

Abstract
Functional somatostatin receptors (SSTR) are lost in human pancreatic cancer. Transfection of SSTR‐1 inhibited pancreatic cancer cell proliferation in vitro. We hypothesize that stable transfection of SSTR‐1 may inhibit pancreatic cancer growth in vivo possibly through cell cycle arrest. In this study, we examined the expression of SSTR‐1 mRNA in human pancreatic cancer tissue specimens, and investigated the effect of SSTR‐1 overexpression on cell proliferation, cell cycle, and tumor growth in a subcutaneous nude mouse model. We found that SSTR‐1 mRNA was downregulated in the majority of pancreatic cancer tissue specimens. Transfection of SSTR‐1 caused cell cycle arrest at the G0/G1 growth phase, with a corresponding decline of cells in the S (mitotic) phase. The overexpression of SSTR‐1 significantly inhibited subcutaneous tumor size by 71% and 43% (n = 5, P < 0.05, Student's t‐test), and inhibited tumor weight by 69% and 47% (n = 5, P < 0.05, Student's t‐test), in Panc‐SSTR‐1 and MIA‐SSTR‐1 groups, respectively, indicating the potent inhibitory effect of SSTR‐1 on pancreatic cancer growth. Our data demonstrate that overexpression of SSTR‐1 significantly inhibits pancreatic cancer growth possibly through cell cycle arrest. This study suggests that gene therapy with SSTR‐1 may be a potential adjuvant treatment for pancreatic cancer. (Cancer Sci 2008; 99: 2218–2223)
Pancreatic cancer has the number one fatality rate of all cancers, and is the fourth leading cause of cancer related deaths in North America. Overall 5‐year survival rate is less than 5%. The risk of developing ductal adenocarcinoma, which constitutes 95% of the pancreas neoplasms, increases with age, cigarette smoking, chronic pancreatitis and a family history of the disease.( 1 ) Unfortunately, there are no effective screening tests, and pancreatic cancer is typically diagnosed at an advanced stage. The vast majority of patients present with non‐specific symptoms, and diagnosis does not occur until the onset of jaundice and weight loss, signs of advanced disease. The only effective treatment of pancreatic cancer is surgical resection; however, 80% of pancreatic adenocarcinomas are unresectable at presentation. In addition, even the minority of patients who undergo resection, often develop local recurrence and die within 15 months of diagnosis.( 2 , 3 ) Clearly, there is a need to understand more about the mechanism of pancreatic cancer and to develop more effective treatments for this deadly disease.
A small cyclic neuropeptide called somatostatin, and its analogs such as octreotide, lantreotide and vapreotide, have been used for treating pancreatic carcinoma in preclinical trials as adjuvants because of their inhibitory effects in a variety of biological processes including cell proliferation and growth hormone release.( 4 , 5 ) Somatostatin interacts with five different types of somatostatin receptors (SSTR) at different affinities to transduce the signals inside the cells. SSTR are G‐protein‐coupled receptors, and the five SSTR play different physiological roles and function via different signaling pathways. Although there are five SSTR subtypes, studies with CHO cells expressing each SSTR subtype reveal a predominant role for SSTR‐1 and SSTR‐2 in mediating the antiproliferative effect.( 4 ) SSTR‐1 activation inhibits hormone secretion and cell viability, and inhibits adenylyl cyclase activity when expressed in CHO‐K1 cells.( 6 , 7 ) SSTR‐2 is the most abundant SSTR subtype detected in many tissues, and most clinically used somatostatin analogs preferentially bind to SSTR‐2, which plays a major role in regulating hormone secretion, apoptosis and cell proliferation in pancreatic cancer. Studies using SSTR‐2 stably transfected cell lines showed significant inhibition of cell growth and induction of apoptosis by SSTR‐2.( 8 , 9 , 10 ) Inhibition of tumor growth in vivo by overexpressed SSTR‐2 was also observed in a nude mouse model.( 11 ) Our study also indicated that after co‐transfection of SSTR‐1 and ‐2 genes in Panc‐1 cells, somatostatin analog treatment augmented the growth inhibitory effect in a dose‐dependent manner beyond that seen with individual SSTR gene transfer alone.( 12 )
Despite promising cell culture and animal studies, clinical trials of somatostatin analogs in the adjuvant treatment of advanced pancreatic cancer have failed to demonstrate a response.( 13 , 14 , 15 , 16 ) Presence of SSTR was not examined in these clinical trials. However, the published work suggests that the reason animal and cell culture studies have shown a response to somatostatin but clinical trials have failed, is a loss of functional SSTR in most human pancreatic cancers. This indicates that increasing the concentration of SSTR on human pancreatic adenocarcinoma cells may render pancreatic cancers vulnerable to growth inhibition, and represent a novel gene therapy strategy to treat pancreatic cancer. Most of the previous studies are focused on SSTR‐2, and little is known about the role of SSTR‐1 in pancreatic cancer suppression. In the present study, we transfected SSTR‐1 gene into Panc‐1 cells, and assessed the cell cycle after the transfection, and also investigated the inhibitory effect of SSTR‐1 in a nude mouse model with subcutaneous xenografts.
Materials and Methods
Chemicals and reagents. Rabbit anti‐SSTR‐1 antibody was purchased from Novus Biologicals (Littleton, CO, USA), and other chemicals were purchased from Sigma (St Louis, MO, USA). Fluorescein isothiocyanate‐conjugate secondary antirabbit immunoglobulin G was purchased from Vector Lab (Burlingame, CA, USA). RNAqueous‐4PCR kit and DNAse I removing reagent were purchased from Ambion (Austin, TX, USA). iQ SYBR Green supermix, and reverse transcription kit were obtained from Bio‐Rad (Hercules, CA, USA). MTS reagent was purchased from Promega (Madison, WI, USA).
Cells and human tissue specimens. Human pancreatic cancer cell lines, Panc‐1 and MIA PaCa‐2, were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). All cells were cultured as previously described.( 17 , 18 ) Human pancreatic adenocarcinoma specimens were collected from patients who underwent surgery according to an approved human protocol at Baylor College of Medicine (Houston, TX, USA).
RNA extraction. Total RNA was extracted from Panc‐1 and MIA Paca‐2 cells and pancreatic cancer tissue specimens, using an Ambion RNAqueous‐4PCR kit following the manufacture's instructions. Briefly, cells or homogenized tissues were lyzed by Ambion lysis buffer for 20 min and the lysates were transferred to an Ambion mini‐column, and centrifuged at 10 000 g for 1 min. The column was washed three times. After incubation with 50 µL of elution buffer, the flow‐through was collected using a new tube. The RNA solution was treated with DNAse I to remove the trace amount of genomic DNA contamination by using an Ambion DNA removing kit. DNAse I (1 µL) was added to 20 µL of RNA solution with proper DNAse I buffer, and incubated at 37°C for 2 h. The DNAse I was removed by adding 0.1–0.2 volume of DNAse removing agent, and the purified RNA was collected by centrifugation at 10 000 g for 1 min.
Primer design and real‐time reverse transcription polymerase chain reaction. Specific primers for the SSTR‐1 gene were designed using Beacon Designer ver. 5.0 software (PREMIER Biosoft International, Palo Alto, CA, USA) as described previously.( 18 , 19 ) The primer sequences for the human SSTR‐1 gene are as following: 5′‐GGCGAAATGCGTCCCAG‐3′ sense and 5′‐CGGAGTA GATGAAAGAGATCAGGA‐3′ antisense. The mRNA levels for SSTR‐1 were analyzed by real‐time reverse transcription polymerase chain reaction (RT‐PCR) using a Bio‐Rad iCycler system (Bio‐Rad). The mRNA were reverse‐transcribed into cDNA using an iScript cDNA synthesis kit. The primer specificity was tested by running a regular PCR for 40 cycles at 95°C for 20 s and 60°C for 1 min, and followed by an agarose gel electrophoresis. The real‐time PCR was performed by using a SYBR supermix kit, and running for 40 cycles at 95°C for 20 s and 60°C for 1 min. Each cDNA sample was run in triplicate and the corresponding non‐reverse transcriptase (non‐RT) mRNA sample was included as a negative control. The β‐actin primer was included in every plate to avoid sample variations. The mRNA level of each sample for each gene was normalized to that of the β‐actin mRNA. Relative mRNA level was presented as 2^(Ct[β‐actin] – Ct[gene of interest]). All data shown were the mean ± standard deviation of three separate experiments.
Cell cycle analysis. Panc‐1 cells transfected with SSTR‐1 gene or vector control were serum starved for 24 h in serum‐free medium and then released using medium with 2% serum. Cells were then collected by trypsinization, and processed using the CycleTEST PLUS DNA Reagent Kit (Beckton Dickinson, San Jose, CA, USA) according to the manufacturer's instructions. Briefly, after trypsinization the cells were centrifuged, and cells were washed in a buffer containing sodium citrate, sucrose and dimethylsulfoxide. Cells were then incubated sequentially for 10 min each in solution A (containing trypsin in a spermine tetrahydrochloride detergent buffer for the enzymatic digestion of cell membranes and cytoskeletons), solution B (containing trypsin inhibitor and ribonuclease A in citrate‐stabilizing buffer with spermine tetrahydrochloride to inhibit the trypsin activity and to digest the RNA) and solution C (containing propidium iodide [PI] and spermine tetrahydrochloride in citrate stabilizing buffer for the stoichiometric binding of PI to the DNA at a final concentration of 125 µg/mL). Flow cytometry analysis was carried out to examine the cell cycle distribution in a Beckton Dickinson FACSCalibur analyzer (Becton Dickinson). Data was further analyzed using the software FLOWJOW ver. 6.1.1 (Tree Star, San Carlos, CA, USA) with the ‘Watson pragmatic model’.
Stable cell line selection. SSTR‐1 overexpression cells were selected in Panc‐1 and MIA PaCa‐2 cells, respectively, with retrovirus vector pBabe (Clontech, Mountain View, CA, USA), following manufacturer's instructions as described previously.( 20 , 21 ) Briefly, full length human SSTR‐1 cDNA was cloned into pBabe vector, and the recombinant plasmid was co‐transfected into 293T cells with plasmid PegPam3 and RDF. Viral supernatants were collected and transduced to the target cells. Stable cell lines were selected by adding 0.5–1 µg/mL puromysin. The overexpression of SSTR‐1 in the stable cell lines was confirmed by real‐time RT‐PCR.
Cell proliferation assay. Cell proliferation was analyzed with the MTS assay. Stable Panc‐1 or MIA PaCa‐2 cells were seeded in 96‐well plates (2 × 103 cells/well), and serum‐starved (0% fetal bovine serum) for 24 h. Cell growth was assessed at 2 days after starvation. MTS reagent (20 µL) mixed with 100 µL growth medium was added to each well, and incubated in 37°C for 2 h. Absorbance was recorded at 490 nm with an EL‐800 universal microplate reader (Bio‐Tek Instruments, Winooski, VT, USA).
Subcutaneous pancreatic cancer mouse models. Subconfluent stable pancreatic cancer cells with SSTR‐1 overexpression were harvested by trypsinization, and resuspended in Dulbecco's modified Eagle's medium. A quantity of 3 × 106 cells were inoculated into the right flank of 5–6‐week‐old male nude mice (NCI‐Charles River) as described previously.( 20 , 21 ) The tumor size was measured weekly using a digital caliper (VWR International, Marietta, GA, USA) and the tumor volume was determined with the formula: tumor volume (mm3) = (length [mm]) × (width [mm])2 × 0.52. The tumor weight was measured at week 6. The animals were euthanized when their tumor size reached 2 cm in diameter or the animals became moribund during the observation period, and the time they were killed was recorded as the time of mortality.
Statistical analysis. Quantitative results are shown as means ± standard deviations. The statistical analysis was performed by Student's t‐test for paired data between control and treated groups or one‐way anova for data from multiple groups. P < 0.05 was considered significant.
Results
SSTR‐1 is downregulated in human pancreatic cancer tissue specimens. SSTR‐1 expression was found to be decreased in pancreatic cancer. To confirm the expression of SSTR‐1 in human pancreatic cancer tissues, we examined SSTR‐1 expression in a large number of human pancreatic cancer tissues with the surrounding normal tissues (n = 26). SSTR‐1 mRNA was substantially decreased in 18 of 26 (70%) clinical pancreatic‐adenocarcinoma samples compared with that in their surrounding normal tissues (Fig. 1). Overall, the average mRNA expression in 26 pancreatic cancer tissues was 61% decreased compared with that in normal tissues. Therefore, decreased expression of SSTR‐1 in the majority of pancreatic cancer tissue specimens suggests that suppressing SSTR‐1 may contribute to pancreatic cancer growth.
Figure 1.
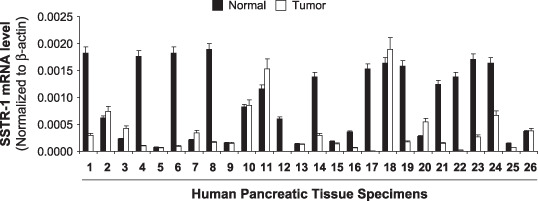
Somatostatin receptor (SSTR)‐1 expression in human pancreatic cancer tissue specimens. Total RNA was extracted from clinical human pancreatic cancer tissue specimens and the mRNA levels for SSTR‐1 were analyzed by real‐time polymerase chain reaction, and normalized to that of the house keeping gene, β‐actin. All data shown are the mean ± standard deviation of three separate experiments.
Transfection of SSTR‐1 caused cell cycle arrest at G0/G1 phase in Panc‐1 cells. Our previous results and other studies have shown that transfer of SSTR‐1 or ‐2 subtype genes inhibited Panc‐1 cell proliferation, indicating a possible role of SSTR genes as tumor suppressors.( 11 , 12 , 22 , 23 ) In the current study, we further examined the effect of SSTR‐1 in cell cycle control of pancreatic cancer cells. We transfected the SSTR‐1 gene into Panc1 cells, and confirmed the expression of SSTR‐1 proteins by flow cytometry. As shown in Fig. 2, only 0.61% of cells showed positive staining for SSTR‐1 in mock‐transfected Panc‐1 cells. After transfection of the SSTR‐1 gene, the expression of SSTR‐1 protein was significantly upregulated in Panc‐1 cells, 22.16% of the cells were stained positive for SSTR‐1 (Fig. 2).
Figure 2.
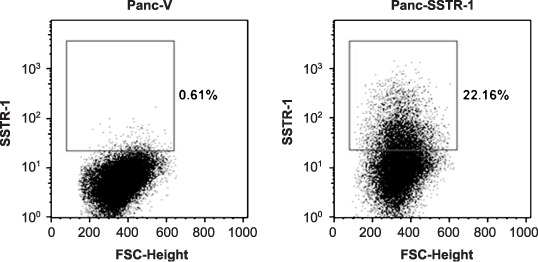
Flow cytometry analysis of somatostatin receptor (SSTR)‐1 in Panc‐1 cells upon transfection. Panc‐1 cells transfected with the SSTR‐1 gene were incubated with anti‐SSTR‐1 antibody. Cells were then stained with fluorescein isothiocyanate‐conjugated secondary antibody. Data were collected by a BD Caliber flow cytometer, and analyzed by FLOWJO software.
To assess the cell cycle in Panc‐1 cells, mock‐transfected and SSTR‐1‐transfected Panc‐1 cells were harvested, stained with PI and subjected to fluorescent‐activated cell sorting analysis. We found that upregulation of SSTR‐1 induced a G0/G1 growth phase arrest in Panc‐SSTR‐1 cells, indicated by accumulation of cells at this stage, with a corresponding decline of cells in the S (mitotic) phase. In Panc‐V cells, 40.6% of cells were in G0/G1 phase and 32.2% were in S phase. On the contrary, 44.8% of Panc‐SSTR‐1 cells were in G0/G1 phase, and only 27.9% of cells entered S phase. That is 10% increased cells population arrested at G0/G1 phase, and 13% decreased cells entering S phase, respectively (Fig. 3 and Table 1). Thus, upregulation of SSTR‐1 in Panc‐1 cells is associated with cell cycle arrest at G0/G1 phase.
Figure 3.
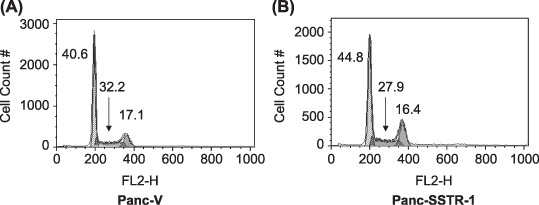
Transfection of somatostatin receptor (SSTR)‐1 caused cell cycle arrest at G0/G1 phase in Panc‐1 cells. Mock transfected (A), or SSTR‐1 transfected (B) Panc‐1 cells were starved and released by growth medium. Cells were stained with propidium iodide, and cell cycle arrest was indicated by accumulation of cells at G0/G1 phase, with a corresponding decline in cells in the S (mitotic) phase.
Table 1.
Cell cycle analysis of somatostatin receptor (SSTR)‐1 transfected Panc‐1 cells
| Sample | RMS † | %G0/G1 | %S | %G2/M |
|---|---|---|---|---|
| Panc‐V | 3.65 | 40.6 | 32.2 | 17.1 |
| Panc‐SSTR‐1 | 4.53 | 44.8 | 27.9 | 16.4 |
RMS, root mean squared, error of the fit, indicates how well the model used fits the data. Lower RMS values indicate a better model fit.
Stable overexpression of SSTR‐1 decreased the proliferation of pancreatic cancer cells. To study the potential functions of SSTR‐1 in pancreatic cancer, stably overexpressing SSTR‐1 cell lines were established in Panc‐1 (Panc‐SSTR‐1) and MIA PaCa‐2 cells (MIA‐SSTR‐1) using a retrovirus vector (pBabe, Clontech). Stable cells containing empty vectors (Panc‐V and MIA‐V) cells were also established in Panc‐1 and MIA PaCa‐2 cells as controls. Overexpression of SSTR‐1 in the stable Panc‐SSTR‐1 and MIA‐SSTR‐1 cells were confirmed and compared with Panc‐V and MIA‐V controls by real‐time RT‐PCR. SSTR‐1 mRNA was overexpressed for 125.8‐fold in Panc‐SSTR‐1 cells and 445.7‐fold in MIA‐SSTR‐1 cells compared with their corresponding vector control cells (P < 0.01, Fig. 4A,B). MTS assay showed that overexpression of SSTR‐1 in Panc‐SSTR‐1 and MIA‐SSTR‐1 cells were associated with decreased cell proliferation by 41% and 20%, respectively, compared with that in Panc‐V and MIA‐V cells (P < 0.05, Fig. 5A,B). These results indicate that SSTR‐1 plays an important role in regulating cell proliferation in these pancreatic cells.
Figure 4.
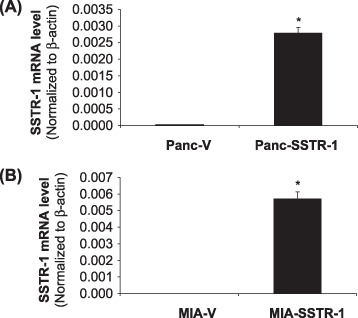
Expression of somatostatin receptor (SSTR)‐1 in stable Panc‐1 and MIA PaCa‐2 cells. Stably overexpressing SSTR‐1 cells were established in Panc‐1 and MIA PaCa‐2 cells. The mRNA levels of SSTR‐1 in Panc‐1 (A), and MIA PaCa‐2 (B) cells were examined with real‐time reverse transcription polymerase chain reaction. Human SSTR‐1 mRNA levels were normalized to that of human β‐actin. SSTR‐1 mRNA levels in Panc‐SSTR‐1 and MIA‐SSTR‐1 cells were significantly higher than that in Panc‐V and MIA‐V cells. *P < 0.01. All data shown are the mean ± standard deviation of three separate experiments.
Figure 5.
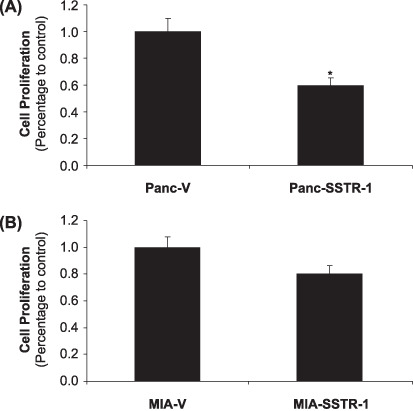
Stable overexpression of somatostatin receptor (SSTR)‐1 decreased the proliferation of pancreatic cancer cells. (A) Panc‐SSTR‐1, (B) MIA‐SSTR‐1, and their corresponding vector control cells were seeded in 96‐well plates (2 × 103 cells/well), and serum‐starved for 24 h before examining the cell proliferation. Absorbance at 490 nm was recorded on day 2 after starvation. Data were expressed as the mean ± standard deviation of triplicate values. *P < 0.05.
SSTR‐1 inhibited pancreatic cancer growth in the nude mouse model of subcutaneous xenograft. We further analyzed the role of SSTR‐1 on tumor growth in vivo using an immunodeficient nude mouse model. Panc‐SSTR‐1 cells showed a dramatic decrease (71%) in tumor volume after 6 weeks compared with Panc‐V control cells in the subcutaneous tumor model (P < 0.01, Fig. 6A). MIA‐SSTR‐1 cells also showed significantly decreased tumor volume by 43% after 6 weeks compared with MIA‐V control cells in the subcutaneous tumor model (P < 0.05, Fig. 6B). The tumor weight was also significantly reduced in Panc‐SSTR‐1 and MIA‐SSTR‐1 groups by 69% and 47%, respectively, compared with the vector control groups (P < 0.05, Fig. 6C,D). These results indicate that overexpression of SSTR‐1 inhibits pancreatic cancer progression.
Figure 6.
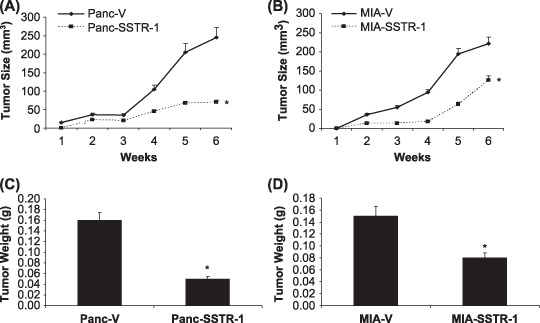
Somatostatin receptor (SSTR)‐1 inhibited pancreatic cancer growth in the nude mouse model of subcutaneous xenograft. (A) Panc‐SSTR‐1, (B) MIA‐SSTR‐1, and their corresponding vector control cells (3 × 106) were subcutaneously inoculated into the right flank of nude mice (n = 5/treatment group). Tumor size was measured weekly for 6 weeks. Tumor volume was calculated by the formula: tumor volume (mm3) = (length [mm]) × (width [mm])2 × 0.52. *P < 0.05. Tumor weight from (C) Panc‐SSTR‐1, (D) MIA‐SSTR‐1 group, and their corresponding vector control groups were also measured at week 6. *P < 0.05.
Discussion
This study revealed that SSTR‐1 may inhibit pancreatic cancer growth by causing cell cycle arrest. We found that SSTR‐1 mRNA was downregulated in the majority of human pancreatic cancer tissues specimens, and transfection of SSTR‐1 gene caused Panc‐1 cell cycle arrest at G0/G1 phase. Overexpression of SSTR‐1 in stable Panc‐SSTR‐1 or MIA‐SSTR‐1 cells led to decreased cell proliferation in vitro and tumor growth in vivo in a subcutaneous nude mice model, indicating the potent inhibitory effect of SSTR‐1 on pancreatic cancer growth.
A fundamental obstacle in our understanding of pancreatic carcinogenesis is the confusion on the histogenesis of this disease. Many investigators think pancreatic cancer arises from pancreatic ductal cells because the vast majority of tumors are ductal adenocarcinomas. Others believe these tumors arise from transdifferentiated acinar cells( 24 ) or from precursor (reserve stem) cells that are distributed along the ductal trees, but are particularly plentiful within islets.( 25 , 26 ) This controversy complicates investigation of the role of SSTR in pancreatic carcinogenesis because the cell of origin cannot be isolated and studied. Important insights are gained by studying the progressive development of precursor lesions and invasive cancers in animal models of pancreatic cancer. Human islet cells express all five SSTR.( 27 , 28 , 29 ) There is no absolute specificity of any SSTR for an islet cell type. Outside the islet, cells expressing SSTR‐2 have been identified in the walls of many small and medium arterioles and scattered non‐islet cells express SSTR‐1 and ‐2.( 29 ) The SSTR status of precursor reserve stem cells in the human pancreas is unknown. In hamsters treated with a pancreatic carcinogen, there is a progressive decrease in SSTR expression, as measured by autoradiography, in normal pancreas, pre‐neoplastic lesions and invasive cancers.( 30 ) In our current study, we found that the SSTR‐1 gene was downregulated in the majority of the pancreatic cancer tissue specimens. Presumably, evolving cancer cells within or near islets would be exposed to a high concentration of islet peptide products, including somatostatin. Loss of SSTR may result in loss of a check on cell proliferation and cell cycle, analogous to loss of other tumor suppressor genes. Multiple tumor suppressor pathways such as Rb/p16, transforming growth factor‐β/DPC4 and p53, are known to be abrogated in most human pancreatic carcinomas.( 31 , 32 ) We hypothesize that SSTR gene expression is another tumor suppressor pathway important in human pancreatic carcinogenesis.
Somatostatin causes antiproliferative effects through SSTR by at least two receptor‐dependent mechanisms. One is by inhibition of mitogenic signaling of growth factor receptor kinases causing growth retardation due to arrest of cell cycle progression. Second, morphological and flow cytometric studies have confirmed the induction of apoptotic cell death. Activation of SSTR‐1 and ‐2 results in induction of the retinoblastoma tumor suppressor protein Rb, p21 and G1 cell cycle arrest.( 33 ) Another group has shown that binding of somatostatin to SSTR‐2 upregulates p27, thus leading to cell cycle arrest in the G0‐G1 phase, and subsequently to apoptosis. In our study, we found that transfection of the SSTR‐1 gene caused cell cycle arrest at the G0‐G1 phase, along with a corresponding decline of cells in the S phase. Although many studies have been done to investigate the mechanism of the growth inhibition by SSTR‐1 and SSTR‐2, the most promising pathway seems to involve SHP1/2, ERK1/2 and p27/p21 signaling. It has been shown that mouse SSTR‐2 interacts with SHP‐1/2, and stimulates ERK1/2 activation through a Ras, Rap‐1 and B‐Raf‐dependent signal pathway. Activated ERK1/2 leads to induction of p27kip1 inhibition of cyclin E‐cdk2 kinase activity and cell cycle arrest.( 23 , 34 , 35 , 36 , 37 ) A similar pathway has been indicated in SSTR‐1 expressing CHO‐K1 cells.( 37 , 38 , 39 ) Upon ligand stimulation, Gβγ‐dependent Src activation leads to SHP‐2 activation and subsequent SHP‐1 recruitment. PI3K is expected to play a role in this pathway as well.
Forced overexpression of SSTR have been shown to inhibit pancreatic cancer progression. Growth of two human pancreatic cancer cell lines, BxPC‐3 and Capan‐1 cells, was inhibited after transfection with SSTR‐2 even without the addition of somatostatin to the growth medium.( 40 ) In addition to in vitro studies, the growth of pancreatic cancer cells stably transfected with SSTR‐2 is decreased in athymic mice.( 10 ) Delivery of a SSTR‐2 transgene by direct injection of adenoviral vector or linear polyethylenimine (PEI) vector into established tumors resulted in significant reduction of pancreatic tumor growth, indicating the feasibility of using SSTR as a novel gene therapy strategy.( 41 ) We have shown that transfection of Panc‐1 cells with SSTR‐1 or SSTR‐2 inhibits cell proliferation in culture.( 11 , 12 , 22 ) However, no reports have indicated the role of SSTR‐1 in inhibiting pancreatic cancer progression in vivo. In this current study, we have demonstrated a significant regression of tumor growth by SSTR‐1 in a subcutaneous nude mouse model. Overexpression of SSTR‐1 in two pancreatic cancer cell lines, Panc‐1 and MIA PaCa‐2, significantly inhibited tumor growth. Further studies are warranted to optimize the delivery efficiency and expression specificity of SSTR‐1 in pancreas.
Taken as a whole, our study suggested a potent inhibitory effect of SSTR‐1 in pancreatic cancer cells, which may serve as potential therapeutic targets for treatment of pancreatic cancer. Given the poor prognosis of patients with pancreatic cancer and the lack of effective therapy, a better understanding of the pathogenesis of pancreas cancer and any novel treatment approach that has translational potential would be of medical and economic importance. This unique approach of using SSTR as a therapeutic target is particularly attractive because of the low toxicity of the naturally occurring peptide, somatostatin, which may allow combination of this antiproliferative signal with standard chemotherapy without synergistic toxicity. With this novel approach, expression of the transgene, SSTR, could also be clinically tracked or used to deliver adjuvant tumor‐specific radiation.
Acknowledgments
This work was supported in part by the American Cancer Society Grant (no. IRG‐93‐034‐09 M. L.), National Institutes of Health (NIH) Grants NIH EB002436, HL08347 (C. C.), RO1 DE15543 and R21 AT003094 (Q. Y.), and the MacDonald Research Fund 05RDM009 (W. E. F.).
References
- 1. Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet 2004; 363: 1049–57. [DOI] [PubMed] [Google Scholar]
- 2. Warshaw AL, Fernandez‐del Castillo C. Pancreatic carcinoma. N Engl J Med 1992; 326: 455–65. [DOI] [PubMed] [Google Scholar]
- 3. Fisher WE, Berger DH. Angiogenesis and antiangiogenic strategies in pancreatic cancer. Int J Gastrointest Cancer 2003; 33: 79–88. [DOI] [PubMed] [Google Scholar]
- 4. Bousquet C, Puente E, Buscail L, Vaysse N, Susini C. Antiproliferative effect of somatostatin and analogs. Chemotherapy 2001; 47 (Suppl 2): 30–9. [DOI] [PubMed] [Google Scholar]
- 5. Hejna M, Schmidinger M, Raderer M. The clinical role of somatostatin analogues as antineoplastic agents: much ado about nothing? Ann Oncol 2002; 13: 653–68. [DOI] [PubMed] [Google Scholar]
- 6. Hershberger RE, Newman BL, Florio T et al . The somatostatin receptors SSTR1 and SSTR2 are coupled to inhibition of adenylyl cyclase in Chinese hamster ovary cells via pertussis toxin‐sensitive pathways. Endocrinology 1994; 134: 1277–85. [DOI] [PubMed] [Google Scholar]
- 7. Zatelli MC, Piccin D, Tagliati F et al . Somatostatin receptor subtype 1 selective activation in human growth hormone (GH)‐ and prolactin (PRL)‐secreting pituitary adenomas: effects on cell viability, GH, and PRL secretion. J Clin Endocrinol Metab 2003; 88: 2797–802. [DOI] [PubMed] [Google Scholar]
- 8. Kikutsuji T, Harada M, Tashiro S et al . Expression of somatostatin receptor subtypes and growth inhibition in human exocrine pancreatic cancers. J Hepatobiliary Pancreat Surg 2000; 7: 496–503. [DOI] [PubMed] [Google Scholar]
- 9. Guillermet J, Saint‐Laurent N, Rochaix P et al . Somatostatin receptor subtype 2 sensitizes human pancreatic cancer cells to death ligand‐induced apoptosis. Proc Natl Acad Sci USA 2003; 100: 155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rochaix P, Delesque N, Esteve JP et al . Gene therapy for pancreatic carcinoma: local and distant antitumor effects after somatostatin receptor sst2 gene transfer. Hum Gene Ther 1999; 10: 995–1008. [DOI] [PubMed] [Google Scholar]
- 11. Celinski SA, Fisher WE, Amaya F et al . Somatostatin receptor gene transfer inhibits established pancreatic cancer xenografts. J Surg Res 2003; 115: 41–7. [DOI] [PubMed] [Google Scholar]
- 12. Li M, Zhang R, Li F et al . Transfection of SSTR‐1 and SSTR‐2 inhibits Panc‐1 cell proliferation and renders Panc‐1 cells responsive to somatostatin analogue. J Am Coll Surg 2005; 201: 571–8. [DOI] [PubMed] [Google Scholar]
- 13. Klijn JG, Hoff AM, Planting AS et al . Treatment of patients with metastatic pancreatic and gastrointestinal tumours with the somatostatin analogue Sandostatin: a phase II study including endocrine effects. Br J Cancer 1990; 62: 627–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Canobbio L, Boccardo F, Cannata D, Gallotti P, Epis R. Treatment of advanced pancreatic carcinoma with the somatostatin analogue BIM 23014. Preliminary results of a pilot study. Cancer 1992; 69: 648–50. [DOI] [PubMed] [Google Scholar]
- 15. Huguier M, Samama G, Testart J et al . Treatment of adenocarcinoma of the pancreas with somatostatin and gonadoliberin (luteinizing hormone‐releasing hormone). The French Associations for Surgical Research. Am J Surg 1992; 164: 348–53. [DOI] [PubMed] [Google Scholar]
- 16. Raderer M, Hamilton G, Kurtaran A et al . Treatment of advanced pancreatic cancer with the long‐acting somatostatin analogue lanreotide: in vitro and in vivo results. Br J Cancer 1999; 79: 535–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li M, Yang H, Chai H et al . Pancreatic carcinoma cells express neuropilins and vascular endothelial growth factor, but not vascular endothelial growth factor receptors. Cancer 2004; 101: 2341–50. [DOI] [PubMed] [Google Scholar]
- 18. Li M, Zhai Q, Bharadwaj U et al . Cyclophilin A is overexpressed in human pancreatic cancer cells and stimulates cell proliferation through CD147. Cancer 2006; 106: 2284–94. [DOI] [PubMed] [Google Scholar]
- 19. Li M, Li W, Kim HJ, Yao Q, Chen C, Fisher WE. Characterization of somatostatin receptor expression in human pancreatic cancer using real‐time RT‐PCR. J Surg Res 2004; 119: 130–7. [DOI] [PubMed] [Google Scholar]
- 20. Li M, Zhang Y, Liu Z et al . Aberrant expression of zinc transporter ZIP4 (SLC39A4) significantly contributes to human pancreatic cancer pathogenesis and progression. Proc Natl Acad Sci USA 2007; 104: 18 636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li M, Bharadwaj U, Zhang R et al . Mesothelin is a malignant factor and therapeutic vaccine target for pancreatic cancer. Mol Cancer Ther 2008; 7: 286–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fisher WE, Wu Y, Amaya F, Berger DH. Somatostatin receptor subtype 2 gene therapy inhibits pancreatic cancer in vitro . J Surg Res 2002; 105: 58–64. [DOI] [PubMed] [Google Scholar]
- 23. Lahlou H, Saint‐Laurent N, Esteve JP et al . sst2 Somatostatin receptor inhibits cell proliferation through Ras‐, Rap1‐, and B‐Raf‐dependent ERK2 activation. J Biol Chem 2003; 278: 39 356–71. [DOI] [PubMed] [Google Scholar]
- 24. Longnecker DS, Shinozuka H, Dekker A. Focal acinar cell dysplasia in human pancreas. Cancer 1980; 45: 534–40. [DOI] [PubMed] [Google Scholar]
- 25. Pour PM, Schmied B. The link between exocrine pancreatic cancer and the endocrine pancreas. Int J Pancreatol 1999; 25: 77–87. [DOI] [PubMed] [Google Scholar]
- 26. Pour PM, Pandey KK, Batra SK. What is the origin of pancreatic adenocarcinoma? Mol Cancer 2003; 2: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nehete PN, Lewis DE, Tang DN, Pollack MS, Sastry KJ. Presence of HLA‐C‐restricted cytotoxic T‐lymphocyte responses in long‐term nonprogressors infected with human immunodeficiency virus. Viral Immunol 1998; 11: 119–29. [DOI] [PubMed] [Google Scholar]
- 28. Reubi JC, Kappeler A, Waser B, Schonbrunn A, Laissue J. Immunohistochemical localization of somatostatin receptor sst2A in human pancreatic islets. J Clin Endocrinol Metab 1998; 83: 3746–9. [DOI] [PubMed] [Google Scholar]
- 29. Kumar U, Sasi R, Suresh S et al . Subtype‐selective expression of the five somatostatin receptors (hSSTR1‐5) in human pancreatic islet cells: a quantitative double‐label immunohistochemical analysis. Diabetes 1999; 48: 77–85. [DOI] [PubMed] [Google Scholar]
- 30. Tang C, Biemond I, Appel MJ, Visser CJ, Woutersen RA, Lamers CB. Expression of receptors for gut peptides in pancreata of BOP‐treated and control hamsters. Carcinogenesis 1996; 17: 2171–5. [DOI] [PubMed] [Google Scholar]
- 31. Caldas C, Hahn SA, da Costa LT et al . Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet 1994; 8: 27–32. [DOI] [PubMed] [Google Scholar]
- 32. Goggins M, Kern SE, Offerhaus JA, Hruban RH. Progress in cancer genetics: lessons from pancreatic cancer. Ann Oncol 1999; 10 (Suppl 4): 4–8. [PubMed] [Google Scholar]
- 33. Kar R, Sen S, Singh A et al . Role of apoptotic regulators in human epithelial ovarian cancer. Cancer Biol Ther 2007; 6. [DOI] [PubMed] [Google Scholar]
- 34. Lopez F, Ferjoux G, Cordelier P et al . Neuronal nitric oxide synthase: a substrate for SHP‐1 involved in sst2 somatostatin receptor growth inhibitory signaling. Faseb J 2001; 15: 2300–2. [DOI] [PubMed] [Google Scholar]
- 35. Lopez F, Esteve JP, Buscail L et al . The tyrosine phosphatase SHP‐1 associates with the sst2 somatostatin receptor and is an essential component of sst2‐mediated inhibitory growth signaling. J Biol Chem 1997; 272: 24 448–54. [DOI] [PubMed] [Google Scholar]
- 36. Bousquet C, Delesque N, Lopez F et al . sst2 somatostatin receptor mediates negative regulation of insulin receptor signaling through the tyrosine phosphatase SHP‐1. J Biol Chem 1998; 273: 7099–106. [DOI] [PubMed] [Google Scholar]
- 37. Ferjoux G, Lopez F, Esteve JP et al . Critical role of Src and SHP‐2 in sst2 somatostatin receptor‐mediated activation of SHP‐1 and inhibition of cell proliferation. Mol Biol Cell 2003; 14: 3911–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Florio T, Yao H, Carey KD, Dillon TJ, Stork PJ. Somatostatin activation of mitogen‐activated protein kinase via somatostatin receptor 1 (SSTR1). Mol Endocrinol 1999; 13: 24–37. [DOI] [PubMed] [Google Scholar]
- 39. Florio T, Thellung S, Arena S et al . Somatostatin receptor 1 (SSTR1)‐mediated inhibition of cell proliferation correlates with the activation of the MAP kinase cascade: role of the phosphotyrosine phosphatase SHP‐2. J Physiol Paris 2000; 94: 239–50. [DOI] [PubMed] [Google Scholar]
- 40. Patel GP, Ma S, Bag J. The autoregulatory translational control element of poly(A)‐binding protein mRNA forms a heteromeric ribonucleoprotein complex. Nucleic Acids Res 2005; 33: 7074–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Benali N, Cordelier P, Calise D et al . Inhibition of growth and metastatic progression of pancreatic carcinoma in hamster after somatostatin receptor subtype 2 (sst2) gene expression and administration of cytotoxic somatostatin analog AN‐238. Proc Natl Acad Sci USA 2000; 97: 9180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]