

Abstract
The ribonuclease III Dicer (Dcr1) has been shown to be required for chromosome segregation and gene silencing in Schizosaccharomyces pombe. These effects are thought to be transcriptional, mediated by formation and maintenance of heterochromatin, and guided by small RNAs derived from Dcr1 along a process known as RNA interference. In order to get further insights into the gene regulatory role of Dcr1, we performed comparative analyses of dcr1 knockout and wild-type fission yeast strains. Analysis of part of the soluble proteomes identified eight cellular proteins whose expression is under Dcr1 control, three of which are integral constituents of the glycolysis pathway. Further correlations with their respective mRNA transcript levels are compatible with the existence of a post-transcriptional gene regulatory mechanism involving Dcr1 or a Dcr1 complex. Experiments designed to identify components of Dcr1 complexes unveiled two novel Dcr1 interactors, namely the zinc finger protein Byr3 and the ribosomal protein L12. Consistently enriched in Dcr1 immune complexes, Byr3 and L12 may link Dcr1 to the transcriptional and translational machineries, respectively, and contribute to post-transcriptional gene regulation in fission yeast.
Keywords: Ribonuclease, Dicer, RNA interference, gene expression, proteome, gene regulation, glycolysis, fission yeast
2. INTRODUCTION
MicroRNA (miRNA)-guided RNA silencing is a recently discovered gene regulatory process by which endogenous miRNAs mediate translational repression of specific mRNAs through imperfect complementarity. Whereas RNA interference (RNAi) refers to the process by which double-stranded RNA (dsRNA) triggers sequence-specific degradation of messenger RNA (mRNA) targets (1). In both cases, the effects are mediated by small 21- to 24-nucleotide (nt) RNAs that are produced upon cleavage of precursor molecules by the ribonuclease (RNase) III Dicer (2). Bearing features characteristic of endogenous Dicer cleavage products, i.e. a 5′ phosphate and 3′ hydroxyl termini with 2-nt 3′ overhangs (3), synthetic small interfering RNAs (siRNAs) exert potent and specific interference with gene expression, and are useful for deciphering gene function, particularly in cultured mammalian cells (4). Designed to be perfectly complementary to their targets, exogenous siRNAs are incorporated into RNA-induced silencing complex (RISC) that will mediate targeting and cleavage of specific mRNAs.
Although it diverged from Metazoa and plants around 1,000 to 1,200 million years ago, the unicellular organism Schizosaccharomyces pombe harbors three components of the RNAi machinery (5). One of these components is Dicer, or Dcr1. We have previously shown that S. pombe Dcr1, as in other species, can cleave a dsRNA substrate into ~23 nt RNAs in vitro (6). Small RNAs of similar size have been identified in vivo and found to originate from centromeric heterochromatic repeats (7). In a model presented by Noma et al. (8), small RNAs produced by Dcr1 are incorporated into the RNA-induced initiation of transcriptional silencing (RITS) complex, guiding it to homologous sequences on heterochromatin to allow its formation and maintenance.
In S. pombe, studies of the RNAi pathway have mainly focused on its control over histone methylation, heterochromatin formation and related transcriptional silencing. Whether protein-coding mRNAs are regulated by small RNAs via a post-transcriptional mechanism in fission yeast remains unknown. Recently, a study by Carmichael et al. (9) showed that the three components of RNAi, i.e. Dcr1, Ago1 and Rdp1, are found in common locations in the cytosolic compartment of fission yeast cells. These observations raise the possibility that Dcr1 may have a role in processes other than RNA silencing at the transcriptional level in S. pombe.
In order to gain further insights into the gene regulatory role of Dcr1, we compared the proteomes of dcr1Δ and wild-type (WT) fission yeast strains. These analyses revealed that the expression of at least eight cellular proteins is controlled by Dcr1. Further monitoring of their respective mRNA levels support the existence of a post-transcriptional regulatory mechanism involving either Dcr1 or a Dcr1 complex. Identified as novel Dcr1 interactors, the zinc finger protein Byr3 and the ribosomal protein L12 may link Dcr1 to the transcriptional and translational machineries, and determine Dcr1 function in post-transcriptional gene silencing (PTGS) in fission yeast.
3. MATERIALS AND METHODS
3.1 Strains and media
A list of S. pombe strains used in this study and their corresponding genotypes are shown in Table 1. Supplemented yeast extract medium (YES) and Edinburgh Minimal Medium Glutamate (EMMG) were used for protein and RNA extraction, and prepared as described in Moreno et al. (10). Geneticin (Invitrogen) was used for selection of stable transformants at a final concentration of 100 μg/ml.
Table 1.
List of S. pombe strains used in this study.
| Strain | Genotype | Reference |
|---|---|---|
| Hu111 | h+ leu1-32 ade6-DN/N ura4-DS/E | (6) |
| Hu676 | h?dcr1Δ::kanMX6 ade6-DN/N ura4-DS/E leu1-32 | (6) |
| Hu679 | h+ dcr1ΔkanMX6 | (6) |
| Hu906 | h? dcr1-HA::kanMX6 leu1-32 ade6-DN/N ura4-DS/E | (6) |
| UL108 | h? L12-1:: ΔkanMX6 leu1-32 ade6-DN/N ura4-DS/E | This study |
| UL109 | h? L12-2:: ΔkanMX6 leu1-32 ade6-DN/N ura4-DS/E | This study |
3.2 Two-dimensional gel electrophoresis
Protein extracts were prepared simultaneously from cells grown in EMMG medium at 30°C and harvested at an optical density (O.D.) 595 nm of 0.25. Cells were washed in MilliQ water, resuspended in 150 μl of lysis buffer (40 mM Tris, 7 M urea, 2 M thiourea and 4% Chaps) containing 1 x protease inhibitors (EDTA-free Complete protease inhibitor cocktail mix, Roche) and 1 mM PMSF (Sigma), and lyzed with 425–600 μm beads (Invitrogen). Protein concentration was measured using the Bradford method (11). Proteins were migrated on an immobilized pH gradient (IPG) band covering pH 5.0 to 6.0 or 5.0 to 7.0 interval, followed by separation on a 10% polyacrylamide gel. Gels were stained with silver, as described in (12), or by using Sypro Ruby (Molecular Probes) following the manufacturer’s instructions. The stained gels were analyzed using Progenesis software to determine spot intensity.
3.3 In-gel protein digestions
Gel plugs containing the proteins of interest were excised with a scalpel and submitted to liquid chromatography-mass spectrometry-mass spectrometry (LC-MS-MS) analysis (Eastern Quebec Proteomics Center, CHUL Research Center, Quebec). In-gel protein digestions were performed on a MassPrep liquid handling station (Micromass Ltd) according to the manufacturer’s specifications using sequencing grade modified trypsin (Promega). Extracted peptides were lyophilized using a speed vac and suspended in 0.1% trifluoroacetic acid or 0.1% formic acid for MALDI-TOF or LC-MS-MS analysis, respectively.
3.4 MALDI-TOF analysis and database searching
The matrix used for MALDI analysis was alpha-cyano-4-hydroxycinnamic acid (1.7 mg/ml in 58% acetonitrile, 0.1% trifluoroacetic acid). Equal volumes of peptide and matrix solution were mixed, and 1 μl of the resulting solution was spotted on a stainless steel MALDI sample plate. The sample/matrix solution was allowed to air-dry at room temperature and was then washed three times with 0.1% trifluoroacetic acid. MALDI-TOF spectra were acquired on a Voyager-DE PRO Biospectrometry Workstation (Applied Biosystems) and analyzed using the DataExplorer software version 4.0 (Applied Biosystems). The instrument was operated in the positive-ion reflector delayed-extraction mode. The ProFound program (version 4.10.5, The Rockefeller University, http://prowl.rockefeller.edu/cgi-bin/ProFound) was used to search the non-redundant NCBI protein database for matching peptide mass fingerprints. Search criteria allowed a maximum of 1 missed cleavage by trypsin, complete carboxyamidomethylation of cysteine, partial methionine oxidation and mass deviation smaller than 60 ppm.
3.5 Ion Trap MS/MS analysis and peptide sequencing
Peptide MS/MS spectra were obtained by microcapillary reverse-phase chromatography coupled to an LCQ DecaXP or LTQ (ThermoFinnigan) quadrupole ion trap mass spectrometer with a nanospray interface. A 10-μl aliquot of the peptide sample was loaded onto a 75-μm internal diameter C18 picofrit column (New Objective). Peptides were eluted with a water-acetonitrile 0.1% formic acid gradient at a flow rate of about 200 nl/min. Resulting peptide spectra were interpreted using the SEQUEST algorithm (13) or MASCOT algorithm (14), and searched against proteins of the NCBI non-redundant protein database.
3.6 mRNA level quantitation by RT-PCR
Total RNA was prepared by the method of hot acidic phenol extraction, as described previously (15). Reverse transcription (RT) was performed using Superscript II Reverse Transcriptase (Invitrogen) according to the manufacturer’s protocol. The 50-μl polymerase chain reaction PCR reactions were as follows: 150 ng cDNA, 50 pmol of each target-specific and actin-specific oligonucleotides (see Table 2), 0.2 mM dNTPs, 3.5 U Expand High Fidelity (Roche) in reaction buffer. The PCR protocol used was: 1 cycle of 2 min at 94°C, 30 cycles of 15 sec at 94°C, 30 sec at 47°C and 3 min at 68 °C and 1 cycle of 10 min at 68°C. Samples were run on a 1% agarose gel and analyzed by densitometry using the Multimage Light Cabanet (Alpha Innotech Corporation). Results were normalized relative to actin mRNA levels.
Table 2.
List of the DNA oligonucleotides used in this study.
| Oligo number | Sequence |
|---|---|
| Cloning of L12 | |
| 1 | GCGCGCGGATCCATGGAGCAAAAGTTAATTTCTGAAGAA GATTTGATGCCTCCTAAATTCGATCCCAATGAAGT |
| 2 | GCGCGCGGATCCTTACTCTTGGGGGATCTCAATTTCGCC |
| Semi-quantitative RT-PCR | |
| 3 | ATGGCCATTCAGAAAGTCTTTGC |
| 4 | CGGAGAAAGAGGGAGCACCGGTGGGG |
| 5 | ATGGAGGCTAATTTTTCAACAAGC |
| 6 | ATGGCCATTCAGAAAGTCTTTGC |
| 7 | ATGTCTTTGTCTACTAAGCTCGC |
| 8 | CGTCGGGGATACCCTCCTCGGCGG |
| 9 | CGACTTCAAGGGTACTGCCGTTGT |
| 10 | CAAGACACCATAGTCACGGGAAAC |
| 11 | TGCTGAAGGACTTGCTCGTCGTAT |
| 12 | GTATTCAGCCAAAGTAGTAGCACC |
| 13 | GCCACTCCTTACCATTTGCCCATC |
| 14 | TGTTCATACCACAGCGAATAGCAG |
| 15 | GGCTTTCCCCCTACTGTCAACGAC |
| 16 | GTAAGTTTTCCGCTATCATAATGG |
| 17 | TGACTGAACACTCATTTAAGCAAA |
| 18 | TCTGGGGTAAATATACGAAGTCTG |
| 19 | CGCAGCGTTGGTTATTGATAATGG |
| 20 | GGGTTGGAAAAGAGCTTCAGGGG |
| Homologous recombination | |
| 21 | TAAATTTTGAGAATTAACTACCATCTCCTTAAATCTTTA GGGTAGGGTTGTGACTGTTGTTTGGTCTTCCCTTTGGACACAC |
| 22 | GTTTGGTCTTCCCTTTGGACACAGAGAATCACTCTTGCT TGATCCCACAGCCACAACCCAAGCGGATCCCCGGGTTAATTAA |
| 23 | GGTTTTATTCAACCAAGTTCTTATACAAAAAGCACCTCT ACAATTAAATCAGAAATCACACGCAACTTTACAGAAAATTGAG |
| 24 | CACGCAACTTTACAGAAAATTGAGAAGGTCACAAATGAG TTATTTTATATCAAATTACGATTGATGAATTCGAGCTCGTTTAAAC |
| 25 | TCATTAAAGAAGTTTTGTTGTGGGTGCCCTCAGGAACAG TCACACTCATTTCGGACAGTCACATAAAACAAAAACAAAACTAAC |
| 26 | CACATAAAACAAAAACAAAACTAACACTTTGCGTATTTT TCTTGAACCAAGCAAGAGTAAGCGGATCCCCGGGTTAATTAA |
| 27 | TATTTTTTGAACCCTATAAAAAGATAGCAAAAACATCAA GATCTAAACATTGCTGTTTTATACAAAACCCGTATACTACCAAGC |
| 28 | TACAAAACCCGTATACTACCAAGCAAGCACATAATCCGT TTACATTAACTTATGTATGCGAGAATTCGAGCTCGTTTAAAC |
3.7 Colony growth assays
Hu111 or Hu676 strains were grown in YES medium at 30°C, washed and resuspended in water at a final concentration of 3 × 106 cells/ml. Five-fold serial dilutions of yeast cells were made and 5 μl drops were plated in triplicate on YES plates containing 10 mM or 700 mM glucose. Plates were incubated at 25°C or 30°C until they reached optimal growth.
3.8 Liquid growth assays
Seventy-five (75) ml of YES media containing 5, 10, 25, 50 or 100 mM glucose were inoculated at an OD(595 nm) of 0.1 with Hu111 or Hu676 strain, and incubated at 25°C until they stopped growing. OD(595 nm) was taken every 2 hours during the exponential growth phase. Generation time was calculated using the Microsoft Excel software.
3.9 Immunoprecipitations
Hu906 or Hu676 cells, transformed or not with the Myc-L12 expression construct, were grown to a density of 0.5 to 2 × 107 cells/ml, harvested and washed in phosphate buffered saline (PBS). Cells were broken in lysis buffer (50 mM Tris•HCl, pH 7.5, 50 mM NaCl and 1% Triton X-100) using 425–650 um glass beads (Sigma). Protein concentration was assessed using the Bradford assay method, with bovine serum albumin as a standard (11). Immunoprecipitations were performed using 1 mg of protein extracts and anti-HA Affinity Matrix (Roche) or anti-Myc antibody and protein G-Agarose beads (Roche). Immunoprecipitates were analyzed by 10% polyacrylamide gel electrophoresis, which was silver-stained or transferred to a Hybond polyvinylidene difluoride membrane (Amersham Pharmacia Biotech). Silver staining was done according to Vorum’s protocol (12). Candidate protein species were excised from the gel and analyzed by LC-MS-MS (Eastern Quebec Proteomics Center, CHUL Research Center, Quebec). Immunoblot analyses were performed using anti-HA or anti-Myc antibodies.
3.10 DNA construct
Genomic DNA was isolated from S. pombe strain Hu111 by using a method adapted from David Amberg’ lab homepage (http://www.upstate.edu/biochem/amberg). The open reading frame of the L12-1 gene was amplified by PCR using oligos 1 and 2 (see Table 2), and cloned as a BamHI fragment into the BamHI site of pREP42X vector (16). The constructs were verified by restriction analysis and DNA sequencing.
3.11 Antibodies
A rabbit anti-L12 antibody was raised against peptide 153KEIDNGEIEIPQE165 and purified by affinity chromatography, according to a standard procedure. Mouse monoclonal anti-HA antibody (clone 12CA5) (Roche), anti-Myc antibody (clone 9E11) (a kind gift from Dr. Louis Flamand, Université Laval) and anti-TAT1 antibody (a kind gift from Dr. Keith Gull, University of Oxford) (17) were used for immunoprecipitation and immunoblot analyses.
3.12 Homologous recombination
The L12-1 or L12-2 gene was deleted via a PCR-based approach using the pFA6a-kanMX6 vector (see Table 2) (18). The PCR products, bearing 120 bp homologous to the target sequence at both extremities, were used to transform S. pombe cells using the lithium acetate method, as described in Bähler et al. (18), and transformants were selected based on their resistance to geneticin (Invitrogen). Homologous genomic integration was confirmed by colony PCR strategies. Expression of L12 protein was confirmed by Western blotting using anti-L12 antibody.
4. RESULTS
4.1 Initial proteomic analysis unveils Hsp16 as a protein regulated by Dcr1
When incubated in the presence of a long dsRNA trigger, Dcr1 catalyzes the formation of small ~23-nt RNA species (6). In order to identify proteins regulated by Dcr1, we performed two-dimensional gel analysis of protein samples from WT and dcr1Δ strains. In a first set of experiments, based on silver staining of the proteins, we identified a spot exhibiting a marked difference in intensity between the two strains (see Figure 1A). After excision from the gel and analysis by LC-MS-MS, the spot was found to contain heat shock protein 16 (Hsp16) (see Figure 1B). Although its function remains unclear, Hsp16 may be involved in the stress response associated with a diminution of nucleotides and DNA damage (19). The identification of Hsp16 as a protein species upregulated in the absence of Dcr1 extends our previous report showing similar variations in Hsp16 at the mRNA level in the dcr1Δ strain (6), thereby validating our approach.
Figure 1.
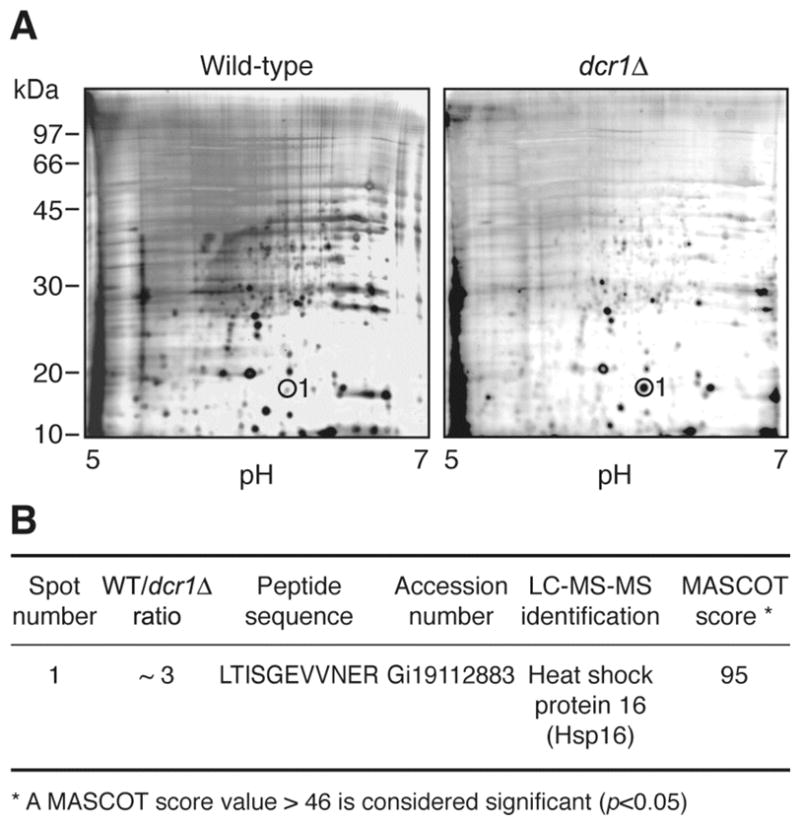
Comparative two-dimensional gel analysis of the WT and dcr1Δ strains using silver staining. (A) Gels showing the protein profiles of Hu303 (WT, left panel) and Hu676 (dcr1Δ, right panel) strains. Protein extracts were separated on an IPG band covering pH 5.0 to 7.0 (first dimension), migrated on a denaturing 10% polyacrylamide gel (second dimension) and stained with silver nitrate. (B) Results of the proteomic analyses. The relative intensity of the spot was estimated. Spot number 1 (circled) was cut from the gels and identified by LC-MS-MS.
4.2 Additional proteins are under Dcr1 control
We next performed a more extensive set of experiments in order to identify additional genes whose regulation is placed under the control of Dcr1. In these experiments, an IPG band covering pH 5 to 6 was used and gels were stained using Sypro Ruby, which allowed a more accurate quantitation of protein levels. These experiments, which were repeated four times to insure reproductibility, allowed us to analyze the intensity of more than 1500 spots (see Figures 2A and 2B). Among these spots, sixteen showed a variation greater than 2 fold between the two strains and were submitted to LC-MS-MS analysis, followed by database searches. A positive identification was obtained for seven of the spots, whereas no peptide sequences could be obtained from the nine remaining spots.
Figure 2.

Comparative two-dimensional gel analysis of the WT and dcr1Δ strains using Sypro Ruby. The virtual gels showing the protein profiles of (A) wild-type (Hu303 strain) and (B) dcr1Δ (Hu676 strain) represent the average of 4 independent experiments. Protein extracts were analyzed as in Figure 1, but the proteins were stained using Sypro Ruby. The spots analyzed by LC-MS-MS are circled on the gel in which they showed the greatest intensity.
4.3 Constituents of the glycolysis pathway are regulated by Dcr1
As depicted in Figure 3A, a total of 11 protein species were downregulated from 40% to more than 80% in the absence of Dcr1. A positive identification was obtained for four of these proteins: the pyrimidine precursor biosynthesis enzyme Thi3 (in spots 1107 and 1154), enolase (ENO) (spot 480) and hexokinase 2 (HK2) (spot 1495) (see Figure 3B). We observed that expression of Thi3 is reduced by approx. 50% in the absence of Dcr1. The Thi3 enzyme, like Thi2 and Thi4, is under the control of thiamine biosynthesis (20); i.e. transcription of the gene is completely repressed by thiamine (21). The identification of Thi3 in two different spots supports the existence of an alternative, post-translationally modified form of the protein. Although such modifications of Thi3 have not been reported yet in the litterature, our data suggest that both forms are regulated to a similar extent by Dcr1 – post-translational modifications did not prevent regulation. As for ENO, its expression was decreased by almost 70% in the absence of Dcr1. ENO is a cytosolic enzyme involved in the conversion of 2-phosphoglycerate into phosphoenolpyruvate. Activated by divalent cations, ENO has Mg2+ as its natural cofactor (22). Concerning HK2, it is expressed in the WT strain, but almost absent from the dcr1Δ strain. As ENO, HK2 is involved in glucose metabolism and catalyzes the first reaction of the glycolysis pathway, converting glucose into glucose-6-phosphate. In S. pombe, the absence of HK2 has been reported to be associated with doubling of the generation time (23).
Figure 3.

Analysis of the protein species downregulated in the dcr1Δ strain. (A) Relative protein expression levels in the WT and dcr1Δ strains were determined by analyzing the intensity of the corresponding spots using the Progenesis software. (B) The protein species contained in the spots indicated were identified by LC-MS-MS. ND, not determined.
In contrast, five of the proteins regulated by Dcr1 showed the opposite trend and were found to be upregulated by more than 2 fold in the dcr1Δ strain (see Figure 4A). Three of them could be identified: a putative chaperonine, thioredoxine peroxidase and phosphoglycerate kinase (PGK) (see Figure 4B). The putative chaperonine is believed to be a heat shock protein of the ATPase family that could be involved in the proper folding of proteins (5). As for thioredoxine peroxidase, it was barely detectable in the WT strain, but expressed at higher levels in the dcr1Δ strain. This enzyme acts as a major peroxyde sensor to activate the Pap1 signaling pathway in states of oxidative stress in S. pombe (24). Finally, PGK is the third enzyme regulated by Dcr1 that is known to be involved in the glycolysis pathway. PGK is known to mediate the conversion of 1,3-biphosphoglycerate into 3-phosphoglycerate to produce the first two ATP molecules (25).
Figure 4.

Analysis of the protein species upregulated in the dcr1Δ strain. (A) Relative protein expression levels in the WT and dcr1Δ strains were determined by analyzing the intensity of the corresponding spots using the Progenesis software. (B) The protein species contained in the spots indicated were identified by LC-MS-MS. ND, not determined. * Present in the dcr1Δ strain, it was barely detectable in the WT strain.
Noticeably, three of the proteins found to be regulated by Dcr1, i.e. HK2, enolase and PGK, are components of the glycolysis pathway. This observation prompted us to examine whether Dcr1 can functionally regulate glycolysis. To this end, we attempted to measure the baseline level of ATP in the WT and dcr1Δ strains, but these experiments were prone to important variations and were inconclusive. A growth defect has previously been reported in the dcr1Δ strain (6), in association with lagging chromosomes during anaphase and abrogated silencing of centromeric repeats. Interestingly, disruption of the hxk2 gene doubles generation time on glucose (23), raising the possibility that the growth defect of the dcr1Δ strain may be explained, at least in part, by downregulation of HK2 expression. This hypothesis was tested by subjecting the WT and dcr1Δ strains to growth assays on solid and liquid media in the presence of various concentrations of glucose. As shown in Figures 5A and 5B, we observed a growth defect of the dcr1Δ strain versus the WT strain. However, growth of the dcr1Δ strain was not restored to WT level under conditions of either limiting or saturating concentrations of glucose in the media. These findings suggest that the observed growth defect of the dcr1Δ strain is unlikely to be related to an altered glycolytic function, but rather linked to a defect in heterochromatin formation.
Figure 5.

Comparative growth analyses of WT and dcr1Δ strains. (A) Colony growth assays on YES media containing 10 mM or 700 mM glucose. Five-fold dilutions of yeast cultures were plated in triplicate and incubated at 25°C or 30°C for 7 or 5 days, respectively. The results are representative of two different experiments. (B) Liquid growth assays. The WT and dcr1Δ strains were grown at 25°C in YES media containing various concentrations of glucose. Generation time during the exponential growth phase was calculated and the results expressed as mean ± SEM (n = 2 to 5 experiments).
4.4 Evidences for a post-transcriptional gene regulatory mechanism involving Dcr1
In order to get further insights into the nature of the mechanisms by which the expression of these proteins is regulated, we quantitated the level of their corresponding mRNAs by semi-quantitative RT-PCR (see Figure 6). Among the proteins downregulated in the dcr1Δ strain, only HK2 showed a concomitant decrease in its mRNA level (see Figure 6A), suggesting that its regulation by Dcr1 is mainly transcriptional. However, the level of mRNAs encoding for ENO and Thi3 was similar between the WT and dcr1Δ strains, despite the fact that the proteins are downregulated in the absence of Dcr1. These data suggest the existence of a Dcr1-dependent, post-transcriptional mechanism regulating ENO and Thi3 mRNA translation.
Figure 6.

Analysis of mRNA levels for candidate proteins exhibiting (A) lower or (B) higher expression levels in the dcr1Δ strain, as assessed by semi-quantitative RT-PCR. Reactions were analyzed by agarose gel electrophoresis and the intensity of the bands was analyzed by densitometry using the MultiImage™ Light Cabanet (Alpha Innotech Corporation). Results were normalized with actin mRNA expression levels, used as a control.
For proteins exhibiting higher expression levels in the dcr1Δ strain, two different patterns were observed. Hsp16 and the putative chaperonine showed a concomitant increase in their mRNA levels (see Figure 6B), suggesting, like HK2, that their regulation by Dcr1 is mainly transcriptional. However, the level of mRNAs encoding for PGK and thioredoxine peroxidase was similar between the WT and dcr1Δ strains, despite the fact that both proteins are upregulated in the dcr1Δ strain. These data suggest the existence of a post-transcriptional mechanism by which PGK and thioredoxine peroxidase mRNA translation may be negatively regulated by Dcr1 or a Dcr1 complex.
4.5 Identification of novel Dcr1 protein interactors
The function of human Dicer is believed to be determined by the nature of its interacting proteins. As in other species, we postulated that Dcr1 also operates within larger protein complexes. In order to identify the components of Dcr1 complexes, we exploited a HA-based immunoprecipitation strategy using a dcr1-HA strain in parallel with a dcr1Δ strain. Dcr1-HA was detected in HA immunoprecipitates prepared from the dcr1-HA strain, but not the dcr1Δ strain (see Figure 7A), as expected. The proteins present in the Dcr1-HA immune complexes were then revealed by SDS-PAGE and silver staining. Despite suboptimal stringent washing conditions that yielded a significant background when using a sensitive staining procedure, two bands were consistently enriched in the Dcr1-HA immunoprecipitates (see Figure 7B). Proteomic analyses of these two candidate protein species by LC-MS-MS yielded positive identifications.
Figure 7.
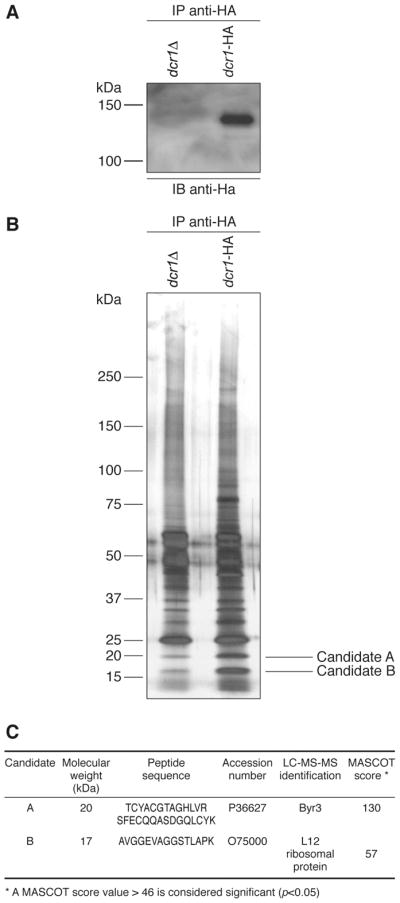
Identification of Dcr1-interacting proteins in S. pombe. HA immunoprecipitates were prepared from dcr1Δ (Hu679) and dcr1-HA (Hu906) strains by incubating protein extracts with an anti-HA affinity matrix. The immunoprecipitates were analyzed by (A) immunoblot analysis using anti-HA antibody, or (B) polyacrylamide gel electrophoresis and silver staining. The protein band slightly above the 75 kDa marker and enriched in the Dcr1-HA immunoprecipitates is unique to this experiment. (C) The protein candidates A and B, enriched in the Dcr1 immunoprecipitate, were excised from the gel and identified by LC-MS-MS.
4.6 The zinc finger protein Byr3
Candidate A was identified as Byr3 (see Figure 7C). This protein, which contains seven zinc finger binding domains (26), is the yeast homolog of human cellular nucleic acid binding protein (CNBP). Functioning as a transcription factor (27), CNBP has also been reported to interact with the 5′ untranslated region of various ribosomal protein mRNAs and to regulate their translation in Xenopus laevis (28).
4.7 The ribosomal protein L12
As for candidate B, it was identified as the ribosomal protein L12 (see Figure 7C), which is homologous to E. coli L11 protein (29). L12 has been shown to take part in the correct assembly of the GTPase site in Saccharomyces cerevisiae and, therefore, proposed to play an important role in ribosomal function (30). Although not essential for yeast viability (31), L12 seems to be required for normal growth. The interaction between Dcr1 and L12 was confirmed by coimmunoprecipitation from dcr1-HA cells expressing the L12-Myc fusion protein by using either an anti-HA affinity matrix (see Figure 8) or, reciprocally, the anti-Myc and protein G-Agarose tandem (L.-A. Gobeil and P. Provost, unpublished data).
Figure 8.
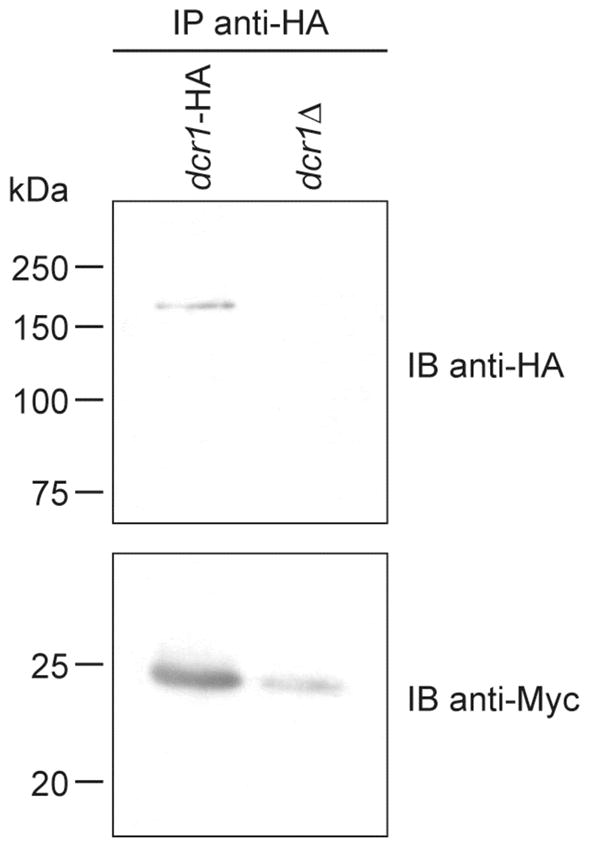
Confirmation of the interaction between Dcr1 and the ribosomal protein L12 by reciprocal coimmunoprecipitation. Dcr1-HA immunoprecipitates were prepared by incubating protein extracts of the dcr1-HA (Hu906) or dcr1Δ (Hu679) strain expressing the L12-Myc fusion protein with an anti-HA affinity matrix. The immunoprecipitates were analyzed by immunoblotting using anti-HA and anti-Myc antibodies.
Considering that L12 could represent a link between Dcr1 and post-transcriptional elements of the gene silencing machinery, we next developed a rabbit polyclonal antibody recognizing S. pombe L12. Although ineffective in immunoprecipitation, the antibody detected bands of the expected size in total cell lysates prepared from S. pombe, but not human cells, in Western blotting (P. Plante and P. Provost, unpublished data). Detection of a doublet by the anti-L12 antibody suggests that the L12 protein may be subjected to post-translational modifications, like its S. cerevisiae counterpart, which is methylated at arginine 67 by RMT2 (32) and at lysine 10 by Rkm2 (33).
Inspection of the S. pombe genome revealed the existence of two genes encoding for L12, namely L12-1 and L12-2. The two genes differ only by the absence or presence of an intron. We thus wished to create L12-1Δ and L12-2Δ strains independently by homologous recombination. The disrupting KanMX6 cassette was correctly inserted into the S. pombe genome and successfully replaced the L12-1 or L12-2 genes, as assessed by colony PCR analyses (L.-A. Gobeil, P. Plante and P. Provost, unpublished data), yielding UL108 and UL109 strains, respectively. Protein extracts prepared from these strains were analyzed by Western blot, in parallel with protein extracts from a WT strain, to monitor L12 protein levels. As shown in Figure 9, the L12-1Δ and L12-2Δ strains displayed levels of L12 protein expression comparable to that observed in the WT strain. From these experiments, we concluded that the two L12 genes are transcriptionally active and may be able to compensate the loss of the other gene. We failed to generate a dual L12-1Δ/L12-2Δ viable strain (L.-A. Gobeil and P. Provost, unpublished data), opening the possibility that L12 may be an essential gene in fission yeast.
Figure 9.
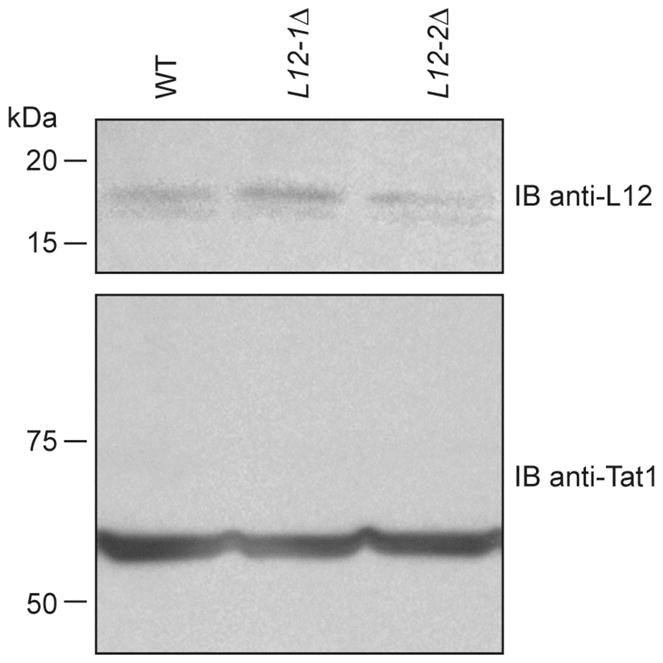
Analysis of L12 protein expression in L12-1Δ, L12-2Δ and WT strains. Protein extracts from L12-1Δ (UL108), L12-2Δ (UL109) and WT (Hu111) strains were analyzed by immunoblotting using anti-L12 and anti-TAT1 antibodies. Tubulin was monitored and used as a loading control.
5. DISCUSSION
In order to study the gene regulatory role of Dcr1 and derived small RNAs in S. pombe, we initially aimed to identify the candidate genes at the protein level through proteomic comparison of the dcr1Δ and WT fission yeast strains. This strategy was preferred over methods based on mRNA detection and quantitation, such as microarray, considering the uncertainty surrounding the exact regulatory mode of these small endogenous RNAs and the fact that some mRNA targets could have gone unnoticed, if their levels were to remain initially unchanged upon small RNA-induced translational repression. For our analysis, we selected the pI (from pH 5.0 to 6.0) and molecular weight (from ~10 to ~75 kDa) range with the aim at obtaining the largest proteome coverage from a single condition.
From the 4,824 protein-coding genes contained in the fission yeast genome, we were able to analyze ~1,500 protein spots by comparative two-dimensional gel electrophoresis. From these proteins, 17 were found to vary significantly by more than 2 fold between the two strains under normal growth conditions. Thus, approximately ~1% of the protein spots analyzed are regulated by Dcr1 and derived small RNAs in fission yeast. In view of the limited proteome coverage of our analysis, this array of RNAi-regulated proteins may be further expanded by examination of a different range of pI and molecular weight, use of alternatives to two-dimensional protein separation approaches, such as liquid-based systems relying on parameters other than pI and molecular weight (eg, hydrophobicity), improved mass spectrometric analyses and assessment of various growth conditions.
Therefore, our initial analyses suggest that RNAi represent a relatively minor contributor to the regulation of gene expression in this unicellular organism, in which the primary function may pertain to the formation and maintenance of heterochromatin associated with telomeres, repetitive DNA elements surrounding centromeres and the silent mating-type loci (34). This is supported by the discovery of small RNAs arising exclusively from centromere heterochromatic repeats in S. pombe (7). Similar figures have been obtained in Drosophila, where Nakahara et al. (35) found that a small percentage (4%) of genes expressed in late-stage oocytes are derepressed when miRNA function is lost. Noticeably, the proportion of genes regulated by RNAi in S. pombe and Drosophila seem to be markedly lower than that predicted for species of other phylogenetic branches. For instance, the fraction of genes regulated by miRNAs in human may reach 30% (36), although a more recent study has proposed that number to attain 92% (37). This apparent gain during the course of evolution is compatible with the hypothesis that the fission yeast RNAi machinery may represent the “ancestor” of the process known as the miRNA-guided RNA silencing in higher eukaryotes.
Among the proteins found to be regulated by Dcr1 are three constituents of the glycolysis pathway, i.e. HK2, PGK and ENO. Regulation of ENO protein expression was previously observed in dcr-1 Drosophila mutants upon comparative two-dimensional gel analysis (35). The authors noted a 34-fold increase in ENO protein level, with the mRNA transcript level remaining unchanged, in dcr-1 mutant versus wild-type oocytes. These findings are compatible with a direct regulatory mode involving miRNAs and suggest that miRNA-mediated regulation of glycolysis may be conserved between species.
However, the observed alterations of HK2, PGK and ENO protein expression levels did not translate into detectable effects on the growth of fission yeast cells. Considering that the absence of HK2 has been associated with doubling of the generation time in S. pombe (23), the relative absence of HK2 in the dcr1Δ strain was expected to lead to a prolongation of the generation time. Visualization of such an effect may have been hampered by the growth defect of the dcr1Δ strain inherent to the documented chromosomal segregation abnormalities (6). Although functional regulation of glycolysis by endogenous RNAi cannot be eliminated, our findings may also be interpreted such as changes in protein expression were not sufficient to alter glycolysis and/or reveal the existence of compensatory mechanisms for glucose metabolism, such as the Krebs cycle. It remains to be determined as to whether functional changes can be quantified at the biochemical level, for instance, through the measurement of key metabolic intermediates, i.e. the respective substrates and products of the regulated enzymes. Our data also suggests that raw proteomic data do not suffice to indicate the biological relevance of documented changes and that they should be interpreted with caution until functionally validated.
Considering the inhibitory role of small regulatory RNAs, genes that are subjected to a direct control by Dcr1-derived small RNAs would be expected to be upregulated in the dcr1Δ strain. That was the case for 6 of the 17 proteins species identified, including Hsp16, a putative chaperonine, thioredoxine peroxidase and PGK. Documentation of their corresponding mRNA levels unveiled the existence of two different gene regulatory mechanisms. There was a marked increase in the level of mRNAs encoding for Hsp16 and putative chaperonine in the dcr1Δ strain. The direct correlation between the protein and mRNA levels of these two genes is in accordance with a regulatory mechanism involving mRNA degradation, similar to that induced by siRNAs. Alternatively, this regulation may be strictly transcriptional and involve small RNA-directed methylation of histone H3 lysine-9 at the specific gene loci, as described previously (38).
In the case of PGK and thioredoxine peroxidase mRNA levels, they remained unchanged upon deletion of Dcr1. These data are in agreement with a post-transcriptional mechanism based on translational repression, reminiscent to that exerted by miRNAs in mammalian cells. Interestingly, two of these proteins, Hsp16 and thioredoxine peroxidase, are associated with specific conditions of cellular stress, such as heat shock and oxidative stress. These observations suggest that the fission yeast may utilize its RNAi machinery in order to modulate expression of specific genes that are required to rapidly adapt to a changing environment.
Among the 17 proteins found to be regulated by Dcr1, 11 of them were downregulated in the strain lacking Dcr1. The dcr1Δ-depleted proteins are presumably regulated one or more steps downstream of small RNA action, because their expression is up-regulated by the silencing machinery. The thiamine biosynthesis precursor protein Thi3, HK2 and ENO exemplify this class of proteins. In the case of Thi3 and ENO, their mRNA levels remain unaffected upon deletion of Dcr1, suggesting that their regulation by RNAi take place at the post-transcriptional level. This situation may emanate from the regulation of Thi3 and ENO protein expression by a repressor of translation, itself downregulated by Dcr1-derived small RNAs.
As for HK2, it is the only gene among the three to show a concurrent downregulation of its mRNA level, suggesting that its regulation by RNAi occurs at the transcriptional level. A plausible scenario would see HK2 expression be normally regulated by a transcriptional repressor, itself downregulated by Dcr1-derived small RNAs. In that context, it is relevant to note that a transcription factor important for the repression of HK2 was identified in S. cerevisiae. Deletion of Rgt1, a glucose sensing transcription factor, was associated with an 18-fold increase in HK2 transcript level (39). Regulation of a similar trancription factor in S. pombe could explain the relatively important change in expression when compared to the other gene targets (present in the WT strain, it is almost absent in Dcr1-lacking cells).
When interpreting the results of our proteomic and transcript analyses, the caveat has to be taken into account that the gene regulatory effects observed, which have been attributed to the loss of Dcr1-derived small RNAs, may also be related to the loss of the Dcr1 protein per se, i.e. independent of small RNAs, a possibility that cannot be dismissed.
Although the gene regulatory role of Dcr1 were first reported in 2002 (6), the cellular context in which Dcr1 functions in S. pombe remains unclear. On the premise that Dcr1 function may be governed by its interacting proteins, a picture that started to emerge from studies on the RNases III Drosha (40, 41) and Dicer (42, 43), we sought and identified L12 and Byr3 as Dcr1-interacting proteins. Although the functional relationship between these components warrants further studies, it may confer particular functionalities to Dcr1 in small RNA biosynthesis and action. For instance, by playing the role of a transcription factor, Byr3 may also contribute to attract Dcr1 in the vicinity of specific Byr3-regulated genes. The ability of Byr3 to bind to RNA could also make it a good candidate for substrate presentation to Dcr1 or small RNA transfer from Dcr1, or a Dcr1 complex, to an effector complex.
As for the ribosomal protein L12, it may help localize Dcr1 to ribosomes and bring it into close proximity with the translational machinery, where mRNAs being translated may be regulated by Dicer-derived small RNAs. A connection between gene silencing and translation has already been reported (44). Moreover, endogenous miRNAs have been reported to inhibit translation initiation in a cap-dependent manner in mammalian cells (45), further militating for that scenario.
In summary, our results support the existence of a post-transcriptional gene regulatory mechanism involving Dcr1 or a Dcr1 complex in fission yeast. We also identified the zinc finger protein Byr3 and the ribosomal protein L12 as novel Dcr1-interacting proteins that may contribute to the RNAi process by linking Dcr1 to the transcriptional and translational machineries. This study may provide the basis of future investigations addressing the exact role and nature of the Dcr1-Byr3-L12 tripartite relationship and verifying the transposability of our findings to other species, including human.
Acknowledgments
We thank Dr. Keith Gull for the generous gift of anti-TAT1 antibody, Dr. Louis Flamand for kindly providing anti-Myc antibody and Vicky Brochu for excellent technical assistance in two-dimensional gel electrophoresis and analysis. L.-A.G. was supported by a Master studentship from Natural Sciences and Engineering Research Council of Canada (NSERC). M.O. is a Burroughs Wellcome Fund Scholar in Molecular Parasitology and holder of a Canada Research Chair in Antimicrobial Resistance. P.P. is a New Investigator of the Canadian Institutes of Health Research (CIHR) and Junior 2 Scholar from the Fonds de la Recherche en Santé du Québec. This work was supported by discovery grant 262938-03 from NSERC.
Abbreviations
- CNBP
cellular nucleic acid binding protein
- Dcr1
Dicer 1
- dsRNA
double-stranded RNA
- EMMG
Edinburgh Minimal Medium Glutamate
- ENO
enolase
- HA
hemaglutinin
- IPG
immobilized pH gradient
- LC-MS-MS
liquid chromatography-mass spectrometry-mass spectrometry
- mRNA
messenger RNA
- microRNA
miRNA
- nt
nucleotide
- O.D
optical density
- PBS
phosphate buffered saline
- PCR
polymerase chain reaction
- PGK
phosphoglycerate kinase
- PTGS
post-transcriptional gene silencing
- RISC
RNA-induced silencing complex
- RITS
RNA-induced initiation of transcriptional silencing
- RNAi
RNA interference
- RNase
ribonuclease
- RT
reverse transcription
- siRNA
small interfering RNA
- WT
wild-type
- YES
yeast extract supplemented
- Δ
knockout
References
- 1.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409(6818):363–6. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 3.Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15(2):188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 5.Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S, Basham D, Bowman S, Brooks K, Brown D, Brown S, Chillingworth T, Churcher C, Collins M, Connor R, Cronin A, Davis P, Feltwell T, Fraser A, Gentles S, Goble A, Hamlin N, Harris D, Hidalgo J, Hodgson G, Holroyd S, Hornsby T, Howarth S, Huckle EJ, Hunt S, Jagels K, James K, Jones L, Jones M, Leather S, McDonald S, McLean J, Mooney P, Moule S, Mungall K, Murphy L, Niblett D, Odell C, Oliver K, O’Neil S, Pearson D, Quail MA, Rabbinowitsch E, Rutherford K, Rutter S, Saunders D, Seeger K, Sharp S, Skelton J, Simmonds M, Squares R, Squares S, Stevens K, Taylor K, Taylor RG, Tivey A, Walsh S, Warren T, Whitehead S, Woodward J, Volckaert G, Aert R, Robben J, Grymonprez B, Weltjens I, Vanstreels E, Rieger M, Schafer M, Muller-Auer S, Gabel C, Fuchs M, Dusterhoft A, Fritzc C, Holzer E, Moestl D, Hilbert H, Borzym K, Langer I, Beck A, Lehrach H, Reinhardt R, Pohl TM, Eger P, Zimmermann W, Wedler H, Wambutt R, Purnelle B, Goffeau A, Cadieu E, Dreano S, Gloux S, Lelaure V, Mottier S, Galibert F, Aves SJ, Xiang Z, Hunt C, Moore K, Hurst SM, Lucas M, Rochet M, Gaillardin C, Tallada VA, Garzon A, Thode G, Daga RR, Cruzado L, Jimenez J, Sanchez M, del Rey F, Benito J, Dominguez A, Revuelta JL, Moreno S, Armstrong J, Forsburg SL, Cerutti L, Lowe T, McCombie WR, Paulsen I, Potashkin J, Shpakovski GV, Ussery D, Barrell BG, Nurse P. The genome sequence of Schizosaccharomyces pombe. Nature. 2002;415(6874):871–80. doi: 10.1038/nature724. [DOI] [PubMed] [Google Scholar]
- 6.Provost P, Silverstein RA, Dishart D, Walfridsson J, Djupedal I, Kniola B, Wright A, Samuelsson B, Radmark O, Ekwall K. Dicer is required for chromosome segregation and gene silencing in fission yeast cells. Proc Natl Acad Sci U S A. 2002;99(26):16648–53. doi: 10.1073/pnas.212633199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reinhart BJ, Bartel DP. Small RNAs correspond to centromere heterochromatic repeats. Science. 2002;297(5588):1831. doi: 10.1126/science.1077183. [DOI] [PubMed] [Google Scholar]
- 8.Noma K, Sugiyama T, Cam H, Verdel A, Zofall M, Jia S, Moazed D, Grewal SI. RITS acts in cis to promote RNA interference-mediated transcriptional and post-transcriptional silencing. Nat Genet. 2004;36(11):1174–80. doi: 10.1038/ng1452. [DOI] [PubMed] [Google Scholar]
- 9.Carmichael JB, Stoica C, Parker H, McCaffery JM, Simmonds AJ, Hobman TC. RNA interference effector proteins localize to mobile cytoplasmic puncta in Schizosaccharomyces pombe. Traffic. 2006;7(8):1032–44. doi: 10.1111/j.1600-0854.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- 10.Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- 11.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 12.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68(5):850–8. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 13.Eng JK, McCormack AL, Yates JRI. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 14.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–67. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 15.Bernstein BE, Tong JK, Schreiber SL. Genomewide studies of histone deacetylase function in yeast. Proc Natl Acad Sci U S A. 2000;97(25):13708–13. doi: 10.1073/pnas.250477697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Craven RA, Griffiths DJ, Sheldrick KS, Randall RE, Hagan IM, Carr AM. Vectors for the expression of tagged proteins in Schizosaccharomyces pombe. Gene. 1998;221(1):59–68. doi: 10.1016/s0378-1119(98)00434-x. [DOI] [PubMed] [Google Scholar]
- 17.Woods A, Sherwin T, Sasse R, MacRae TH, Baines AJ, Gull K. Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies. J Cell Sci. 1989;93(Pt 3):491–500. doi: 10.1242/jcs.93.3.491. [DOI] [PubMed] [Google Scholar]
- 18.Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, 3rd, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14(10):943–51. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 19.Taricani L, Feilotter HE, Weaver C, Young PG. Expression of hsp16 in response to nucleotide depletion is regulated via the spc1 MAPK pathway in Schizosaccharomyces pombe. Nucleic Acids Res. 2001;29(14):3030–40. doi: 10.1093/nar/29.14.3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schweingruber AM, Dlugonski J, Edenharter E, Schweingruber ME. Thiamine in Schizosaccharomyces pombe: dephosphorylation, intracellular pool, biosynthesis and transport. Curr Genet. 1991;19(4):249–54. doi: 10.1007/BF00355050. [DOI] [PubMed] [Google Scholar]
- 21.Maundrell K. nmt1 of fission yeast. A highly transcribed gene completely repressed by thiamine. J Biol Chem. 1990;265(19):10857–64. [PubMed] [Google Scholar]
- 22.Kustrzeba-Wojcicka I, Golczak M. Enolase from Candida albicans--purification and characterization. Comp Biochem Physiol B Biochem Mol Biol. 2000;126(1):109–20. doi: 10.1016/s0305-0491(00)00169-3. [DOI] [PubMed] [Google Scholar]
- 23.Petit T, Blazquez MA, Gancedo C. Schizosaccharomyces pombe possesses an unusual and a conventional hexokinase: biochemical and molecular characterization of both hexokinases. FEBS Lett. 1996;378(2):185–9. doi: 10.1016/0014-5793(95)01451-9. [DOI] [PubMed] [Google Scholar]
- 24.Vivancos AP, Castillo EA, Biteau B, Nicot C, Ayte J, Toledano MB, Hidalgo E. A cysteine-sulfinic acid in peroxiredoxin regulates H2O2-sensing by the antioxidant Pap1 pathway. Proc Natl Acad Sci U S A. 2005;102(25):8875–80. doi: 10.1073/pnas.0503251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watson HC, Walker NP, Shaw PJ, Bryant TN, Wendell PL, Fothergill LA, Perkins RE, Conroy SC, Dobson MJ, Tuite MF, et al. Sequence and structure of yeast phosphoglycerate kinase. Embo J. 1982;1(12):1635–40. doi: 10.1002/j.1460-2075.1982.tb01366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu HP, Rajavashisth T, Grewal N, Jung V, Riggs M, Rodgers L, Wigler M. A gene encoding a protein with seven zinc finger domains acts on the sexual differentiation pathways of Schizosaccharomyces pombe. Mol Biol Cell. 1992;3(7):721–34. doi: 10.1091/mbc.3.7.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konicek BW, Xia X, Rajavashisth T, Harrington MA. Regulation of mouse colony-stimulating factor-1 gene promoter activity by AP1 and cellular nucleic acid-binding protein. DNA Cell Biol. 1998;17(9):799–809. doi: 10.1089/dna.1998.17.799. [DOI] [PubMed] [Google Scholar]
- 28.Pellizzoni L, Lotti F, Maras B, Pierandrei-Amaldi P. Cellular nucleic acid binding protein binds a conserved region of the 5′ UTR of Xenopus laevis ribosomal protein mRNAs. J Mol Biol. 1997;267(2):264–75. doi: 10.1006/jmbi.1996.0888. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki K, Olvera J, Wool IG. The primary structure of rat ribosomal protein L12. Biochem Biophys Res Commun. 1990;172(1):35–41. doi: 10.1016/s0006-291x(05)80169-x. [DOI] [PubMed] [Google Scholar]
- 30.Uchiumi T, Kominami R. Binding of mammalian ribosomal protein complex P0.P1.P2 and protein L12 to the GTPase-associated domain of 28 S ribosomal RNA and effect on the accessibility to anti-28 S RNA autoantibody. J Biol Chem. 1997;272(6):3302–8. doi: 10.1074/jbc.272.6.3302. [DOI] [PubMed] [Google Scholar]
- 31.Briones E, Briones C, Remacha M, Ballesta JP. The GTPase center protein L12 is required for correct ribosomal stalk assembly but not for Saccharomyces cerevisiae viability. J Biol Chem. 1998;273(48):31956–61. doi: 10.1074/jbc.273.48.31956. [DOI] [PubMed] [Google Scholar]
- 32.Chern MK, Chang KN, Liu LF, Tam TC, Liu YC, Liang YL, Tam MF. Yeast ribosomal protein L12 is a substrate of protein-arginine methyltransferase 2. J Biol Chem. 2002;277(18):15345–53. doi: 10.1074/jbc.M111379200. [DOI] [PubMed] [Google Scholar]
- 33.Porras-Yakushi TR, Whitelegge JP, Clarke S. A novel SET domain methyltransferase in yeast: Rkm2-dependent trimethylation of ribosomal protein L12ab at lysine 10. J Biol Chem. 2006;281(47):35835–45. doi: 10.1074/jbc.M606578200. [DOI] [PubMed] [Google Scholar]
- 34.Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, Moazed D. RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004;303(5658):672–6. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakahara K, Kim K, Sciulli C, Dowd SR, Minden JS, Carthew RW. Targets of microRNA regulation in the Drosophila oocyte proteome. Proc Natl Acad Sci U S A. 2005;102(34):12023–8. doi: 10.1073/pnas.0500053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 37.Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126(6):1203–17. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 38.Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002;297(5588):1833–7. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- 39.Palomino A, Herrero P, Moreno F. Rgt1, a glucose sensing transcription factor, is required for transcriptional repression of the HXK2 gene in Saccharomyces cerevisiae. Biochem J. 2005;388(Pt 2):697–703. doi: 10.1042/BJ20050160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432(7014):235–40. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 41.Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125(5):887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 42.Haase AD, Jaskiewicz L, Zhang H, Laine S, Sack R, Gatignol A, Filipowicz W. TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep. 2005;6(10):961–7. doi: 10.1038/sj.embor.7400509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436(7051):740–4. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kennerdell JR, Yamaguchi S, Carthew RW. RNAi is activated during Drosophila oocyte maturation in a manner dependent on aubergine and spindle-E. Genes Dev. 2002;16(15):1884–9. doi: 10.1101/gad.990802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309(5740):1573–6. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]