalities in IBM muscle [9••]. Initially stated as hypothesis in 1992 and 1993, 27 papers as of October 2007 have made this temporal claim, which spontaneously transformed from hypothesis to fact through citation alone, a process called citation transmutation. Authors have repeatedly stated it as fact (eg, “the appearance of Aβ-positive noncongophilic deposits precedes vacuolization in IBM muscle fibers”), supporting these statements to citations to papers in which it had only been proposed as hypothesis (“may represent early changes of IBM”). This invented temporal claim has been used to justify National Institutes of Health funding for animal experimentation [9••].
Until such time as IBM temporally and causally defined events are defined through further study of human disease biomarkers, animal models are highly unlikely to be productive endeavors for individuals with IBM. However, paradoxically, animal models, with little dependence on their validity, are very productive for biomedical researchers, aiding the ability to publish papers and obtain research funding. Because they allow for hypothesis-driven research (even on the wrong hypotheses), they allow for the use of key ingredients for successful grant proposals: carefully controlled scientific experiments with exciting novel technologies.
The Crucial Role of Biomarkers
Biomarkers are biological observations associated with disease. For example, dystrophin DNA sequence variants in blood cells are biomarkers of Duchenne muscular dystrophy, and serum creatine kinase is a biomarker in some disorders of cardiac and skeletal muscle.
All theories of IBM pathogenesis have relied critically on muscle biomarkers. Biomarker specificity is the key issue for the future. Finding molecular biomarkers that distinguish IBM muscle from both other inflammatory myopathy muscle (polymyositis and dermatomyositis) and from a range of other myopathies containing rimmed vacuoles would advance mechanistic understanding. Great care needs to be taken in establishing the validity of biomarkers, and three issues need particular attention: immunohistochemical methodology, the use of controls, and quantitation.
Immunohistochemical Methodology
Immunohistochemical approaches have dominated the field. Assuming that immunoreactive signal indeed represents target can be very problematic, as just because an antibody was raised against a specific protein does not ensure that its binding in muscle indicates the presence of that protein. Interpreting antibodies known to react to multiple proteins as indicative of the presence of a specific protein is an invalid practice that has pervaded the field (eg, 6E10 indicative of Aβ, though it reacts also to βAPP; SMI-31 indicative of tau, though it reacts to neurofilament H and is stated as such on its manufacturer’s datasheet).
The interpretation of artifact as immunohistochemical signal is a major problem in the field. Many published images in the IBM literature appear to me to show artifact, not immunohistochemical signal. The problem of autofluorescing material visible in both red and green channels without the presence of primary antibodies is particularly concerning (Fig. 1), as many published images have been interpreted as demonstrating this material as immunoreactive and have been used to conclude that certain proteins aggregated in IBM myofibers. It is not sufficient to simply state in methods that autofluorescence was excluded; claims of fluorescent immunoreactive material need to be supported by single fluorochrome staining accompanied by images using both red and green fluorescent filter sets (Fig. 1).
Fig. 1.
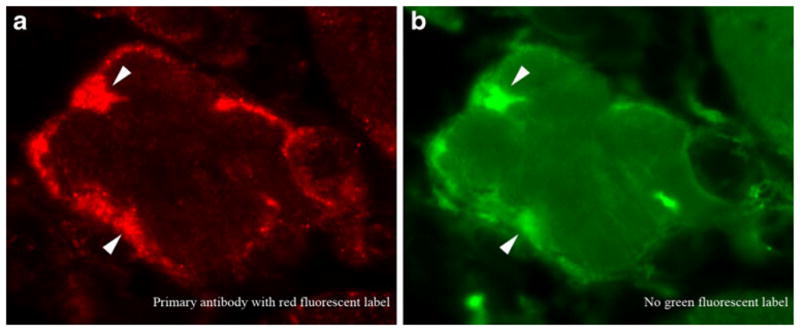
The potential for misinterpretation of artifact as protein aggregates in inclusion body myositis (IBM) myofibers. Immunofluorescence microscopy using a primary antibody (not disclosed here) with a red fluorochrome (a). The IBM literature is filled with similar images interpreted as demonstrating the accumulation of a specific protein. The same image viewed through a green fluorescent filter set (b), however, confirms that the material producing most, if not all, red fluorescence seen in (a) also emits green fluorescence. Because no green fluorescent molecules were used in this experiment, this material is autofluorescent. No protein aggregates can be concluded to be present. Had these studies been performed with a second green fluorescent-labeled antibody, they may have been interpreted incorrectly as showing colocalization of two different proteins in IBM aggregates. Arrowheads highlight some of the autofluorescent regions
Disease Controls
Appropriate controls in all studies are essential. Demonstrating that a given biomarker is increased in IBM compared with normal muscle is of limited value. What needs to be demonstrated is that the biomarker differs in IBM from an appropriate range of other diseases. How else can we know whether the biomarker is key to IBM? The consequences of not performing disease control studies are substantial for IBM research. Why are not transgenic muscle-overproducing βAPP animals reported as models of amyotrophic lateral sclerosis rather than IBM, as βAPP was easily demonstrated as fourfold increased in amyotrophic lateral sclerosis muscle Western blots [49]? Why are RNF5 transgenic animals interpreted as models of IBM based on immunohistochemical studies of RNF5 in IBM alone, when RNF5 transcript elevation has not been reported in IBM, and RNF5 transcript and protein not even studied in other inflammatory myopathies [29]?
Quantitation
Is it sufficient to report that a molecule is “present” in immunohistochemical studies of IBM myofibers and interpret it as mechanistically important, but not to report that it was seen in only 3 of 1,000 myofibers examined? Frequently ignored issues of quantitation are crucial to understanding the value of a biomarker to disease mechanism. For example, reporting on studies of muscle from IBM patients that Aβ was present in “almost 100% of their vacuolated fibers” is difficult to interpret when unaccompanied by a statement as to how many vacuolated fibers were seen in those same samples. As some of the same laboratory’s samples contain “only two or three vacuolated muscle fibers” [9••] (see discussion at http://www.bmj.com/cgi/data/bmj.b2680/DC1/1, page 10), and typical biopsy sections contain 1,000 or more myofiber cross-sections, the percentage of Aβ-containing myofibers in such patients may be actually less than 0.3%. Does this degree of presence justify the status of Aβ as a disease-causing biomarker? Does it support claims of “the universal presence of Aβ in IBM” and that it is a “hallmark” of the disease [11]? Can studies reporting no visible βAPP or Aβ in three of five IBM patient muscle biopsy samples and its presence in “only a few fibres” in the other two samples be validly cited as showing “βAPP in IBM fibers has been confirmed by others” [9••]? Quantitation matters for biomarker significance.
Trends: Good for Researchers, Bad for Patients
Two roads lie before researchers contemplating the pursuit of relevant disease mechanisms in IBM: pursue unbiasedly generated or fortuitously found clues, or join a trend. The former is more likely to produce scientific advances helpful to patients, the latter safer and more likely to produce grant funding and successful publication helpful to the researcher.
Trends are very powerful mechanisms for successful publication and funding. For example, at the time of this writing, there were 100,802 PubMed-indexed papers containing the search term nitric oxide, with 42,735 papers having nitric oxide in their title. Such widespread interest leaves opportunity for exploration of this molecule’s presence in IBM [50]. The more diseases in which a biomarker is found to be abnormal, the lower its specificity for any particular disease and the less likely it is to be critically involved in that disease’s pathogenesis. Widespread interest in a molecule should make it less valuable an area of study for IBM, yet paradoxically, the greater the interest in the molecule, the more likely peers are to approve grant funding and manuscript acceptance. Every new trend allows opportunity for scientific studies in many unrelated diseases. Although no one can know whether the pursuit of any given trend will turn out to be productive for people with IBM, in my opinion, the odds are strongly against the benefit of adapting ideas from other fields into models of IBM disease mechanism unless they are driven by independent justification. Trends that IBM research has seen include concepts of “stress” (“endoplasmic reticulum stress,” “oxidative stress”), myofiber breakdown (“autophagy,” “proteasome dysfunction,” “unfolded protein response”), and “molecular toxicity” (Aβ, tau, prion protein).
Conclusions
After two to three decades of intense interest in IBM, there is almost no understanding as to the cause of this disease and the mechanisms by which myofibers are injured. Large gaps exist between the identification of disease biomarkers and their causal relationships to muscle injury. Currently advocated animal models do not represent this disease and should not be relied upon for therapeutic development. Careful attention to biomarker specificity, quantitation, and methodology is crucial to progress for patients with IBM.
Acknowledgments
This work is supported in part by a grant to Dr. Greenberg from the National Institutes of Health (R21NS057225).
Footnotes
Disclosure Dr. Greenberg has served as a consultant regarding clinical trial planning for MedImmune and has a sponsored research agreement with MedImmune. He is also an inventor of intellectual property pertaining to myositis diagnostics.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Yunis EJ, Samaha FJ. Inclusion body myositis. Lab Invest. 1971;25:240–248. [PubMed] [Google Scholar]
- 2•.Machado P, Miller A, Holton J, Hanna M. Sporadic inclusion body myositis: an unsolved mystery. Acta Reumatol Port. 2009;34:161–182. This is a comprehensive review of many aspects of IBM. [PubMed] [Google Scholar]
- 3.Karpati G, O’Ferrall EK. Sporadic inclusion body myositis: pathogenic considerations. Ann Neurol. 2009;65:7–11. doi: 10.1002/ana.21622. [DOI] [PubMed] [Google Scholar]
- 4.Greenberg SA. Inclusion body myositis: review of recent literature. Curr Neurol Neurosci Rep. 2009;9:83–89. doi: 10.1007/s11910-009-0013-x. [DOI] [PubMed] [Google Scholar]
- 5.Askanas V, Engel WK, Nogalska A. Inclusion body myositis: a degenerative muscle disease associated with intra-muscle fiber multi-protein aggregates, proteasome inhibition, endoplasmic reticulum stress and decreased lysosomal degradation. Brain Pathol. 2009;19:493–506. doi: 10.1111/j.1750-3639.2009.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6•.Needham M, Mastaglia FL. Sporadic inclusion body myositis: a continuing puzzle. Neuromuscul Disord. 2008;18:6–16. doi: 10.1016/j.nmd.2007.11.001. This is a thoughtful review of IBM mechanistic theories. [DOI] [PubMed] [Google Scholar]
- 7•.Greenberg SA. Inflammatory myopathies: disease mechanisms. Curr Opin Neurol. 2009;22:516–523. doi: 10.1097/WCO.0b013e3283311ddf. This is a review of inflammatory myopathy disease mechanisms that focuses on dermatomyositis and IBM. [DOI] [PubMed] [Google Scholar]
- 8.Askanas V, Engel WK. Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology. 2006;66(2 Suppl 1):S39–S48. doi: 10.1212/01.wnl.0000192128.13875.1e. [DOI] [PubMed] [Google Scholar]
- 9•.Greenberg SA. How citation distortions create unfounded authority: analysis of a citation network. BMJ. 2009;339:b2680. doi: 10.1136/bmj.b2680. This article rigorously demonstrates how distortions in the scholarly process of citation may create belief in unfounded claims, with particular attention paid to claims regarding Aβin IBM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muth IE, Barthel K, Bahr M, et al. Proinflammatory cell stress in sporadic inclusion body myositis muscle: overexpression of alphaB-crystallin is associated with amyloid precursor protein and accumulation of beta-amyloid. J Neurol Neurosurg Psychiatry. 2009;80:1344–1349. doi: 10.1136/jnnp.2009.174276. [DOI] [PubMed] [Google Scholar]
- 11.Greenberg SA. Comment on ‘Interrelation of inflammation and APP in sIBM: IL-1{beta} induces accumulation of {beta}-amyloid in skeletal muscle’. Brain. 2009;132:e106. doi: 10.1093/brain/awn163. [DOI] [PubMed] [Google Scholar]
- 12.Askanas V, Sarkozi E, Bilak M, et al. Human muscle macrophages express beta-amyloid precursor and prion proteins and their mRNAs. Neuroreport. 1995;6:1045–1049. doi: 10.1097/00001756-199505090-00024. [DOI] [PubMed] [Google Scholar]
- 13.Sarkozi E, Askanas V, Johnson SA, et al. Expression of beta-amyloid precursor protein gene is developmentally regulated in human muscle fibers in vivo and in vitro. Exp Neurol. 1994;128:27–33. doi: 10.1006/exnr.1994.1109. [DOI] [PubMed] [Google Scholar]
- 14.Sarkozi E, Askanas V, Johnson SA, et al. beta-Amyloid precursor protein mRNA is increased in inclusion-body myositis muscle. Neuroreport. 1993;4:815–818. doi: 10.1097/00001756-199306000-00055. [DOI] [PubMed] [Google Scholar]
- 15.Nalbantoglu J, Karpati G, Carpenter S. Conspicuous accumulation of a single-stranded DNA binding protein in skeletal muscle fibers in inclusion body myositis. Am J Pathol. 1994;144:874–882. [PMC free article] [PubMed] [Google Scholar]
- 16.Greenberg SA, Sanoudou D, Haslett JN, et al. Molecular profiles of inflammatory myopathies. Neurology. 2002;59:1170–1182. doi: 10.1212/wnl.59.8.1170. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt J, Barthel K, Wrede A, et al. Interrelation of inflammation and APP in sIBM: IL-1 beta induces accumulation of beta-amyloid in skeletal muscle. Brain. 2008;131:1228–1240. doi: 10.1093/brain/awn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukuchi K, Pham D, Hart M, et al. Amyloid-beta deposition in skeletal muscle of transgenic mice: possible model of inclusion body myopathy. Am J Pathol. 1998;153:1687–1693. doi: 10.1016/s0002-9440(10)65682-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin LW, Hearn MG, Ogburn CE, et al. Transgenic mice over-expressing the C-99 fragment of betaPP with an alpha-secretase site mutation develop a myopathy similar to human inclusion body myositis. Am J Pathol. 1998;153:1679–1686. doi: 10.1016/s0002-9440(10)65681-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugarman MC, Yamasaki TR, Oddo S, et al. Inclusion body myositis-like phenotype induced by transgenic overexpression of beta APP in skeletal muscle. Proc Natl Acad Sci U S A. 2002;99:6334–6339. doi: 10.1073/pnas.082545599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strazielle C, Dumont M, Fukuchi K, Lalonde R. Transgenic mice expressing the human C99 terminal fragment of betaAPP: effects on cytochrome oxidase activity in skeletal muscle and brain. J Chem Neuroanat. 2004;27:237–246. doi: 10.1016/j.jchemneu.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 22.Kitazawa M, Green KN, Caccamo A, LaFerla FM. Genetically augmenting Abeta42 levels in skeletal muscle exacerbates inclusion body myositis-like pathology and motor deficits in transgenic mice. Am J Pathol. 2006;168:1986–1997. doi: 10.2353/ajpath.2006.051232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moussa CE, Fu Q, Kumar P, et al. Transgenic expression of beta-APP in fast-twitch skeletal muscle leads to calcium dyshomeo-stasis and IBM-like pathology. FASEB J. 2006;20:2165–2167. doi: 10.1096/fj.06-5763fje. [DOI] [PubMed] [Google Scholar]
- 24.Sugarman MC, Kitazawa M, Baker M, et al. Pathogenic accumulation of APP in fast twitch muscle of IBM patients and a transgenic model. Neurobiol Aging. 2006;27:423–432. doi: 10.1016/j.neurobiolaging.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 25.Kitazawa M, Trinh DN, LaFerla FM. Inflammation induces tau pathology in inclusion body myositis model via glycogen synthase kinase-3beta. Ann Neurol. 2008;64:15–24. doi: 10.1002/ana.21325. [DOI] [PubMed] [Google Scholar]
- 26.Rebolledo DL, Minniti AN, Grez PM, et al. Inclusion body myositis: a view from the Caenorhabditis elegans muscle. Mol Neurobiol. 2008;38:178–198. doi: 10.1007/s12035-008-8041-0. [DOI] [PubMed] [Google Scholar]
- 27•.Salajegheh M, Pinkus JL, Nazareno R, et al. Nature of “Tau” immunoreactivity in normal myonuclei and inclusion body myositis. Muscle Nerve. 2009;40:520–528. doi: 10.1002/mus.21471. The identity of SMI-31 immunoreactive material widely interpreted as phosphorylated tau was shown to likely be neurofilament H. [DOI] [PubMed] [Google Scholar]
- 28.Maurage CA, Bussière T, Sergeant N, et al. Tau aggregates are abnormally phosphorylated in inclusion body myositis and have an immunoelectrophoretic profile distinct from other tauopathies. Neuropathol Appl Neurobiol. 2004;30:624–634. doi: 10.1111/j.1365-2990.2004.00577.x. [DOI] [PubMed] [Google Scholar]
- 29.Delaunay A, Bromberg KD, Hayashi Y, et al. The ER-bound RING finger protein 5 (RNF5/RMA1) causes degenerative myopathy in transgenic mice and is deregulated in inclusion body myositis. PLoS ONE. 2008;3:e1609. doi: 10.1371/journal.pone.0001609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greenberg SA. Proposed immunologic models of the inflammatory myopathies and potential therapeutic implications. Neurology. 2007;69:2008–2019. doi: 10.1212/01.WNL.0000291619.17160.b8. [DOI] [PubMed] [Google Scholar]
- 31.Engel AG, Arahata K. Monoclonal antibody analysis of mononu-clear cells in myopathies. II: Phenotypes of autoinvasive cells in polymyositis and inclusion body myositis. Ann Neurol. 1984;16:209–215. doi: 10.1002/ana.410160207. [DOI] [PubMed] [Google Scholar]
- 32.Greenberg SA, Bradshaw EM, Pinkus JL, et al. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology. 2005;65:1782–1787. doi: 10.1212/01.wnl.0000187124.92826.20. (Published erratum appears in Neurology 2006, 66:493.) [DOI] [PubMed] [Google Scholar]
- 33•.Bradshaw EM, Orihuela A, McArdel SL, et al. A local antigen-driven humoral response is present in the inflammatory myopathies. J Immunol. 2007;178:547–556. doi: 10.4049/jimmunol.178.1.547. The intramuscular B-cell response in IBM was shown to be antigen driven, laying the groundwork for future antibody-based antigen discovery. [DOI] [PubMed] [Google Scholar]
- 34.Salajegheh M, Lin YY, Parker KC, et al. Using humoral immunity for the identification of candidate antigens in inclusion body myositis [abstract] Neurology. 2007;68(Suppl 1):A361. [Google Scholar]
- 35.Banwell BL, Engel AG. AlphaB-crystallin immunolocalization yields new insights into inclusion body myositis. Neurology. 2000;54:1033–1041. doi: 10.1212/wnl.54.5.1033. [DOI] [PubMed] [Google Scholar]
- 36.Chou SM. Myxovirus-like structures in a case of human chronic polymyositis. Science. 1967;158:1453–1455. doi: 10.1126/science.158.3807.1453. [DOI] [PubMed] [Google Scholar]
- 37.Chou SM. Myxovirus-like structures and accompanying nuclear changes in chronic polymyositis. Arch Pathol. 1968;86:649–658. [PubMed] [Google Scholar]
- 38.Carpenter S, Karpati G, Heller I, Eisen A. Inclusion body myositis: a distinct variety of idiopathic inflammatory myopathy. Neurology. 1978;28:8–17. doi: 10.1212/wnl.28.1.8. [DOI] [PubMed] [Google Scholar]
- 39.Carpenter S. Inclusion body myositis, a review. J Neuropathol Exp Neurol. 1996;55:1105–1114. doi: 10.1097/00005072-199611000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Karpati G, Carpenter S. Pathology of the inflammatory myopathies. Baillieres Clin Neurol. 1993;2:527–556. [PubMed] [Google Scholar]
- 41.Greenberg SA, Pinkus JL, Amato AA. Nuclear membrane proteins are present within rimmed vacuoles in inclusion-body myositis. Muscle Nerve. 2006;34:406–416. doi: 10.1002/mus.20584. [DOI] [PubMed] [Google Scholar]
- 42•.Weihl CC, Temiz P, Miller SE, et al. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2008;79:1186–1189. doi: 10.1136/jnnp.2007.131334. This article found sarcoplasmic TDP-43, a protein normally seen only in nuclei, in biopsy samples from patients with IBM and valosin-containing protein mutation inclusion body myopathies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43••.Salajegheh M, Pinkus JL, Taylor JP, et al. Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve. 2009;40:19–31. doi: 10.1002/mus.21386. TDP-43 presence in IBM was quantitated in relation to other reported biomarkers. The relation between this nucleic acid-binding protein and nuclear abnormalities is discussed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Küsters B, van Hoeve BJ, Schelhaas HJ, et al. TDP-43 accumulation is common in myopathies with rimmed vacuoles. Acta Neuropathol. 2009;117:209–211. doi: 10.1007/s00401-008-0471-2. [DOI] [PubMed] [Google Scholar]
- 45.Olivé M, Janué A, Moreno D, et al. TAR DNA-binding protein 43 accumulation in protein aggregate myopathies. J Neuropathol Exp Neurol. 2009;68:262–273. doi: 10.1097/NEN.0b013e3181996d8f. [DOI] [PubMed] [Google Scholar]
- 46.Capsoni S, Ruberti F, Di Daniel E, Cattaneo A. Muscular dystrophy in adult and aged anti-NGF transgenic mice resembles an inclusion body myopathy. J Neurosci Res. 2000;59:553–560. doi: 10.1002/(SICI)1097-4547(20000215)59:4<553::AID-JNR11>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 47.Chen X, Ghribi O, Geiger JD. Rabbits fed cholesterol-enriched diets exhibit pathological features of inclusion body myositis. Am J Physiol Regul Integr Comp Physiol. 2008;294:R829–R835. doi: 10.1152/ajpregu.00639.2007. [DOI] [PubMed] [Google Scholar]
- 48.Kitazawa M, Vasilevko V, Cribbs DH, LaFerla FM. Immunization with amyloid-beta attenuates inclusion body myositis-like myopathology and motor impairment in a transgenic mouse model. J Neurosci. 2009;29:6132–6141. doi: 10.1523/JNEUROSCI.1150-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koistinen H, Prinjha R, Soden P, et al. Elevated levels of amyloid precursor protein in muscle of patients with amyotrophic lateral sclerosis and a mouse model of the disease. Muscle Nerve. 2006;34:444–450. doi: 10.1002/mus.20612. [DOI] [PubMed] [Google Scholar]
- 50.Yang CC, Alvarez RB, Engel WK, Askanas V. Increase of nitric oxide synthases and nitrotyrosine in inclusion-body myositis. Neuroreport. 1996;8:153–158. doi: 10.1097/00001756-199612200-00031. [DOI] [PubMed] [Google Scholar]