

Abstract
Most human tumors result from the accumulation of multiple genetic and epigenetic alterations in a single cell. Mutations that confer a fitness advantage to the cell are known as driver mutations and are causally related to tumorigenesis. Other mutations, however, do not change the phenotype of the cell or even decrease cellular fitness. While much experimental effort is being devoted to the identification of the functional effects of individual mutations, mathematical modeling of tumor progression generally considers constant fitness increments as mutations are accumulated. In this paper we study a mathematical model of tumor progression with random fitness increments. We analyze a multi-type branching process in which cells accumulate mutations whose fitness effects are chosen from a distribution. We determine the effect of the fitness distribution on the growth kinetics of the tumor. This work contributes to a quantitative understanding of the accumulation of mutations leading to cancer.
Keywords: cancer evolution, branching process, fitness distribution, beneficial fitness effects, mutational landscape
1 Introduction
Tumors result from an evolutionary process occurring within a tissue (Nowell, 1976). From an evolutionary point of view, tumors can be considered as collections of cells that accumulate genetic and epigenetic alterations. The phenotypic changes that these alterations confer to cells are subjected to the selection pressures within the tissue and lead to adaptations such as the evolution of more aggressive cell types, the emergence of resistance, induction of angiogenesis, evasion of the immune system, and colonization of distant organs with metastatic growth. Advantageous heritable alterations can cause a rapid expansion of the cell clone harboring such changes, since these cells are capable of outcompeting cells that have not evolved similar adaptations. The investigation of the dynamics of cell growth, the speed of accumulating mutations, and the distribution of different cell types at various timepoints during tumorigenesis is important for an understanding of the natural history of tumors. Further, such knowledge aids in the prognosis of newly diagnosed tumors, since the presence of cell clones with aggressive phenotypes lead to less optimistic predictions for tumor progression. Finally, a knowledge of the composition of tumors allows for the choice of optimum therapeutic interventions, as tumors harboring pre-existing resistant clones should be treated differently than drug-sensitive cell populations.
Mathematical models have led to many important insights into the dynamics of tumor progression and the evolution of resistance (Goldie and Coldman, 1983 and 1984; Bodmer and Tomlinson, 1995; Coldman and Murray, 2000; Knudson, 2001; Maley and Forrest, 2001; Michor et al., 2004; Iwasa et al., 2005; Komarova and Wodarz, 2005; Michor et al., 2006; Michor and Iwasa, 2006; Frank 2007; Wodarz and Komarova, 2007; Schweinsberg, 2008; Durrett, Schmidt, and Schweinsberg, 2009). These mathematical models generally fall into one of two classes: (i) constant population size models, and (ii) models describing exponentially growing populations. Many theoretical investigations of exponentially growing populations employ multi-type branching process models (e.g., Iwasa et al., 2006; Haeno et al., 2007; Durrett and Moseley, 2009), while others use population genetic models for homogeneously mixing exponentially growing populations (e.g., Beerenwinkel et al., 2007; Durrett and Mayberry, 2010). In this paper, we focus on branching process models. In these models, cells with i ≥ 0 mutations are denoted as type-i cells, and Zi(t) specifies the number of type-i cells at time t. Type-i cells die at rate bi, give birth to one new type-i cell at rate ai, and give birth to one new type-(i + 1) cell at rate ui+1. Some authors (e.g., Haeno et al., 2007) consider an alternate version of our model in which mutations occur with probability μi+1 during birth events which occur at rate αi, but the two versions are equivalent provided ui+1 = αiμi+1 and ai = αi (1 − μi+1). This relationship between the parameters must be kept in mind when comparing results across papers.
One biologically unrealistic aspect of this model as presented in the literature is that all type-i cells are assumed to have the same birth and death rates. This assumption describes situations during tumorigenesis in which the order of mutations is predetermined, i.e. the genetic changes can only be accumulated in a particular sequence and all other combinations of mutations lead to lethality. Furthermore, in this interpretation of the model, there cannot be any variability in phenotype among cells with the same number of mutations. In many situations arising in biology there is marked heterogeneity in phenotype even if genetically, the cells are identical (Elowitz et al., 2002; Becskei et al., 2005; Kaern et al., 2005; Feinerman et al., 2008). This variability may be driven by stochasticity in gene expression or in post-transcriptional or post-translational modifications. In this paper, we modify the branching process model so that mutations alter cell birth rates by a random amount.
An important consideration for this endeavor is the choice of the mutational fitness distribution. The exponential distribution has become the preferred candidate in theoretical studies of the genetics of adaptation. The first theoretical justification of this choice was given by Gillespie (1983, 1984), who argued that if the number of possible alleles is large and the current allele is close to the top of the rank ordering in fitness values, then extreme value theory should provide insight into the distribution of the fitness values of mutations. For many distributions including the normal, Gamma, and lognormal distributions, the maximum of n independent draws, when properly scaled, converges to the Gumbel or double exponential distribution, Λ(x) = exp(−e−x). In the biological literature, it is generally noted that this class of distributions only excludes exotic distributions like the Cauchy distribution, which has no moments. However, in reality, it eliminates all distributions with P(X > x) ~ Cx−α. For distributions in the domain of attraction of the Gumbel distribution, and if Y1 > Y2 ··· >Yk are the k largest observations in a sample of size n, then there is a sequence of constants bn so that the spacings Zi = i(Yi−Yi+1)/bn converge to independent exponentials with mean 1, see e.g., Weissman (1978). Following up on Gillespie’s work, Orr (2003) added the observation that in this setting, the distribution of the fitness increases due to beneficial mutations has the same distribution as Z1 independent of the rank i of the wild type cell.
To infer the distribution of fitness effects of newly emerged beneficial mutations, several experimental studies were performed; for examples, see Imhoff and Schlotterer (2001), Sanjuan et al. (2004), and Kassen and Bataillon (2006). The data from these experiments is generally consistent with an exponential distribution of fitness effects. However, there is an experimental caveat that cannot be neglected (Rozen et al., 2002): if only those mutations are considered that reach 100% frequency in the population, then the exponential distribution is multiplied by the fixation probability. By this operation, a distribution with a mode at a positive value develops. In a study of a quasi-empirical model of RNA evolution in which fitness was based on secondary structures, Cowperthwaite et al. (2005) found that fitnesses of randomly selected genotypes appeared to follow a Gumbel-type distribution. They also discovered that the fitness distribution of beneficial mutations appeared exponential only when the vast majority of small-effect mutations were ignored. Furthermore, it was determined that the distribution of beneficial mutations depends on the fitness of the parental genotype (Cowperthwaite et al., 2005; MacLean and Buckling, 2009). However, since the exceptions to this conclusion arise when the fitness of the wild type cell is low, these findings do not contradict the picture based on extreme value theory.
In contrast to the evidence above, recent work of Rokyta et al. (2008) has shown that in two sets of beneficial mutations arising in the bacteriophage ID11 and in the phage φ6 – for which the mutations were identified by sequencing – beneficial fitness effects are not exponential. Using a statistical method developed by Biesal et al. (2007), they tested the null hypothesis that the fitness distribution has an exponential tail. They found that the null hypothesis could be rejected in favor of a distribution with a right truncated tail. Their data also violated the common assumption that small-effect mutations greatly outnumber those of large effect, as they were consistent with a uniform distribution of beneficial effects. A possible explanation for the bounded fitness distribution may be found in the culture conditions utilized in the experiments: they evolved ID11 on E. coli at an elevated temperature (37° C instead of 33° C). There may be a limited number of mutations that will enable ID11 to survive in increased temperatures. The latter situation may be similar to scenarios arising during tumorigenesis, where, in order to develop resistance to a drug or to progress to a more aggressive stage, the conformation of a particular protein must be changed or a certain regulatory network must be disrupted. If there is a finite, but large, number of possible beneficial mutations, then it is convenient to use a continuous distribution as an approximation.
In this paper, we consider both bounded distributions and unbounded distributions for the fitness advance and derive asymptotic results for the number of type-k individuals at time t. We determine the effects of the fitness distribution on the growth kinetics of the population, and investigate the rates of expansion for both bounded and unbounded fitness distributions. This model provides a framework to investigate the accumulation of mutations with random fitness effects.
The remainder of this section is dedicated to statements and discussion of our main results. Proofs of these results can be found in Sections 2–5.
1.1 Bounded distributions
The model we consider is a multi-type branching process in which type-i cells have accumulated i ≥ 0 advantageous mutations. All cells in the population die at rate b0. The initial population consists entirely of type-0 cells that give birth at rate a0 > b0 to new type-0 cells and produce type-1 cells at rate u1. We assume that the population of type-0 cells starts at a sufficiently large population V0 so that we can approximate its size by Z0(t) = V0eλ0t, where λ0 = a0 − b0. When a type-0 cell produces a type-1 cell, the new cell gives birth to type-1 cells at rate a0 + x, where x ≥ 0 is drawn according to a probability distribution ν and produces type-2 cells at rate u2. In general, a type-k cell with birth rate a produces a new type-(k + 1) cell at rate uk and the new type-(k + 1) cell assumes an increased birth rate a + x where x ≥ 0 is drawn according to ν. We let Zk(t) denote the total number of type-k cells in the population at time t. When we refer to the kth generation of mutants, we mean the set of all type-k cells.
We begin by considering situations in which the distribution of the increase in the birth rate is concentrated on [0, b]. In particular, suppose that ν has density g with support in [0, b] and assume that g satisfies:
| (*) |
Our first result describes the mean number of first generation mutants at time t, EZ1(t).
Theorem 1
If (*) holds, then
where a(t) ~ b(t) means a(t)/b(t) → 1.
The next result shows that the actual growth rate of type-1 cells is slower than the mean. Here, and in what follows, we use ⇒ to indicate convergence in distribution.
Theorem 2
If (*) holds and p = b/λ0, then for θ ≥ 0,
| (1.1) |
where c1(λ0, b) is a constant whose value will be given in (3.8). In particular, we have
where V1 has Laplace transform given by the righthand side of (1.1).
Theorem 2 is similar to Theorem 3 in Durrett and Moseley (2009) which assumes a deterministic fitness distribution so that all type-1 cells have growth rate λ1 = λ0 + b. There, the asymptotic growth rate of the first generation is exp(λ1t). In contrast, the continuous fitness distribution we consider here has the effect of slowing down the growth rate of the first generation by the polynomial factor t1+p. To explain this difference, we note that the calculation of the mean given in Section 3 shows that the dominant contribution to Z1(t) comes from growth rates x = b − O(1/t). However, mutations with this growth rate are unlikely until the number of type-0 cells is O(t), i.e., roughly at time r1 = (1/λ0) log t. Thus at time t, the number of type-1 cells will be roughly exp((λ0+b)(t − r1)) = exp((λ0 +b)t)/t1+p.
To prove Theorem 2, we look at mutations as a point process in [0, t] × [0, b]: there is a point at (s, x) if there was a mutant with birth rate a0 + x at time s. This allows us to derive the following explicit expression for the Laplace transform of Z1(t):
where and is a continuous-time branching process with birth rate a0+x, death rate b0, and initial population . In Figure 1, we compare the exact Laplace transform of t1+p exp(−(λ0+b)t)Z1(t) with the results of simulations and the limiting Laplace transform from Theorem 2, illustrating the convergence as t → ∞.
Figure 1.

Plot of the exact Laplace transform (LT) for t(1+p) e−(λ0+b)t Z1 (t) at times t = 60, 80,100,120, the approximations from Monte Carlo (MC) simulations at the corresponding times, and the asymptotic Laplace transform from Theorem 2. Parameter values: a0 = 0.2, b0 = 0.1, b = 0.01, and u1 = 10−3. g is uniform on [0, .01].
Notice that the Laplace transform of V1 has the form exp(C θα) where α = λ0/(λ0 + b) which implies that P(V1 > v) ~ v−α as v → ∞ (see, for example, the argument in Section 3 of Durrett and Moseley (2009)). To gain some insight into how this limit comes about, we give a second proof of the convergence that tells us the limit is the sum of points in a nonhomogeneous Poisson process. Each point in the limiting process represents the contribution of a different mutant lineage to Z1(t). More precisely, we define a three dimensional point process 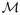 (t) on [0, t] × [0, b] × (0, ∞) by the following rule: there is a point at (s, x, v) if there was a type-1 mutant with birth rate a0 + x at time s and the number of its type-1 descendants at time t,
, has
as t → ∞ with v > 0. We define F : [0, ∞)3 → [0, ∞) by
(t) on [0, t] × [0, b] × (0, ∞) by the following rule: there is a point at (s, x, v) if there was a type-1 mutant with birth rate a0 + x at time s and the number of its type-1 descendants at time t,
, has
as t → ∞ with v > 0. We define F : [0, ∞)3 → [0, ∞) by
i.e. F maps a point in (t) onto its contribution to V1 = limt→∞ t1+pe−(λ0+b)tZ1(t).
Theorem 3
As t → ∞, F((t)) ⇒ Λ where Λ is a Poisson process on (0, ∞) with mean measure μ(z, ∞) = A1(λ0, b)u1V0z−λ0/(λ0+b) and A1(λ0, b) is a constant whose value is given in (3.9). In particular, V1 = limt→∞ t1+pe−(λ0+b)tZ1(t) is the sum of the points in Λ.
A similar result can be obtained for deterministic fitness distributions, see the Corollary to Theorem 3 in Durrett and Moseley (2009). However, the new result shows that the point process limit is not an artifact of assuming that all first generation mutants have the same growth rate. Even when the fitness advances are random, different mutant lines contribute to the limit. This result is consistent with observations of Maley et al. (2006) and Shah et al. (2009) that tumors contain cells with different mutational haplotypes. Theorem 3 also gives quantitative predictions about the relative contribution of different mutations to the total population. These implications will be explored further in a follow-up paper currently in progress.
With the behavior of the type-1 individuals analyzed, we are ready to proceed to the study of type-k individuals. The computation of the mean is straightforward.
Theorem 4
If (*) holds, then
As in the k = 1 case, the mean involves a polynomial correction to the exponential growth and again, does not give the correct growth rate for the number of type-k cells. To state the correct limit theorem describing the growth rate of Zk(t), we will define pk and u1,k by
for all k ≥ 1.
Theorem 5
If (*) holds, then for θ ≥ 0
| (1.2) |
where ck(λ0, b) is a constant whose value will be given in (4.9). In particular, we have
where Vk has Laplace transform given by the righthand side of (1.2).
If we let
be the number of type-k descendants at time t of the 1 mutant at (s, x, v) ∈ (t) where (t) is the three dimensional point process described in the paragraph preceding Theorem 3, then
is the same as a process in which the initial type (here type-1 cells) behaves like ve(λ0 + x)(t − s) instead of Z0(t) = V0eλ0t. Therefore, Theorem 5 can be proved by induction. To explain the form of the result we consider the case k = 2. Breaking things down according to the times and the sizes of the mutational changes, we have
As in the result for Z1(t) the dominant contribution comes from x1, x2 = b − O(1/t) and as in the discussion preceding the statement of Theorem 2, the time of the first mutation to b − O(1/t) is ≈ r1 = (log t)/λ0. The descendants of this mutation grow at exponential rate λ0 + b − O(1/t), so the time of the first mutation to 2b − O(1/t) is ≈ r2 = r1 + (log t)/(λ0 + b). Noticing that
tells us what to guess for the polynomial term: t−(2+p2) where
In Figure 2, we compare the asymptotic Laplace transform from Theorem 5 with the results of simulations in the case k = 2. To explain the slow convergence to the limit, we note that if we take account of the mutation rates u1, u2 in the heuristic from the previous paragraph (which becomes important when u1, u2 are small), then the first time we see a type-1 cell with growth rate b − O(1/t) will not occur until time when the type-0 cells reach O(t/u1) and so the first type-2 cell with growth rate 2b − O(1/t) will not be born until time when the descendants of the type-1 cells with growth rate b − O(1/t) reach size O(t/u2). When u1 = u2 = 10−3, λ0 = .1, and b = .01, r ≈ 223. The mutations created at this point will need some time to grow and become dominant in the population. It would be interesting to compare simulations at time 300, but we have not been able to do this due to the large number of different growth rates in generation 1.
Figure 2.

Plot of the approximations to the Laplace transform of t2+p2e−(λ0+2b)t Z2(t) from Monte Carlo (MC) simulations at times t = 80,100,120 along with the asymptotic Laplace transform from Theorem 5. Parameter values: a0 = 0.2, b0 = 0.1, b = 0.01, and u1 = u2 = 10−3. g is uniform on [0, 0.01].
1.2 Unbounded distributions
In this section, we consider situations in which the fitness distribution is unbounded. We will suppose that the fitness distribution ν has tail
| (1.3) |
as x → ∞ for some α, γ, K > 0, and β ∈ ℝ. Our assumption (1.3) on the tail of ν is satisfied by a number of natural distributions including the gamma(β +1, γ) distribution which has α = 1 (and includes the exponential distribution as the special case β = 0) and the normal distribution which has α = 2, β = −1.
To analyze this situation, we will again take a Poisson process viewpoint and look at the contribution from a mutation at time s with increased growth rate x. A mutation that increases the growth rate by x at time s will, if it does not die out, grow to e(λ0 + x)(t − s) ζ at time t where ζ has an exponential distribution. The growth rate (λ0 + x)(t − s) ≥ z when
Therefore,
where
| (1.4) |
as x → ∞ and
| (1.5) |
The size of this integral can be found by maximizing the exponent φ over s for fixed z. Since
| (1.6) |
and
| (1.7) |
we can see that ∂2 φ/∂s2(s, z) < 0 when αz > λ0(t − s) so that for all z in this range, φ(s, z) is concave as a function of s and achieves its maximum at a unique value sz.
When α = 1, it is easy to set (1.6) to 0 and solve for sz. This in turn leads to an asymptotic formula for μ(z, ∞) and allows us to derive the following limit theorem for Z1(t).
Theorem 6
Suppose α = 1 and let c0 = λ0/4γ. Then t−2 log Z1(t) → c0 and
where y* is the rightmost point in the point process with intensity given by
| (1.8) |
When α ≠ 1, solving for sz becomes more difficult, but we are still able to prove the following limit theorem for Z1(t).
Theorem 7
For any integer α > 1, there exist explicitly calculable constants ck = ck(α, γ), 0 ≤ k < α, and κ = κ(β, α, γ) so that t−(α+1)/α log Z1(t) → c0 and
where y* is the rightmost particle in a point process with explicitly calculable intensity.
The complicated form of the result is due to the fact that the fluctuations are only of order t1/α so we have to be very precise in locating the maximum. The explicit formulas for the constants and the intensity of the point process are given in (5.11) and (5.12). With more work this result could be proved for general α > 1, but we have not tried to do this or prove Conjecture 1 below because the super-exponential growth rates in the unbounded case are too fast to be realistic.
We conclude this section with two comments. First, the proof of Theorem 7 shows that in contrast to the bounded case, in the unbounded case, most type-1 individuals are descendants of a single mutant. Second, the proof shows that the distribution of the mutant with the largest growth rate is born at time s ~ t/(α + 1) (see Remark 1 at the end of Section 5) and has growth rate z = O(t(α+1)/α). The intuition behind this is that since the type-0 cells have growth rate eλ0s and the distribution of the increase in fitness has tail ≈ e−γxα, the largest advance x attained by time t should occur when s = O(t) and satisfy
The growth rate of its family is then (λ0 + x)(t − s) = O(t(α+1)/α).
Since the type-1 cells grow at exponential rate c1t(α+1)/α, if we apply this same reasoning to type-2 mutants, then the largest additional fitness advance x attained by type-2 individuals should satisfy
and the growth rate of its family will be O(t1+1/α+1/α2). Extrapolating from the first two generations, we make the following
Conjecture 1
Let . As t → ∞,
Note that in the case of the exponential distribution, q(k) = k + 1.
The rest of the paper is organized as follows. Sections 2–5 are devoted to proofs of our main results. After some preliminary notation and definitions in Section 2, Theorems 1–3 are proved in Section 3, Theorems 4–5 in Section 4, and Theorems 6–7 in Section 5. We conclude with a discussion of our results in Section 6.
2 Preliminaries
This section contains some preliminary notation and definitions which we will need for the proofs of our main results. We denote by 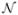 (t) the points in a two-dimensional Poisson process on [0, t] × [0, ∞) with mean measure
(t) the points in a two-dimensional Poisson process on [0, t] × [0, ∞) with mean measure
where in Sections 3–4, ν(dx) = g(x)dx with g satisfying (*) and in Section 5, ν has tail satisfying (1.3). In other words, we have a point at (s, x) if there was a mutant with birth rate a0 + x at time s. Define a collection of independent birth/death branching processes
indexed by (s, x) ∈ (t) with
, individual birth rate a0 + x, and death rate b.
is the contribution of the mutation at (s, x) and
It is well known that
where ζ ~ exp((λ0 + x)/(a0 + x)) (see, for example, equation (1) in Durrett and Moseley (2009)). In several results, we shall make use of the three-dimensional Poisson process (t) on [0, t] × [0, ∞) × (0, ∞) with intensity
In words, (s, x, v) ∈ (t) if there was a mutant with birth rate a0 + x at time s and the number of its descendants at time t,
, has
. It is also convenient to define the mapping z: [0, ∞) × [0, t] → [0, ∞) which maps a point (s, x) ∈ (t) to the growth rate of the induced branching process if it survives: z(s, x) = (λ0 + x)(t − s) and let
for A ⊂ [0, ∞).
We shall use C to denote a generic constant whose value may change from line to line. We write f(t) ~ g(t) if f(t)/g(t) → 1 as t → ∞ and f(t) = o(g(t)) is f(t)/g(t) → 0. f(t) ≫ (≪)g(t) means that f(t)/g(t) → ∞ (resp. 0) as t → ∞ and f(t) = O(g(t)) means |f(t)| ≤ Cg(t) for all t > 0. We also shall use the notation f(t) ≃ g(t) if f(t) − g(t) → 0 as t → ∞.
3 Bounded distributions, Z1
In this section, we prove Theorems 1–3.
Proof of Theorem 1
Mutations to type-1 cells occur at rate V0u1eλ0s so
| (3.1) |
We begin by showing that the contribution from x ∈ [0, b − (1 + k) (log t)/t] can be ignored for any k ∈ [0, ∞). The Mean Value theorem implies that
| (3.2) |
Using this and the fact that for any c < d, we can see that
| (3.3) |
To handle the other piece of the integral, we take k = 1 and note that
After changing variables y = (b − x)t, dx = −dy/t, the last integral
which proves the result.
The above proof tells us that the dominant contribution to the type-1 cells comes from mutations with fitness increase x ≥ bt = b − 2log t/t. To describe the times at which the dominant contributions occur, let S(t) = (2/b) log log t. Then the contribution to the mean from x ∈ [bt, b] and s ≥ S(t) is by (3.1)
Since btS(t) ≥ (3/2) log log t for all t sufficiently large, this quantity is o(t−1e(λ0+b)t). In words, the dominant contribution to the mean comes from points close to (0, b) or more precisely from [0, (2/b) log log t] × [b − (2 log t)/t, b].
Proof of Theorem 2
It suffices to prove (1.1). The computation in (3.3) with k = 2 + p implies that the contribution from mutations with x ≤ bt = b − (3 + p)(log t)/t can be ignored. Therefore, we have
where At = {(s, x) ∈ (t): x > bt}. Lemma 2 of Durrett and Moseley (2009) implies that
where and is a birth/death branching process with birth rate a0 + x, death rate b0, and initial population . Using
| (3.4) |
we have
Changing variables s = rx + r where on the inside integral, y = (b − x)t, dy/t = −dx on the outside, and continuing to write x as short hand for b − y/t, the above
| (3.5) |
Formula (20) in Durrett and Moseley (2009) implies that as u → ∞,
| (3.6) |
and therefore, letting t → ∞ and using (1 + p) λ0/(λ0 + b) = 1, we can see that the expression in (3.5)
Changing variables , dr = dq/(λ0 + b) gives
To simplify the first integral we note that
For the second integral, we prove
Lemma 1
If 0 < c < 1
| (3.7) |
Proof
We can rewrite the integral as
so that after interchanging the order of integration and changing variables w = e−qx, dw = −dqe−qx so that w/x = e−q, dw/x = −dqe−q, we have
which is = Γ(c)Γ(1 − c).
Taking c = λ0/(λ0 + b) and letting
| (3.8) |
we have proved Theorem 2.
Recall that we have assumed Z0(t) = V0eλ0t is deterministic. This assumption can be relaxed to obtain the following generalization of Theorem 2 which is used in Section 4.
Lemma 2
Suppose that Z0(t) is a stochastic process with Z0(t) ~ eλ0tV0 for some constant V0 as t → ∞. Then the conclusions of Theorem 2 remain valid.
To see why this is true, we can use a variant of Lemma 2 from Durrett and Moseley (2009) to conclude that
where is the σ-field generated by Z0(s) for s ≤ t. Therefore,
Given ε > 0, we can choose tε > 0 so that
for all t > tε. Since the contribution from t ≤ tε will not affect the limit and the term inside the expectation is bounded, the rest of the proof can be completed in the same manner as the proof of Theorem 2.
We conclude this section with the
Proof of Theorem 3
Let (t) be the three dimensional Poisson process defined in Section 2. Recall that
i.e. F maps a point in (t) to its contribution to the limit t1+pe−(λ0+b)tZ1(t). Using (3.4), we see that in order to have F(s, x, v) > z we need
Therefore, the expected number of mutations that contribute more than z to the limit is
The exponential term can be simplified by making the change of variables
ds = dr/r(λ0 + x) yielding the equivalent expression
where α(x, t) = zt−(1+p)e(b−x)t(λ0 + x)/(a0 + x) and β(x, t) = α(x, t) e(λ0+x)t. As in the previous proof, the main contribution comes from x ∈ [bt, b] so when we change variables y = (b − x)t, dx = −dy/t, replace the x’s by b’s and use 1 = (1 + p)λ0/(λ0 + b) we convert the above into
Performing the integrals gives the result with
| (3.9) |
4 Bounded distributions, Zk
We now move on to the proofs of Theorems 4 and 5. Recall that we have defined pk by the relation
Proof of Theorem 4
Breaking things down according to the times and the sizes of the mutational changes we have
| (4.1) |
The first step is to show
Lemma 3
Let bt = b − (2k + 1)(log t)/t. The contribution to EZk(t) from points (x1,…xk) with some xi ≤ bt is o(t−2ke(λ0+kb)t).
Proof
(3.2) implies that
Applying this and working backwards in the above expression for EZk(t), we get
and the desired result follows.
With the Lemma established, when we work backwards
From this and induction, we see that the contribution from points {x1, … xk) with xi ∈ [bt, b] for all i is
Changing variables yi = t(b − xi) the above
which proves the desired result.
In the proof of the last result, we showed that the dominant contribution comes from mutations with xi > bt. To prove our limit theorem we will also need a result regarding the times at which the mutations to the dominant types occur.
Lemma 4
Let . The contribution to EZk{t) from points with s1 ≥ αk log t is o(t−2ke(λ0+kb)t).
Proof
Replace the Xi’s in the exponents by b’s, we can see from (4.1) that the expected contribution from points with s1 ≥ αk log t is
and the desired result follows.
Recall that
For the induction used in the next proof, we will also need the corresponding quantity with λ0 replaced by λ0 + x and k by k − 1
which means
The limit will depend on the mutation rates through
Again we will need the corresponding quantity with k − 1 terms
We shall write u2,k = u2,k(b) and note that
| (4.2) |
Proof of Theorem 5
We shall prove the result under the more general assumption that Z0(t) ~ V0eλ0t for some constant V0. The result then holds for k = 1 by Lemma 2. We shall prove the general result by induction on k. To this end, suppose the result holds for k −1. Let
be the type-k descendants at time t of the 1 mutant at (s,x,v) ∈ (t). Since
compared to Z0(t) ~ V0eλ0t, it follows from the induction hypothesis that
| (4.3) |
Integrating over the contributions from the three-dimensional point process we have
where . To prove the desired result we need to replace θ by θtk+pke−(λ0+kb)t. Doing this with (4.3) in mind we have
| (4.4) |
By Lemmas 3 and 4, we can restrict attention to x ∈ [bt, b] and s ≤ αk log t. The first restriction implies that all of the x’s except the one in (b − x) can be set equal to b and the second that we can replace t by t − s. Since (k + pk) −(k − 1 + pk−1 (b)) = (λ0 + kb)/λ0, the term in the exponential on the righthand side of (4.4) is
Changing variables s = R(t) + r where R(t) = (1/λ0)(log t), and y = (b − x)t, dy = −tdx the above becomes
Using (4.3) now we have that the 1 − φ term converges to
To simplify this expression, we let
dr = dq/(λ0 + b). Plugging this into eλ0r results in
so we conclude that the term in the exponential on the righthand side of (4.4) converges to
| (4.5) |
To obtain a cleaner expression for the constants, we begin by noting that the change of variables w = v (λ0 + b)/(a0 + b), dw = dv(λ0 + b)/(a0 + b) implies that
| (4.6) |
Furthermore, we also have
| (4.7) |
Finally, to evaluate the third integral in (4.5), we make the change of variables x = e−q, dx = −e−q dq, or dq = −dx/x to show that it is
Integrating by parts f (x) = 1−e−x, g′(x) = x−1−λ0/(λ0+b), f′ (x) = e−x, g(x) = x−λ0/(λ0+b) (λ0+ b)/λ0 shows that the previous expression is
| (4.8) |
Combining (4.6) – (4.8) and using (4.2), we conclude that the expression in (4.5) is
| (4.9) |
Setting ck(λ0,b) equal to the quantity in (4.9) divided by V0u1,kθλ0/(λ0+kb) we have proved the result.
To work out an explicit formula for the constant and to compare with Durrett and Moseley (2009), it is useful to let λj = λ0 + jb, aj = a0 + jb and
From this we see that
and hence
In Durrett and Moseley (2009) if we let 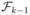 be the σ-field generated by Zj(t) for j ≤ k and all t ≥ 0 then
be the σ-field generated by Zj(t) for j ≤ k and all t ≥ 0 then
Iterating we have
and hence
where .
5 Proofs for unbounded distributions
In this Section, we prove Theorems 6 and 7. The first step is to show that unlike in the case of bounded mutational advances, for unbounded distributions, the main contribution to the limit is given by the descendants of a single mutation. We will later show that the largest growth rate will come from z = O(t(α+1)/α) so the next result is enough. Recall that z(s, x) = (λ0 + x) (t − s) is the growth rate of the family descended from a mutant at (s, x).
Lemma 5
For any z̄, t > 0, we have
Proof
z(s, y) ≤ z̄ if and only if we have a mutation at time s which increases fitness by y ≤ z̄/(t − s) − λ0 and hence, the expected number of individuals produced by mutations with growth rates ≤ z̄ is
since .
To motivate the proof of the general result, we begin with the case when α = 1. Recall that the mean number of mutations with growth rate larger than z has
Proof of Theorem 6
Since
for any z > 0, we have
as t → ∞. Lemma 5 tells us that if there is a mutation with growth rate z = O(t2), then the contribution from mutations with growth rates smaller than z − ε can be ignored so it suffices to describe the distribution of the largest growth rates. We will show that if
then
| (5.1) |
so that the largest growth rate is O(t2) and comes from the rightmost particle in the point process with intensity given by (1.8).
To prove (5.1), we first need to locate the maximum of φ. Let z > λ0t so that there exists a unique maximum sz. Solving φs(s, z) = 0 and using the expression for φs in (1.6) yields
where a0 = (γ/λ0)1/2 = (4c0)−1/2 which leads to the expression
| (5.2) |
If we take
in (5.2) and use (1 + y)1/2 = 1 + y/2 + O(y2), we obtain
| (5.3) |
as t → ∞. Furthermore, (1.7) implies that
as t → ∞ with . Since φs(sz, z) = 0, taking a Taylor expansion around sz yields
| (5.4) |
where |g(s, z)| ≤ C|s − sz|3/t2 for all s. Also note that letting
and recalling (1.4), we have
where ct → 1 as t → ∞ so that
where |g2(s, z)||s − sz|−1t−β = o(1).
Write
where A = {s: |s − szt| ≤ C(t log t)1/2} ∩ [0, t]. Since concavity implies that for s ∈ Ac and C sufficiently large, we have
the contribution of the second integral is negligible. After the change of variables s = szt + (t/a)1/2r, when t is large, the first integral becomes
and therefore since |g(s, zt)| ≤ C(t log t)3/2/t2 when s ∈ A, we have
| (5.5) |
where . Since
we can conclude that
which proves (5.1).
When α ≠ 1, we no longer have an explicit formula for the maximum value sz which complicates the process of identifying the largest growth rate. We shall assume for convenience that α > 1 is an integer.
Proof of Theorem 7
As in the proof of Theorem 6, it suffices to describe the distribution for the largest growth rates. Let z > λ0t so the maximum sz exists. To find a useful expression for the value of φ(sz, z), we write
Using the definition of sz as the solution to φs(sz, z) = 0 yields the condition that
i.e.,
If we substitute the right side of this equation back in for t − sz in the parenthesis, then writing a0 = (αγ/λ0)1/(α+1) we have
Repeating this α times and then using the approximation (1 − x)n = 1 − nx + O(x2) with n = (α − 1)/(α + 1) yields
| (5.6) |
where
for j ≥ 1. The error term is O(z−1) because
for all z > λ0t and s ≤ t. Factoring out a0 in (5.6) and using (1 + x)−1 = Σ(−x)j when |x| < 1, we have that
| (5.7) |
for large z where the bj are given by
and in general,
(5.7) implies that
and therefore,
| (5.8) |
where the dj can be calculated explicitly, for example:
To figure out the distribution of the growth rate for the largest mutant, we let c0 = (−λ0/d0)(α+1)/α and then search for κj, j = 1, …, α − 1 and κ so that plugging
into (5.8) yields
| (5.9) |
for some constants k1, k2, k3. Substituting zt into (5.8) and writing κ0 = 1, κα = x/c0 to ease the notation we obtain
Since λ0t + d0(−λ0t/d0) = 0, the first order terms in this expansion is t(α−1)/α and after using the Taylor series expansion
we obtain
| (5.10) |
where
and in general
j = 1, 1, …, α where for each i and k, in the inner product, i1, …, ik are always chosen to satisfy i1 + i2 + ··· + ik = j − i. Since ρj depends only on κi, i ≤ j, then after noting that the coefficient of κj in ρj is −αλ0/(α + 1), we can use forward substitution to solve the system ρj = 0, j = 1, 2, …, α − 1 for κj to obtain the recursive formulas
| (5.11) |
for i = 1, 2, …, α − 1. Setting ρ = −k3 yields
and for this choice of cj, κ, we obtain (5.9) with
and k1 = −(ρα − k2x). Since
choosing k3 = (2β/α + 1)/2 replaces (5.3) in the proof of Theorem 6.
Now substituting (5.6) and (5.7) in (1.7) yields
where in the second to last line we have used the fact that . When z = zt, this becomes
where
Since φs(sz, z) = 0 and a calculation similar to the one above shows that φsss(szt, zt) = O(t−2), we have
where |g(s,z)| ≤ C|s − sz|3/t2 for all s. This replaces (5.4) from the α = 1 proof and the rest of the proof is the same. Note that the intensity for the limiting point process is given by
| (5.12) |
Remark 1
From (5.6), we have
which tells us that the time at which the mutant with largest growth rate is born is ~ t/(α + 1).
6 Discussion
In this paper, we have analyzed a multi-type branching process model of tumor progression in which mutations increase the birth rates of cells by a random amount. We studied both bounded and unbounded distributions for the random fitness advances and calculated the asymptotic rate of expansion for the kth generation of mutants.
In the bounded setting, we found that there are only two parameters of the distribution that affect the limiting growth rate of the kth generation (see Theorems 1, 2, 4, and 5): the upper bound for the support of the distribution and the value of its density at the upper bound. This is a rather intuitive result since one would expect that in the long run, the kth generation will be dominated by mutants with the maximum possible fitness. In addition, we found that there is a polynomial correction to the exponential growth of the kth generation. This correction is not present in the case where the fitness advances are deterministic. We have discussed this point in further detail in Section 1.1 and after the proof of Theorem 5 in Section 4. Finally, we showed that the limiting population is descended from several different mutations (see Theorem 3).
In the unbounded setting, we assumed that the distribution of the fitness advance has tail
| (6.1) |
where α, β, γ, and K are parameters. We found that the population of cells with a single mutation grows asymptotically at a super-exponential rate exp(t(α+1)/α) (see Theorems 6 and 7) and at large times, most of the first generation is derived from a single mutation (see Lemma 5 and the preceding paragraph). The super-exponential growth rate suggests that distributions of the form (6.1), which includes the exponential distribution that is often used to model fitness advances of organisms under selective pressure, is not a good choice for modeling the mutational advances in the progression to cancer where there is very little evidence for populations growing at a super-exponential rate.
These conclusions provide several interesting contributions to the existing literature on evolutionary models of cancer progression. First, our model generalizes previous multi-type branching models of tumor progression by allowing for random fitness advances as mutations are accumulated and provides a mathematical framework for further investigations into the role played by the fitness distribution of mutational advances in driving tumorigenesis. Second, we have discovered that bounded distributions lead to exponential growth whereas unbounded distributions lead to super-exponential growth. This dichotomy might provide a new method for testing whether a tumor population has evolved with an unbounded distribution of mutational advances. Third, we observe that in the case of bounded distributions, the growth rate of the tumor is somewhat ‘robust’ with respect to the mutational fitness distribution and depends only on its upper endpoint. Finally, our calculations of the growth rates for the kth generation of mutants serve as a groundwork for studying the evolution and role of heterogeneity in tumorigenesis. These implications will be explored further in future work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Becskei A, Kaufmann BB, van Oudenaarden A. Contributions of low molecule number and chromosomal positioning to stochastic gene expression. Nature Genetics. 2005;9:937–944. doi: 10.1038/ng1616. [DOI] [PubMed] [Google Scholar]
- Beerenwinkel N, Antal T, Dingli D, Traulsen A, Kinzler KW, Velculescu VE, Vogelstein B, Nowak MA. Genetic progression and the waiting time to cancer. PLoS Computational Biology. 2007;3 doi: 10.1371/journal.pcbi.0030225. paper e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisel CJ, Rokyta DR, Wichman HA, Joyce P. Testing the extreme value domain of attraction for distributions of beneficial fitness effects. Genetics. 2007;176:2441–2449. doi: 10.1534/genetics.106.068585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer W, Tomlinson I. Failure of programmed cell death and differentiation as causes of tumors: some simple mathematical models. Proc Natl Acad Sci USA. 1995;92:11130–11134. doi: 10.1073/pnas.92.24.11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coldman AJ, Murray JM. Optimal control for a stochastic model of cancer chemotherapy. Mathematical Biosciences. 2000;168:187–200. doi: 10.1016/s0025-5564(00)00045-6. [DOI] [PubMed] [Google Scholar]
- Cowperthwaite MC, Bull JJ, Meyers LA. Distributions of beneficial fitness effects in RNA. Gentics. 2005;170:1449–1457. doi: 10.1534/genetics.104.039248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrett R, Mayberry J. Traveling waves of selective sweeps. Ann Appl Prob. 2009 to appear. [Google Scholar]
- Durrett R, Moseley S. Evolution of resistance and progression to disease during clonal expansion of cancer. Theor Pop Biol. 2009 doi: 10.1016/j.tpb.2009.10.008. to appear. [DOI] [PubMed] [Google Scholar]
- Durrett R, Schmidt D, Schweinsberg J. A waiting time problem arising from the study of multi-stage carcinogenesis. Ann Appl Prob. 2009;19:676–718. [Google Scholar]
- Elowitz MB, et al. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Feinerman O, et al. Variability and robustness in T cell activation from regulated heterogeneity in protein levels. Science. 2008;321:1081. doi: 10.1126/science.1158013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SA. Dynamics of Cancer: Incidence, Inheritance and Evolution. Princeton Series in Evolutionary Biology. 2007 [PubMed] [Google Scholar]
- Gillespie JH. A simple stochastic gene substitution model. Theor Pop Biol. 1983;23:202–215. doi: 10.1016/0040-5809(83)90014-x. [DOI] [PubMed] [Google Scholar]
- Gillespie JH. Molecular evolution over the mutational landscape. Evolution. 1984;38:1116–1129. doi: 10.1111/j.1558-5646.1984.tb00380.x. [DOI] [PubMed] [Google Scholar]
- Goldie JH, Coldman AJ. Quantitative model for multiple levels of drug resistance in clinical tumors. Cancer Treatment Reports. 1983;67:923–931. [PubMed] [Google Scholar]
- Goldie JH, Coldman AJ. The genetic origin of drug resistance in neoplasms: implications for systemic therapy. Cancer Research. 1984;44:3643–3653. [PubMed] [Google Scholar]
- Haeno H, Iwasa Y, Michor F. The evolution of two mutations during clonal expansion. Genetics. 2007;177:2209–2221. doi: 10.1534/genetics.107.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasa Y, Michor F, Komorova NL, Nowak MA. Population genetics of tumor suppressor genes. J Theor Biol. 2005;233:15–23. doi: 10.1016/j.jtbi.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Iwasa Y, Nowak MA, Michor F. Evolution of resistance during clonal expansion. Genetics. 2006;172:2557–2566. doi: 10.1534/genetics.105.049791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassen R, Bataillon T. Distribution of fitness effects among beneficial mutations before selection in experimental populations of bacteria. Nature Genetics. 2006;38:484–488. doi: 10.1038/ng1751. [DOI] [PubMed] [Google Scholar]
- Kaern M, et al. Stochasticity in gene expression: from theories to phenotypes. Nature Reviews Genetics. 2005;6:451. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- Knudson AD. Two genetic hits (more or less) to cancer. Nature Reviews Cancer. 2001;1:157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- Komarova NL, Wodarz D. Drug resistance in cancer: principles of emergence and prevention. Proc Natl Acad Sci USA. 2005;102:9714–9719. doi: 10.1073/pnas.0501870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maley CC, et al. Genetic clonal diveresity predicts progression to esophageal adenocarcinoma. Nature Genetics. 2006;38:468–473. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- Maley CC, Forrest Exploring the relationship between neutral and selective mutations in cancer. Artif Life. 2001;6:325–345. doi: 10.1162/106454600300103665. [DOI] [PubMed] [Google Scholar]
- Michor F, Iwasa Y, Nowak MA. Dynamics of cancer progression. Nature Reviews Cancer. 2004;4:197–205. doi: 10.1038/nrc1295. [DOI] [PubMed] [Google Scholar]
- Michor F, Nowak MA, Iwasa Y. Stochastic dynamics of metastasis formation. J Theor Biol. 2006;240:521–530. doi: 10.1016/j.jtbi.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Michor F, Iwasa Y. Dynamics of metastasis suppressor gene inactivation. J Theor Biol. 2006;241:676–689. doi: 10.1016/j.jtbi.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Nowak MA, Michor F, Iwasa Y. Genetic instability and clonal expansion. J Theor Biol. 2006;241:26–32. doi: 10.1016/j.jtbi.2005.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell PC. The cloncal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- Orr HA. The distribution of fitness effects among beneficial mutations. Genetics. 2003;163:1519–1526. doi: 10.1093/genetics/163.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP, Jones CD. Detecting the undetected: Estimating the total number of loci underlying a quantitative trait. Genetics. 2002;156:2093–2107. doi: 10.1093/genetics/156.4.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokyta DR, Beisel CJ, Joyce P, Ferris MT, Burch CL, Wichman HA. Beneficial fitness effects are not exponential in two viruses. J Mol Evol. 2008;67:368–376. doi: 10.1007/s00239-008-9153-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen DE, de Visser JAGM, Gerrish PJ. Fitness effects of fixed beneficial mutations in microbial populations. Curret Biology. 2002;12:1040–1045. doi: 10.1016/s0960-9822(02)00896-5. [DOI] [PubMed] [Google Scholar]
- Sanjuán R, Moya A, Elena SF. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc Natl Acad Sci, USA. 2004;101:8396–8401. doi: 10.1073/pnas.0400146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweinsberg J. The waiting time for m mutations. Electron J Probab. 2008;13:1442–1478. [Google Scholar]
- Shah SP, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- Weissman I. Estimation of parameters and large quantiles based on the k largest observations. j Amer Stat Assoc. 1978;73:812–815. [Google Scholar]
- Wodarz D, Komarova NL. Can loss of apoptosis protect against cancer? Trends Genet. 2007;23:232–237. doi: 10.1016/j.tig.2007.03.005. [DOI] [PubMed] [Google Scholar]