

Abstract
Tobacco smoke exhaled from smokers is a key component of secondhand smoke, contributing to lung alveolar wall destruction seen in chronic lung diseases. Although mainstream and sidestream tobacco smoke are cyto-toxic to lung cells, it is unclear whether exhaled smoke induces lung cell injury or even death. We sought to establish an in vitro model to examine the effects of exhaled smoke on lung cells. Phosphate-buffered saline-conditioned cigarette smoke (CCS) derived from a blow-by system was used to mimic exhaled tobacco smoke exposure. Exposure of medium to CCS leads to dose-dependent increases in nicotine/cotinine levels. Scanning spectrophotometric analysis of the CCS-exposed medium reveals an absorption peak at 290 nm wavelength. The OD values at 290 nm are correlated with nicotine levels in the exposed medium, indicating that a simple measurement of OD at 290 nm can be used to monitor CCS exposure. Tobacco smoke contacts the microvascular endothelium located at lung alveoli, before it enters the blood stream. Hence, human lung microvascular endothelial cells (hMVEC) were exposed to CCS and assessed for cell injury and death. Exposure of hMVEC to CCS equivalent to burning 12-16 cigarettes leads to increased LDH release from the cells into the medium. This suggests that CCS can induce lung cell injury. CCS at a low level increases cell growth, whereas the high level of CCS decreases cell viability. In addition, CCS exposure induces cell detachment and morphological changes. Our results demonstrate that exposure of buffer-conditioned mainstream cigarette smoke leads to increased nicotine/cotinine levels and cell injury/death, which may contribute to the pathophysiology of passive smoking-associated lung diseases.
Keywords: Cellular model, secondhand smoke, chronic lung diseases, lung microvascular endothelial cells, passive smoking-associated lung diseases
Introduction
Secondhand smoke is a serious health risk, which causes about 50,000 deaths in the United States each year [1]. Growing evidences suggest that exposure to secondhand smoke is associated with the pathophysiology of chronic obstructive pulmonary disease including emphysema, a deadly lung disorder associated with alveolar cell death and destruction [2-6]. Secondhand smoke consists of exhaled mainstream smoke and sidestream smoke derived from burning a tobacco product. Non-smokers are exposed to secondhand smoke when close to the smoker or in the indoor space where smoking occurs. Abundant studies have demonstrated that mainstream and sidestream smoke are cytotoxic to lung cells in vitro and in vivo [7-11]. For instance, exposure of cultured cells to smoke extracts or condensates result in the damage and death of lung alveolar cells including endothelial and epithelial cells [7-8]. Furthermore, the numbers of apoptotic cells are increased in the emphysematous lungs and the lungs of animals exposed to smoke [9-11]. When mainstream smoke passes through aqueous solutions such as lung fluid or medium, some ingredients in the gas and solid phase are absorbed. It has been speculated that exhaled smoke, a component of secondhand smoke, may be less toxic to humans than whole smoke is, since it has been “conditioned” or “detoxified” by smokers’ lungs.
In order to determine the direct effects of exhaled smoke on the induction of lung cell injury and death, a cellular model is needed. However, there are challenges related to mimicking exhaled smoke. First, many factors can affect the contents of exhaled smoke. For instance, the length of smoke that is inhaled influences the amount of chemicals that are absorbed by the lung fluid. Furthermore, it is very difficult to expose humans or animals to tobacco smoke and then collect exhaled smoke. Given the complex nature of exhaled smoke, an in vitro model would help closely mimic exhaled smoke under controllable conditions.
Exhaled smoke is a complex mixture of substances, and as such, a surrogate measure of exposure that is representative of exhaled smoke as a whole is essential. Several components have been used as surrogates or markers. Most cigarettes (in inhaled smoke) contain 1 to 3 milligrams of nicotine. Nicotine is most commonly found in the gas phase in the environment and has been widely used as a potential marker of tobacco smoke exposure. When tobacco is smoked, the lungs are directly exposed to smoke. Nicotine-rich blood in the vas-culature passes from the lungs to the brain within a few seconds. Nicotine stays in the blood for several hours to two days. The levels of blood nicotine in individual smokers range from 25 to 444 nmol/L (4 to 72 ng/mL) [12]. The levels of blood nicotine in habitual tobacco users have been reported to be as high as 3.09 μg/mL by gas chromatography [13].
The metabolites of smoke ingredients in physiological fluids are also used as accurate methods for estimating tobacco smoke exposure in subjects. Cotinine, a metabolite of nicotine, leaves its traces in blood and urine for up to 7 days. Cotinine is the biological marker of choice in most epidemiological studies. Cotinine assays are sensitive enough to distinguish individuals without tobacco exposure from those persons with low exposure. The current optimal plasma cotinine cut-point to distinguish smokers from non-smokers in the general US population is 3 ng/ mL [14]. The plasma cotinine levels are largely varied from 16 ng/mL to 1180 ng/mL in the individuals who smoke 20 cigarettes per day [15]. Other markers such as solanesol, 3-ethenylpyridine, carbon monoxide, iso- and an-teisoalkanes (C29-C34), fluorescent particulate matters, respirable suspended particles, and ultraviolet particulate matters are also used as the markers of tobacco smoke exposure.
Asmoke exposure protocol has been developed in our laboratory to closely mimic in vivo exposure of exhaled smoke. In the system, mainstream cigarette smoke is passed through phosphate-buffered saline (PBS, pH 7.4) to mimic lung-conditioned smoke. A peak absorbance has been identified, correlated to nicotine/ cotinine levels, and used to monitor the exposure of conditioned cigarette smoke (CCS). The cytotoxic effects of buffer-condition cigarette smoke on human lung endothelial cells have been assessed.
Materials and methods
Cell culture
Human pulmonary microvascular endothelial cells (hMVEC) were purchased from Lonza and propagated in monolayer cultures as described by Zhang et al [16-17]. Cells that were subcul-tured less than 5 times in post-confluent monolayers and maintained in EGM-2 MV medium (Lonza) were used for all experiments.
Modular exposure of hMVEC to CCS
Mainstream cigarette smoke was passed through PBS in a blow-by system as shown in Figure 1, thus closely mimicking the process of tobacco smoke that passes through the airways of smoker's lungs. Mainstream smoke traveled 10 cm through PBS since the range of an average tracheal length is from 5.4 cm to 13.1 cm for a 2-20 year old person [18]. The inlet of a modular incubator chamber (Billups-Rothenberg) was connected to a 1,000 mL-flask containing 1,000 mL of PBS. A cigarette holder was attached to the flask, and the outlet of the holder was submerged 10 cm below the PBS surface. Then, the monolayers of cultured hMVEC on the bottom of a 35-mm dish were covered by 2 mL of medium to mimic in vivo lung alveolar fluid. The thickness of the medium layer above the endothelium layer in the center of the dish is about 2 mm. Although the thickness of the alveolar epithelial fluid is estimated to be about 20-150 μm [19], 2 mm medium coverage was used in the cell model of exhaled smoke for the following reasons. First, the lung fluid contains organic substances and lung cells that may scavenger and/or metabolite substances in tobacco smoke. Therefore, more PBS is needed to achieve similar effects as the alveolar fluid does. Second, smoke must pass the alveolar epithelial layer to reach the endothe-lium. Third, too low level of PBS may cause direct exposure and cell injury. Finally, insufficient medium in the dish can result in the alteration of nutrient concentrations due to evaporation during the exposure process, which could affect the cells. The dishes in the chamber were exposed to CCS derived directly from burning 2R4F reference research cigarettes (the second run of the 1R4F reference cigarette developed by the NIH-NCI, the USDA and the Tobacco and Health Research Institute of the University of Kentucky) that was passed or filtered through PBS solution. Cigarettes were burned at a rate of one cigarette per 4 minutes for 16 min (4 cigarettes). The smoke was passed through 10 cm of PBS, and then delivered to the exposure chamber. The CCS-exposed cells were returned to normal conditions, i.e. room air-5% CO2 at 37°C, and kept for a 44 min to mimic the intervals between smokings in vivo. This was defined as one exposure cycle or exposure unit (EU), i.e. burning 4 cigarettes in 16 min plus 44 min recovery. For exposure of cells to 8-16 cigarettes or 2-4 EU, the exposure cycle had been repeated 2-4 times. Control hMVEC in a parallel chamber were treated under identical conditions with an unlit cigarette (0 EU). After CCS exposure, 1 mL of medium was taken from the dish and used for the spectrophotometric analysis. The exposed cells were either used immediately or incubated in fresh medium under normal conditions for an additional 12-14 hrs.
Figure 1.

A diagram of the flow-by system for exposure of hMVEC to CCS. The inlet of a modular incubator chamber was connected to a flask containing 1,000 mL of PBS, and the outlet was connected to a vacuum line through a valve for adjustment of flow rates and an absorption unit. Culture dishes in the exposure chamber were exposed to CCS generated by burning 2R4F cigarettes and passing through PBS at a rate of one cigarette per four minutes. Cells in the control chamber were treated under identical conditions with an unlit cigarette.
LC-MS/MS analysis of nicotine and related derivatives
The liquid chromatography - tandem mass spectrometry (LC-MS/MS) method for assessments of nicotine, cotinine, trans-3'-hydroxy co-tinine, nornicotine, and anabasine was modified from a fully validated method previously reported for biological matrices [20]. In brief, a mixed mode solid phase extraction (SPE) procedure was modified since the matrix of all study samples was PBS. The calibrators were prepared by freshly spiking into PBS. The calibrators and the CCS-exposed samples were simply diluted with acetonitrile containing deuterium-labeled internal standards (IS) before subjecting to instrument analysis. Deuterium-labeled IS were used for all analytes. The analysis was performed on a Quattro Micro triple quadrupole mass spectrometer with 2795 Alliance HTâ LC system, both from Waters. An Atlantis HILIC silica column (100 by 2 mm from Waters) was used in hydrophilic interaction mode. The mass spectrometer was operated in a multiple reaction monitoring (MRM) mode with positive elec-trospray ionization. Two MRM transitions were monitored for each analyte or IS. The lower limit of quantification for all analytes was 1 ng/mL, and the total imprecision (coefficient of variation, CV %) was <10% for the analytical measurement range from 1 ng/mL to 5000 ng/mL.
Scanning spectrophotometry for assessments of CCS exposure
The CCS-exposed PBS/medium without the presence of cells was subjected to the scanning spectrophotometric analysis using the wavelengths from 200 to 800 nm on a spectropho-tometer (SpectraMax M5, Molecular Devices, CA). The OD values of the CCS-exposed PBS/ medium at 200-800 nm wavelengths were measured and recorded. The wavelength that was corresponded to the maximal OD was identified.
Cell injury and viability assessments
Plasma membrane damage leads to the leakage of lactate dehydrogenase (LDH) from injured cells. hMVEC in the wells of a 96-well plate were exposed to CCS. The exposed cells were rinsed and incubated in Hank's Buffered Salt Solution (HBSS) for 3 hr. Then, the levels of LDH activity in the conditioned HBSS were assessed using the Cytotoxicity Detection kit (Roche). The OD values at 490 nm wavelength were subtracted by the values at the reference wavelength (600 nm). In a separate set of CCS-exposed cells, cell proliferation was measured using the Cell Proliferation Reagent, WST-1 (Roche). Mitochondrial dehydrogenase in viable cells cleaves WST-1, a water-soluble tetrazolium salts, and causes colorimetric change. The amount of the cleaved formazan dye is directly proportional to the metabolically active cells. hMVEC were exposed to CCS and then incubated in HBSS containing WST-1. The OD values of conditioned HBSS at 450 nm were measured and subtracted by the values at the reference wavelength (600 nm).
Morphological examination of the endothelial monolayer
After hMVEC were exposed to 0-4 EU of CCS, the cell morphology was examined under a phase contrast microscope as described earlier [21]. Microphotographs were taken using a digital camera attached to the microscope.
Statistical analysis
Significance for the effects of CCS on nicotine/ cotinine levels, absorbance at λ290, LDH release, and cell viability were determined by analysis of standard deviation and t-Test/ANOVA using the data analysis tools of Microsoft Excel [22].
Results
CCS exposure leads to increased nicotine levels
To closely mimic exhaled tobacco smoke, mainstream cigarette smoke is passed through PBS as described in the section of Materials and Methods. Nicotine is an essential element of tobacco smoke and is widely used as a marker to monitor smoke exposure. First, we determine whether CCS exposure leads to the elevation of nicotine levels. The accumulative level of nicotine in the buffer that is exposed to conditioned smoke derived from 4 cigarettes or 1 EU, is 800 ng/mL under our experimental conditions (Figure 2A). This implies that passive inhalation of exhaled smoke could lead to increased nicotine levels in the lung. Next, we examine the effects of prolonged and interrupted exposure to CCS on nicotine accumulation. The levels of exhaled smoke in the environment over time are not constant. To mimic this, a modular exposure protocol is used. Exposure to conditioned smoke derived from 4-16 cigarettes over 1-4 hrs, i.e. 1-4 EU or exposure cycles, increases the nicotine levels from 800 to 5000 ng/mL in the exposed buffer; whereas the nicotine level was not detectable in the medium exposed to 0 EU (Fig 2A). Finally, the levels of cotinine, the metabolic products of nicotine, are assessed. To our surprise, CCS increases the cotinine levels from 4.5 to 25 ng/mL in CCS-exposed buffers (Figure 2B). The cotinine level in the control PBS (0 EU) is 0.15 ng/mL. The source of this cotinine is unclear since PBS without the presence of cells used for conditioning and exposure is unlikely to catalyze the conversion of nicotine to cotinine. It remains to be determined whether the mainstream smoke contains cotinine derived from burning cigarettes. Nevertheless, the presence of cotinine in CCS indicates that cotinine may be used as a marker to monitor exhaled smoke. The levels of trans-3'-hydroxy cotinine, nornico-tine, and anabasine in CCS-exposed PBS are not significantly changed, compared to controls (data not shown).
Figure 2.
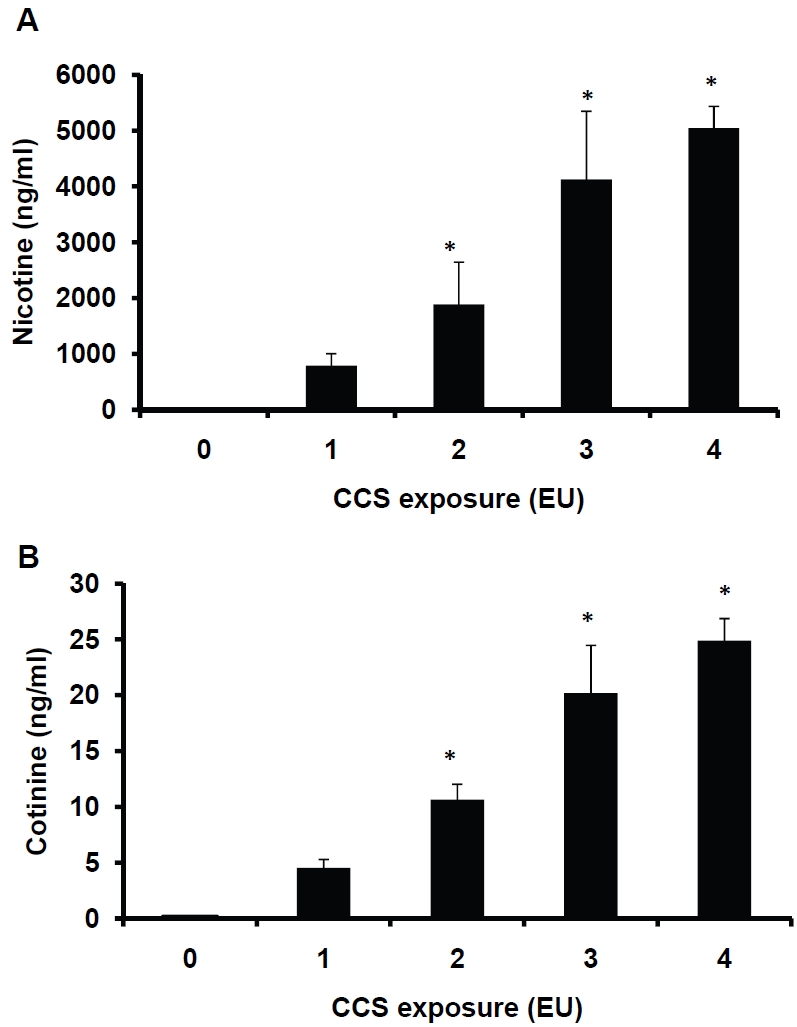
Assessments of nicotine and cotinine levels in CCS-exposed PBS. Two milliliters of PBS in each 35-mm dish were exposed to air (0 EU), or 1-4 EU (4-16 cigarettes) of CCS as described in the Materials and Methods. The CCS-exposed PBS was collected and subjected to LC-MS/MS analysis. The levels of nicotine (Fig 2A) and cotinine (Fig 2B) were determined. Data represent the means ± SD of triplicates. *:P<0.05vs. 1EU, n=3.
Monitoring CCS exposure
To determine whether exposure to CCS alters chemical characteristics of the exposed PBS and EGM-2, the scanning spectrophotometric analysis using the wavelengths from 200 to 800 nm was performed. PBS or EGM-2 exposed to 0 EU was used as a blank or a reference for the analysis, hence the OD value of 0 EU-exposed PBS or EGM-2 was 0. The pattern of the absorbance (spectrogram) between λ200nm to λ800nm is similar among the samples exposed to 1-4 EU of CCS (Figure 3). All spectrograms show a CCS exposure-associated peak at λ290nm (Figure 3). To determine whether CCS exposure increases the OD values at 290 nm (OD290nm) in a dose-dependent manner, 1-4 EU CCS-exposed PBS were assessed for the values of OD290nm. CCS exposure induces dose-dependent increases in the OD values at the absorbance peak (Figure 4A). In addition, the OD290nm values are correlated to the levels of nicotine in the CCS-exposed buffer (R2=0.9885, Figure 4B). The chemicals that are responsible for the absorbance peak at λ290nm are unclear. However, the assessment of OD290nm can be used to monitor CCS exposure.
Figure 3.

The scanning spectrophotometric analysis. PBS/EBM-2 (2 mL) in dishes (35-mm) was exposed to air (0 EU), or 1-4 EU (4-16 cigarettes) of CCS as described in the Materials and Methods. The absorption spectrum of the CCS-exposed sample was determined on a scanning spectrophotometer using the wavelengths from 200 to 800 nm. PBS or EGM-2 exposed to 0 EU was used as a blank or a reference for the scanning analysis to eliminate the background absorbance. The representatives of scanning spectrum of 1-4 EU-exposed PBS and 4 EU-exposed EGM-2 are shown, indicating an absorption peak at λ290 nm (Figure 3).
Figure 4.
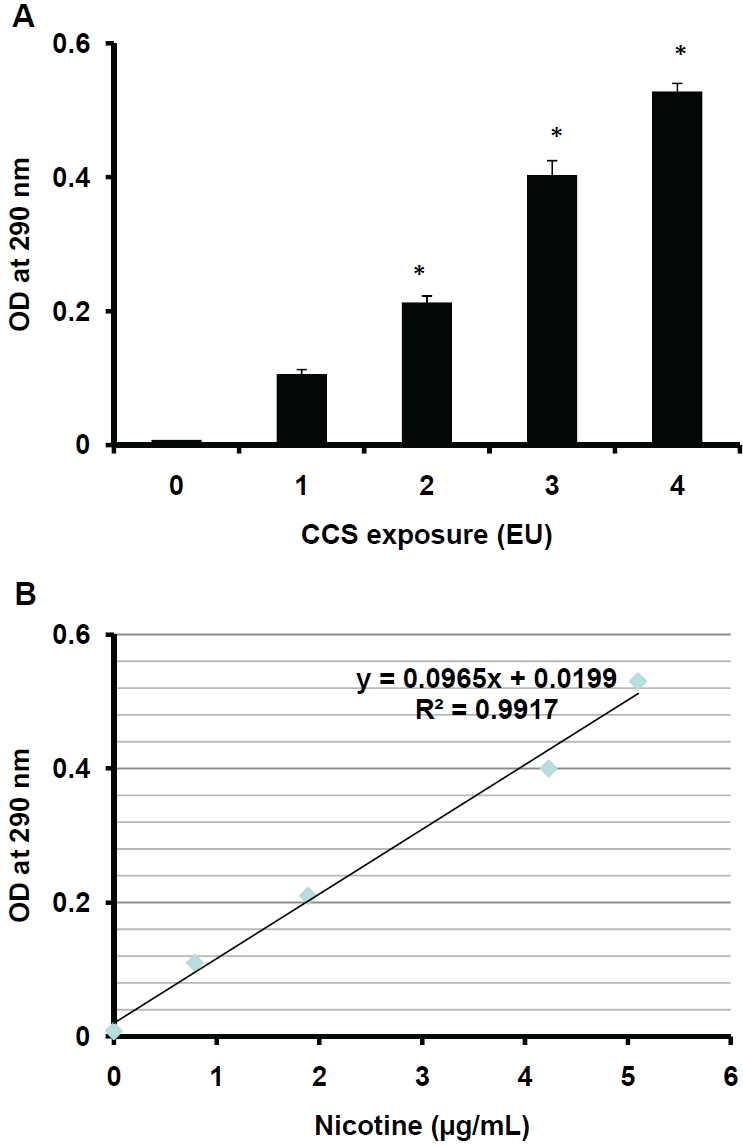
The correlation of OD290nm and nicotine levels. PBS (2 mL) in dishes (35-mm) was exposed to air (0 EU), or 1-4 EU (4-16 cigarettes) of CCS as described in the Materials and Methods. The OD290nm values of the exposed PBS was assessed (Fig 4A). The correlation of OD290nm values and nicotine levels in CCS-exposed PBS is shown (Fig 4B). Data represent the means ± SD of triplicates. *: P<0.05 vs. 1 EU, n=3.
CCS causes cell injury and impacts cell viability
To examine the effects of CCS on cell injury, LDH levels in the medium are measured. The relative levels of LDH in the medium containing the cells exposed to 3-4 EU of CCS are significantly higher than those in the medium containing control cells (0 EU), indicating CCS-induced cell injury (Figure 5A). Furthermore, the effects of CCS on cell viability are assessed. The relative level of WST-1 in the cells exposed to 1 EU of smoke is higher than that in the control cells (0 EU, Figure 5B). This suggests that exposure to low levels of CCS stimulates cell growth. The relative levels of WST-1 in the cells exposed to 2 -4 EU of CCS are significantly decreased in a dose-dependent manner, indicating that exposure of hMVEC to high levels of CCS decreases cell viability. Release of LDH and decreases in WST-1 levels indicate CCS-induced cell injury and death.
Figure 5.
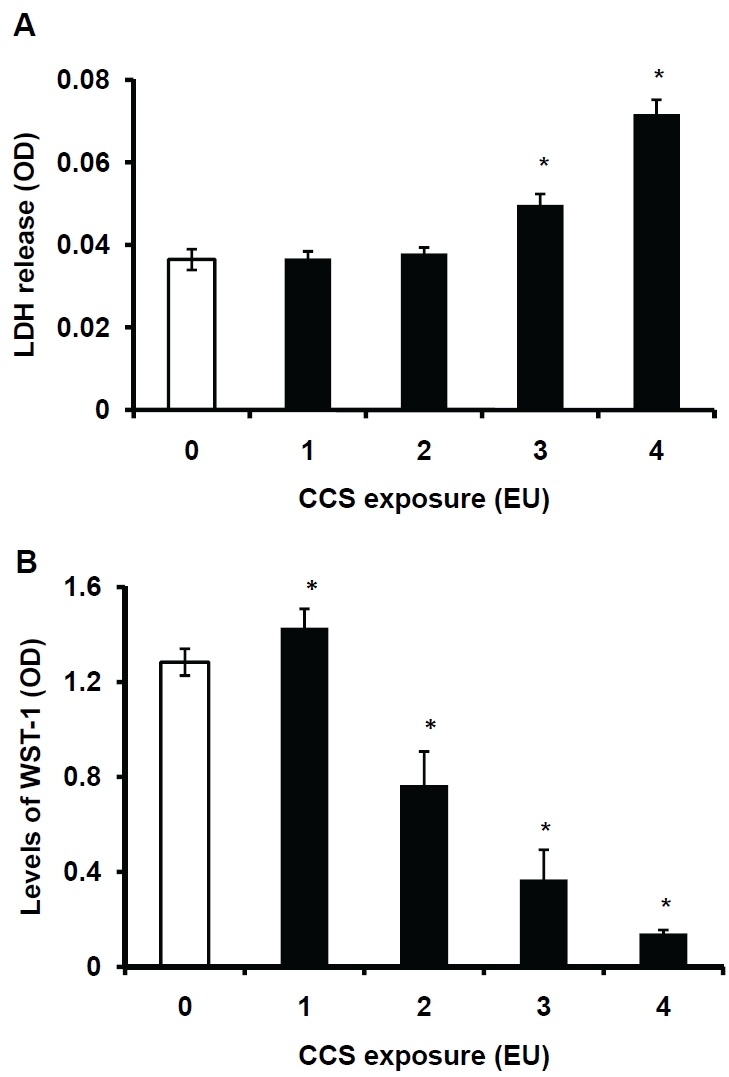
Assessments of CCS-induced LDH release and cell viability. hMVEC were exposed to air (0 EU), or 1-4 EU (4-16 cigarettes) of CCS as described in the Materials and Methods. The exposed cells were rinsed and incubated in HBSS. The levels of LDH activity in the conditioned HBSS were assessed using the rapid enzymatic assay kit (Fig 5A). Some treated cells were subjected to the viability assay using the WST-1 cell proliferation kit (Figure 5B). Data represent the means ± SD. *: P<0.05 vs. 0 EU, n=4.
Conditioned smoke alters cell morphology
hMVEC are exposed to CCS and examined under a microscope. The morphology of 1-2 EU-exposed hMVEC is comparable to that of 0 EU-exposed cells (Figure 6). Exposure of hMVEC to 3-4 EU of CCS decreases the number of attached cells on the dish, indicating the CCS-induced detachment of cells from the dish. The remaining cells on the dish show irregular shapes. This observation indicates that conditioned smoke can cause endothelial cell detachment and death.
Figure 6.

Morphology of CCS-exposed cells. hMVEC were exposed to air (0 EU), or 1-4 EU (4-16 cigarettes) of CCS as described in the Materials and Methods. Cell morphology was examined under a phase contrast microscope (Figure 6).
Discussion
An exposure protocol has been established to closely mimic the effects of exhaled mainstream smoke. Plasma nicotine/cotinine levels in the subjects exposed to tobacco smoke range from ng/mL to μg/mL. The levels of exhaled smoke exposure can be affected by many factors such as lung fluid, the number of cigarettes smoked, and the length of exposure. We attempted to quantify the effects of exhaled smoke exposure by setting up an exposure system. Mainstream smoke generated from burning cigarettes at a rate of 4 min per cigarette was passed through 10 cm distance in 1,000 mL of fresh PBS. Exposure of PBS (2 mL) in a 35-mm dish to CCS for 4 cigarettes or 1 EU leads to elevation of nicotine to about 0.8 μg/mL, or 0.2 μg/mL per cigarette. This is equivalent to the mean values of plasma nicotine/cotinine levels in individuals smoking 20 cigarettes per day [12-13]. The capillary region of the lungs is in immediate contact with smoke. Therefore, nicotine levels in the lung microvessels may be higher than those in the peripheral vessels. Exposure of PBS to conditioned smoke for 4 EU increases the nicotine level to 5µg/mL. This may mimic the conditions in the lung alveoli when exhaled smoke is inhaled. The nicotine levels in the exposed buffer are accumulative. It seems that nicotine levels in PBS are relatively stable during CCS exposure. Although this exposure protocol can be used to mimic exhaled tobacco smoke in a controllable way, several limitations should be considered. First, the lung fluid contains biological substances such as proteins, which may absorb or react with exhaled smoke in a different way than PBS does. Hence, the nicotine levels in the system may be different from passive smokers’ lungs. Second, smoke aging in the air and lungs often alters the levels and compositions of exhaled smoke. The in vitro system mimics a specific condition. Finally, lung airways are far more complex than our exposure system. It is expected that the lungs may metabolite more smoke ingredients including nicotine than PBS. Therefore, the nicotine levels estimated in this system may be higher than those in the lungs, when they are exposed to the similar amounts of exhaled smoke.
Although the identity of OD290 product remains unknown, OD290values correlate with nicotine levels in CCS-exposed buffer/medium. Therefore, the value of OD at 290 nm wavelength can be used as a marker to monitor conditioned smoke exposure. Nicotine and its derivatives are used to assess mainstream and sidestream tobacco smoke exposure. Our results suggest that the assessment of OD290 and/or the level of nicotine may help determine the degree of lung cell injury caused by conditioned smoke exposure. The spectrum of smoke-exposed medium (EGM-2) shows a similar pattern and reveals absorbance peak at 290 nm. OD290 of EGM-2 is used to assess the relative levels of nicotine in the medium to monitor CCS exposure. Tobacco smoke consists of more than five thousand substances. It is unclear which ingredient of CCS causes the absorbance at 290 nm. However, the levels of 290 nm absorbed sub-stance(s) increased in PBS or medium are proportional to the levels of nicotine.
Exposure to CCS increases LDH release and decreases cell viability. Exposure of hMVEC to 3-4 EU of CCS leads to a LDH leak from the cells, indicating smoke-induced injury. CCS increases cell growth at a low level (1 EU) and decreases cell viability at higher levels (2-4 EU). Furthermore, morphological examination supports the notion that conditioned smoke can cause cell detachment/death. Exhaled tobacco smoke is thought to be less cytotoxic since it is filtered by the lungs of smokers. However, our current studies demonstrate that exposure of cultured lung vascular endothelial cells to CCS under conditions close to in vivo exposure to exhaled tobacco smoke results in cell injury and death. This is in agreement with the observations that exposure of infant monkey lungs to a component of sidestream tobacco smoke leads to activation of caspases, cleavage of PARP, and increased numbers of apoptotic cells [23]. In addition, mainstream smoke has been shown to enhance cell death in rat lungs [24]. Different constituents of tobacco smoke such as the gas phase, extracts, and condensates have been reported to induce endothelial cell death [25-28].
Conclusions
Our present studies demonstrate an exposure protocol/system to closely mimic in vivo exposure to exhaled tobacco smoke, the key component of secondhand smoke. In addition, exhaled smoke can be cytotoxic to lung cells based on observations using this cell model. Compromise of pulmonary vascular endothelial function represents a critical component of the patho-physiology in tobacco smoke-related lung diseases including emphysema [29-30]. Conditioned smoke-mediated loss of lung endothelial cells supports a causal link between secondhand smoke exposure and pathophysiological changes reported in lung diseases.
Acknowledgments
This work was supported in part by a Career Investigator Award from the American Lung Association of Florida, Inc (JZ), by a Clinical Innovator Award from the Flight Attendant Medical Research Institute (JZ), by the Medical Research Service of the Department of Veterans Affairs (JMP) and by NIH grants HL68666 and HL85133(JMP).
References
- 1. Agency, California Environmental Protection. 2005. Proposed Identification of Environmental Tobacco Smoke as a Toxic Air Contaminant: Executive Summary.
- 2.Eisner MD, Balmes J, Yelin EH, Katz PP, Hammond SK, Benowitz N, Blanc PD. Directly measured secondhand smoke exposure and COPD health outcomes. BMC Pulm Med. 2006;6:12. doi: 10.1186/1471-2466-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hedley AJ, Lam TH, McGhee SM, Leung GM, Pow M. Passive smoking: secondhand smoke does cause respiratory disease. Bmj. 2003;327:502. doi: 10.1136/bmj.327.7413.502-a. author reply 504-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jindal SK, Aggarwal AN, Chaudhry K, Chhabra SK, D'Souza GA, Gupta D, Katiyar SK, Kumar R, Shah B, Vijayan VK. A multicentric study on epidemiology of chronic obstructive pulmonary disease and its relationship with tobacco smoking and environmental tobacco smoke exposure. Indian J Chest Dis Allied Sci. 2006;48:23–29. [PubMed] [Google Scholar]
- 5.Eisner MD, Balmes J, Katz PP, Trupin L, Yelin EH, Blanc PD. Lifetime environmental tobacco smoke exposure and the risk of chronic obstructive pulmonary disease. Environ Health. 2005;4:7. doi: 10.1186/1476-069X-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nurminen MM, Jaakkola MS. Mortality from occupational exposure to environmental tobacco smoke in Finland. J Occup Environ Med. 2001;43:687–693. doi: 10.1097/00043764-200108000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Hoshino Y, Mio T, Nagai S, Miki H, Ito I, Izumi T. Cytotoxic effects of cigarette smoke extract on an alveolar type II cell-derived cell line. Am J Physiol Lung Cell Mol Physiol. 2001;281:L509–516. doi: 10.1152/ajplung.2001.281.2.L509. [DOI] [PubMed] [Google Scholar]
- 8.Wickenden JA, Clarke MC, Rossi AG, Rahman I, Faux SP, Donaldson K, MacNee W. Cigarette smoke prevents apoptosis through inhibition of caspase activation and induces necrosis. Am J Respir Cell Mol Biol. 2003;29:562–570. doi: 10.1165/rcmb.2002-0235OC. [DOI] [PubMed] [Google Scholar]
- 9.Petrache I, Fijalkowska I, Medler TR, Skirball J, Cruz P, Zhen L, Petrache HI, Flotte TR, Tuder RM. alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol. 2006;169:1155–1166. doi: 10.2353/ajpath.2006.060058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res. 2006;7:53. doi: 10.1186/1465-9921-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuder RM, Zhen L, Cho CY, Taraseviciene-Stewart L, Kasahara Y, Salvemini D, Voelkel NF, Flores SC. Oxidative Stress and Apoptosis Interact and Cause Emphysema Due to Vegf Receptor Blockade. Am J Respir Cell Mol Biol. 2003;29:88–97. doi: 10.1165/rcmb.2002-0228OC. [DOI] [PubMed] [Google Scholar]
- 12.Russell MA, Jarvis M, Iyer R, Feyerabend C. Relation of nicotine yield of cigarettes to blood nicotine concentrations in smokers. Br Med J. 1980;280:972–976. doi: 10.1136/bmj.280.6219.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choudhury B. Studies on the Blood Nicotine Level .in the Oral and Oropharyngeal Cancers of Habitual Tobacco Users. IJO & HNS. 1998;50:230–232. doi: 10.1007/BF03006997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benowitz NL, Hukkanen J, Jacob P., 3rd Nicotine chemistry, metabolism, kinetics and bio-markers. Handb Exp Pharmacol. 2009:29–60. doi: 10.1007/978-3-540-69248-5_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muscat JE, Stellman SD, Caraballo RS, Richie JP., Jr Time to first cigarette after waking predicts cotinine levels. Cancer Epidemiol Bio-markers Prev. 2009;18:3415–3420. doi: 10.1158/1055-9965.EPI-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Patel JM, Block ER. Enhanced apoptosis in prolonged cultures of senescent porcine pulmonary artery endothelial cells. Mech Ageing Dev. 2002;123:613–625. doi: 10.1016/s0047-6374(01)00412-2. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Xia SL, Block ER, Patel JM. NO upregulation of a cyclic nucleotide-gated channel contributes to calcium elevation in endothelial cells. Am J Physiol Cell Physiol. 2002;283:C1080–1089. doi: 10.1152/ajpcell.00048.2002. [DOI] [PubMed] [Google Scholar]
- 18.Griscom NT, Wohl ME. Dimensions of the growing trachea related to age and gender. AJR Am J Roentgenol. 1986;146:233–237. doi: 10.2214/ajr.146.2.233. [DOI] [PubMed] [Google Scholar]
- 19.Widdicombe J. Airway and alveolar permeability and surface liquid thickness: theory. J Appl Physiol. 1997;82:3–12. doi: 10.1152/jappl.1997.82.1.3. [DOI] [PubMed] [Google Scholar]
- 20.Yue B, Kushnir MM, Urry FM, Rockwood AL. Quantitation of nicotine, its metabolites, and other related alkaloids in urine, serum, and plasma using LC-MS-MS. Methods Mol Biol. 2010;603:389–398. doi: 10.1007/978-1-60761-459-3_38. [DOI] [PubMed] [Google Scholar]
- 21.Patel JM, Zhang J, Block ER. Nitric oxide-induced inhibition of lung endothelial cell nitric oxide synthase via interaction with allosteric thiols: role of thioredoxin in regulation of catalytic activity. Am J Respir Cell Mol Biol. 1996;15:410–419. doi: 10.1165/ajrcmb.15.3.8810647. [DOI] [PubMed] [Google Scholar]
- 22.Winer BJ. New York: McGrawHill; 1971. Statistical Principles in Experimental Design. [Google Scholar]
- 23.Zhong CY, Zhou YM, Joad JP, Pinkerton KE. Environmental tobacco smoke suppresses nuclear factor-kappaB signaling to increase apoptosis in infant monkey lungs. Am J Respir Crit Care Med. 2006;174:428–436. doi: 10.1164/rccm.200503-509OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D'Agostini F, Balansky RM, Izzotti A, Lubet RA, Kelloff GJ, De Flora S. Modulation of apoptosis by cigarette smoke and cancer chemopre-ventive agents in the respiratory tract of rats. Carcinogenesis. 2001;22:375–380. doi: 10.1093/carcin/22.3.375. [DOI] [PubMed] [Google Scholar]
- 25.Yang YM, Liu GT. Damaging effect of cigarette smoke extract on primary cultured human umbilical vein endothelial cells and its mechanism. Biomed Environ Sci. 2004;17:121–134. [PubMed] [Google Scholar]
- 26.Tuder RM, Wood K, Taraseviciene L, Flores SC, Voekel NF. Cigarette smoke extract decreases the expression of vascular endothelial growth factor by cultured cells and triggers apoptosis of pulmonary endothelial cells. Chest. 2000;117:241S–242S. doi: 10.1378/chest.117.5_suppl_1.241s. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Wilcken DE, Wang XL. Cigarette smoke activates caspase-3 to induce apoptosis of human umbilical venous endothelial cells. Mol Genet Metab. 2001;72:82–88. doi: 10.1006/mgme.2000.3115. [DOI] [PubMed] [Google Scholar]
- 28.Pouli AE, Hatzinikolaou DG, Piperi C, Stavridou A, Psallidopoulos MC, Stavrides JC. The cytotoxic effect of volatile organic compounds of the gas phase of cigarette smoke on lung epithelial cells. Free Radic Biol Med. 2003;34:345–355. doi: 10.1016/s0891-5849(02)01289-3. [DOI] [PubMed] [Google Scholar]
- 29.Finkelstein R, Fraser RS, Ghezzo H, Cosio MG. Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med. 1995;152:1666–1672. doi: 10.1164/ajrccm.152.5.7582312. [DOI] [PubMed] [Google Scholar]
- 30.Niewoehner DE. Cigarette smoking, lung inflammation, and the development of emphysema. J Lab Clin Med. 1988;111:15–27. [PubMed] [Google Scholar]