

Abstract
Congenital generalized lipodystrophy (CGL) is a rare autosomal recessive disorder characterized by near total absence of body fat since birth with predisposition to insulin resistance, diabetes, hypertriglyceridemia and hepatic steatosis. Three CGL loci, AGPAT2, BSCL2 and CAV1 have been identified previously. Recently, mutations in polymerase I and transcript release factor (PTRF) were reported in five Japanese patients presenting with myopathy and CGL (CGL4). We report on novel PTRF mutations and detailed phenotype of two male and three female patients with CGL4 belonging to two pedigrees of Mexican origin (CGL7100 and CGL178) and one pedigree of Turkish origin (CGL180). All patients had near total loss of body fat and congenital myopathy manifesting as weakness, percussion-induced muscle mounding and high serum creatine kinase levels. Four of them had hypertriglyceridemia. Three of them had atlantoaxial instability. Two patients belonging to CGL178 pedigree required surgery for pyloric stenosis in the first month of life. None of them had prolonged QT interval on electrocardiography but both siblings belonging to CGL7100 had exercise-induced arrhythmias. Three of them had mild acanthosis nigricans but had normal glucose tolerance. Two of them had hepatic steatosis. All patients had novel null mutations in PTRF gene. In conclusion, mutations in PTRF result in a novel phenotype that includes generalized lipodystrophy with mild metabolic derangements, myopathy, cardiac arrhythmias, atlantoaxial instability and pyloric stenosis. It is unclear how mutations in PTRF, which plays an essential role in formation of caveolae, affect a wide variety of tissues resulting in a variable phenotype.
Keywords: congenital generalized lipodystrophy, myopathy, PTRF, cardiac arrhythmias, pyloric stenosis, atlantoaxial instability, caveolae
INTRODUCTION
Congenital generalized lipodystrophy (CGL) is a rare autosomal recessive disorder characterized by near total loss of body fat from birth. Mutations in three genes, 1-acylglycerol 3-phosphate-O-acyltransferase 2 (AGPAT2), Berardinelli-Seip congenital lipodystrophy 2 (BSCL2), and caveolin-1 (CAV1) were known at the time of our recent report of two Mexican-American siblings with a novel subtype of CGL associated with muscular weakness and cervical spine instability [Simha et al., 2008]. Since that publication, mutations in the polymerase I and transcript release factor (PTRF) gene were reported by Hayashi et al., [2009] resulting in a phenotype similar to our patients including generalized lipodystrophy and muscular dystrophy with elevated creatine kinase levels (CGL4). The report consists of five patients ranging in age from 8 years to 24 years old and three of them had muscle weakness with muscle mounding and muscle hypertrophy. None of the patients had mental retardation or acanthosis nigricans. Two of them had hepatosplenomegaly and one additional patient was reported to have fatty liver. Two of the older male patients had acromegaloid features with no androgynism. Two patients had arrhythmias and one of them developed atrial fibrillation. . In addition, other variable manifestations such as constipation, nephrosis, umbilical prominence, scoliosis, recurrent pneumonias and transient immunoglobulin A deficiency were also reported [Hayashi et al., 2009].
In this report, we describe the phenotype of two new Mexican siblings and one Turkish female harboring novel homozygous null mutations in PTRF. In addition, we report novel compound heterozygous null mutations in PTRF in the two Mexican siblings previously described by us [Simha et al., 2008].
CLINICAL REPORTS
CGL178.5
This is an 11-year-old Hispanic boy, the son of nonconsanguineous Mexican parents (Fig. 1A, 1B, 2A and 2B). Reportedly, he was a normal looking baby at birth. He was born by cesarean at term gestation with a birth weight of 3 kg. The only complication during terminal part of pregnancy was oligohydramnios. Two weeks after birth, he developed projectile vomiting and was diagnosed with pyloric stenosis. He then began losing sc fat from all over the body. He had surgery for pyloric stenosis at the age of 40 days. Following the surgery, he developed chronic diarrhea, especially after high fat meals, until the age of three years. He was noticed to be “stiff since birth” with reduced range of movement in all his joints. He was slower than other children of his age. He is currently in the 6th grade and doing well in school. He gets tired easily on riding a bicycle. He has had hypercholesterolemia since the age of 5 years for which he has not received any drug therapy thus far. He also has a right sided hearing loss which is being evaluated and is currently not wearing a hearing aid.
Fig. 1.
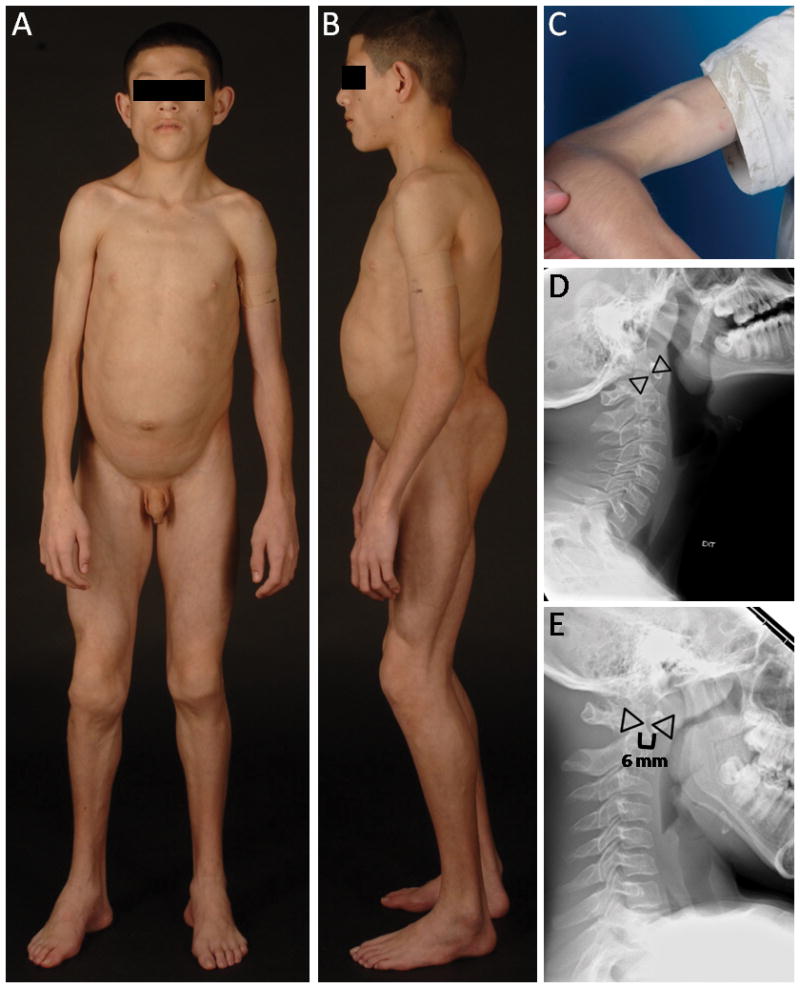
A. CGL 178.5, anterior view of a 11-year-old boy showing generalized lipodystrophy, prominent muscularity, acromegalic features such as large hands and feet. B. CGL 178.5, lateral view showing generalized lipodystrophy and protuberant abdomen. C. Percussion muscle mounding on the biceps of patient CGL 178.5. D and E, Lateral radiographs of the cervical spine of CGL 178.5 during extension and flexion of cervical spine, respectively showing atlantoaxial instability. The interdental distance (shown by space between the triangles) was 4 mm during extension but was 6 mm during flexion.
Fig 2.
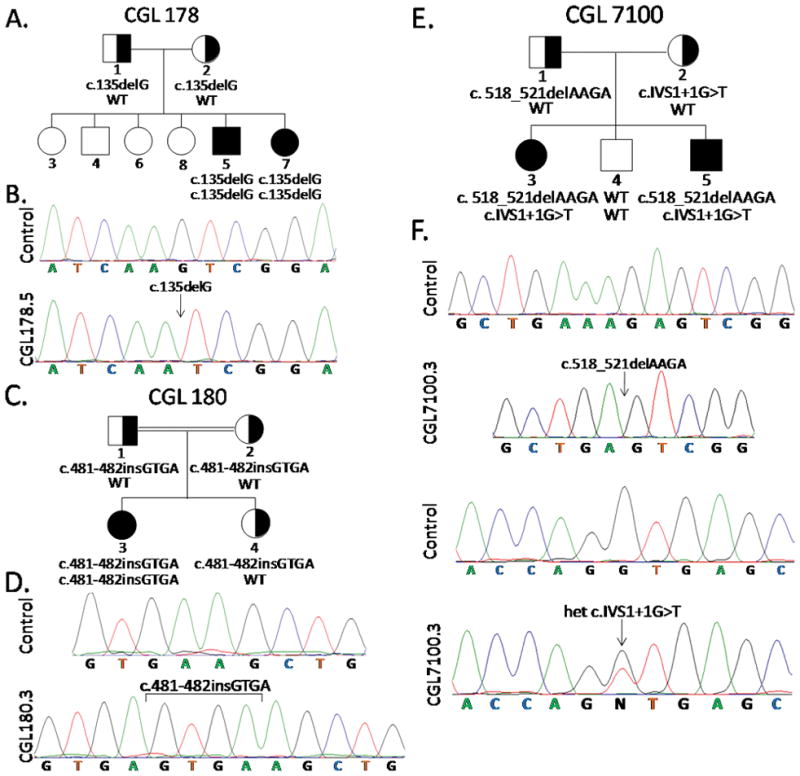
Pedigrees and Sequence Chromatograms of Patients. A. 1. CGL 178 pedigree. B. Normal chromatogram and that from CGL 178.5 showing homozygous c.135delG PTRF mutation. C. CGL 180 pedigree D. Normal chromatogram and that from CGL 180.3 showing homozygous insertion in exon 2 c.481-482insGTGA. E. CGL 7100 pedigree. F. Chromatograms showing two normal sequences and those from CGL 7100.3 showing heterozygous PTRF mutations c.518-521delAAGA (cloned chromatogram is shown) and c.IVS1+1G>T. WT, wild type genotype. Circles denote females and squares denote males. Half filled indicates heterozygotes. Filled symbols indicate affected and unfilled symbols indicate unaffected subjects.
On examination he was 153.5 cm tall and weighs 41.9 kg and head circumference in the 40th centile. His fundus examination was normal. Oral examination revealed enlarged tonsils bilaterally. Thyroid, lung and cardiac examinations were normal with the exception of a short systolic murmur, grade 1/6. Liver was palpable 1 cm below the right costal margin and he did have umbilical prominence with a protuberant abdomen. His genitalia were prepubertal and testes were approximately 2 mL bilaterally. He had cavus-varus feet bilaterally and decreased range of motion at the ankles. Otherwise, joint examination was normal with no contractures. Painless muscle mounding was easily elicited by finger percussion or reflex hammer on several muscles over the thenar, forearm, arm and thigh regions (Fig. 1C). He displayed good strength of 4/5 in the extremities. Muscle-stretch reflexes were normal. He was able to stand and ambulate independently with a “stiff” gait and calcaneo varus gait pattern. He had marked fat loss from his face, extremities, palms and soles. He had an apparent muscularity and prominent veins consistent with generalized lipodystrophy. There was no acanthosis noted in the neck or groin, however, he had some minimal acanthosis in the axilla.
The boy underwent a standard 2-hour oral glucose tolerance test with 1.75 g/kg body weight glucose load, which was normal, but he had extreme fasting (74 μU/mL) and postprandial hyperinsulinemia (217–365 μU/mL). He had markedly low levels of high density lipoprotein cholesterol (14 mg/dL) and had high serum triglycerides (1074 mg/dL). Glycosylated hemoglobin A1c was normal (5.7%) but he had elevated creatine kinase (CK) level (625 U/L). Alanine aminotransferase level was 54 U/L and aspartate amiotransferase level was 35 U/L. Serum leptin level was 0.30 ng/mL and adiponectin level was 0.53 mg/L. His electrocardiograms did not show any signs of arrhythmia or evidence of QT prolongation (QTc=433 ms). His liver fat on 1H magnetic resonance spectroscopy (MRS) was 33.8% and intramyocellular soleus fat was 0.82%. Dual energy X-ray absorptiometry (DEXA) study revealed total body fat of 12.7% (normal values, mean ± SD, being 30.0 ± 8.4, in age matched Hispanic boys)[Kelly et al., 2009]. His cervical spine x-ray did show evidence of atlantoaxial instability with predental space measuring 4 mm in extension and increasing to 6 mm during flexion (Fig. 1D and 1E). He was however asymptomatic and had no neck pain or evidence of myelopathy.
Nerve conduction study of the upper and lower extremities was normal with the exception of a delayed distal latency of sensory action potentials in the ulnar and sural nerves. Needle electromyography revealed increased insertional activity and frequent bursts of rhythmic high-frequency (60–200 Hz) bursts of simple or complex motor unit potentials with variable amplitude lasting 60–750 ms consistent with neuromyotonia (Fig. 3A). They were mostly elicited by needle insertion.
Fig. 3.

A, An electromyographic tracing from the biceps brachii showing neuromyotonic discharge. B, Holter monitor tracing of CGL 7100.3 showing baseline normal rhythm with four consecutive beats of ventricular tachycardia. C, Electrocardiogram, lead V2 on treadmill stress test of CGL 7100.3 after three minutes on Bruce Protocol at heart rate of 178 beats per minute, 10 METs, speed of 3.4 mph and 14% grade, showing bidirectional ventricular tachycardia suggestive of catecholaminergic polymorphic ventricular tachycardia (CPVT).
CGL178.7
She is a 16-month-old Hispanic girl, the younger sister of patient 178.5 (Fig 2A). At birth, she appeared normal although mother’s pregnancy was complicated by hypertension. She was born at 36 weeks of gestation with a birth weight of 3 kg. Her peripartum course was complicated by respiratory distress and she was admitted to the neonatal intensive care unit for a period of three days where she received antibiotic therapy. She subsequently developed vomiting and was diagnosed with pyloric stenosis and had surgery at one month of age. She had evidence of patent ductus arteriosus and patent foramen ovale on echocardiogram done at birth to evaluate a heart murmur. Her developmental milestones were delayed. She started sitting at 12 months and started walking at 15 months. Her vocabulary consists of about 5–7 words. Her appetite was not very good. Fat in her diet induces diarrhea. She receives a low fat diet with high caloric supplements because of failure to thrive.
On examination she was 78.7 cm tall (25th centile) and weighed 8.3 kg (<5th centile). She had normal appearing eyes and low set ears. There was no acanthosis in the neck, axilla or groin. Respiratory examination was normal. Cardiac examination revealed a 2/6 grade ejection systolic murmur. She had a protuberant abdomen with liver palpable 1 cm below the right costal margin. Genital examination revealed normal female anatomy with no clitoromegaly. Percussion muscle mounding was elicited in several upper and lower extremity muscles and she displayed no limitation in range of motion of her joints. Muscle-stretch reflexes were reduced throughout. She reached for objects with either hand and was able to ambulate independently with a wide based toddler gait. She had marked fat loss from the face and extremities; however, she did have some subcutaneous fat in her palms and soles.
She had low levels of HDL cholesterol (19 mg/dL) and had slightly elevated serum triglycerides (142 mg/dL). Serum insulin level was 8.9 μU/ml. Glycosylated hemoglobin A1c was normal (5.3%) but she had elevated CK level (571 U/L). Alanine aminotransferase level was 42 U/L and aspartate amiotransferase level was 37 U/L. Her serum leptin level was 0.44 ng/mL and serum adiponectin was 2.27 mg/L. Electrocardiogram showed no arrhythmias or prolongation of QT (QTc = 394 ms).
CGL 180.3
She is an 11-year-old Turkish girl, the daughter of consanguineous parents (Fig. 2C and Fig. 2D). Reportedly, a generalized pattern of lipodystrophy was noted at birth. She was born full-term with a birth weight of 2200 g. She had developmental delay and started walking at age 2. She has some difficulty walking due to scoliosis and has a tendency to walk on her toes. She has pes cavus.
On examination she was 138 cm tall and weighed 30.5 kg. She had percussion myoedema in her upper extremities. She has umbilical prominence and hepatomegaly. She has hyperextensibility of fingers. She did not have acanthosis nigricans. Her fasting serum glucose was 88 mg/dL and she had a normal glucose tolerance test. Serum triglyceride level was 109 mg/dL. She had markedly elevated serum CK levels (2337 U/L).
CGL7100.3
This pedigree and the affected patients were previously reported by us [Simha et al., 2008] (Fig. 2E and 2F). Currently, she is a 14-year-old Hispanic girl who has recently developed worsening hypertension requiring an increase in lisinopril dose from 2.5 mg twice daily to 5 mg twice daily. She recently achieved menarche and her periods are somewhat irregular.
On examination she was 160 cm tall and weighs 46.2 kg. We were able to elicit percussion induced muscle mounding in the biceps. Since the last report, we determined her liver fat content using 1H MRS which was elevated at 16.64%. In addition, 1H MRS of soleus revealed muscle fat content of 1.04%. Her serum leptin level was 0.58 ng/mL and adiponectin level was 0.53 mg/L. On electrocardiography, there was no evidence of QT prolongation (QTc=429 ms). Her echocardiogram was normal for ventricular size and function. Holter monitoring for 24 hours revealed several nonsustained runs of a fast polymorphic ventricular tachycardia up to a heart rate of 300 beats per minute (Fig. 3B). She had a prior Holter monitor at three years of age, which was essentially normal with the exception of rare premature ventricular contractions and sinus tachycardia. Therefore, an exercise study was conducted which revealed several runs of asymptomatic polymorphic ventricular tachycardia (Fig. 3C). This arrhythmia resembles catecholaminergic polymorphic ventricular tachycardia (CPVT). She has been started on nadolol to treat this condition.
CGL7100.5
This 8-year-old male was also previously reported by us [Simha et al., 2008] (Fig 2E). He continues to have some learning disabilities. He has been unable to read at the level of his peers and has difficulty counting.
On examination he was 134.5 cm tall and weighs 28.2 kg. We were able to elicit percussion induced muscle mounding in his biceps muscle. His liver fat on 1H MRS is 2.26% and intramyocellular soleus fat is 0.18%. His serum leptin level was 0.83 ng/mL and serum adiponectin level was 0.35 mg/L. On electrocardiography, there was no evidence of QT prolongation (QTc = 403 ms). His echocardiogram was normal for left ventricular size and function. Holter monitoring for 24 hours revealed short runs of atrial tachycardia at rates of 180–220 beats per minute. In addition, there appeared to be aberrant conduction of the atrial tachycardia versus occasional short runs of polymorphic ventricular tachycardia. Therefore, an exercise study was conducted which revealed asymptomatic short runs of wide complex tachycardia consistent with a polymorphic ventricular tachycardia. In addition, occasional atrial ectopy was noted. This arrhythmia also resembles CPVT. He has also been started on nadolol to treat this condition.
MATERIALS AND METHODS
All patients were evaluated in the Clinical and Translational Research Center at the University of Texas Southwestern Medical Center at Dallas. Informed consent was obtained from the patients and parents. The study protocol was approved by the Institutional Review Board. Blood was collected after a 12 hour overnight fast for analysis of serum lipoproteins, insulin, glucose, and a chemistry profile as previously described [Garg 2000] and for genotypin.
Genomic DNA was isolated from blood using the Easy DNA kit from Invitrogen (Carlsbad, CA) according to the manufacturer’s protocol. The coding region of the PTRF gene was amplified using gene specific primers (available on request). The exon 1 was amplified in one fragment, while exon 2 was amplified in two overlapping fragments. The PCR conditions employed have been described before [Simha et al., 2003a]. The purified PCR products were sequenced to determine the nucleotide alternations. The splice site prediction was made using web program NetGene (http://www.cbs.dtu.dk/services/NetGene2/).
Serum leptin and adiponectin were measured using radioimmunoassay kits (Millipore, Billerica, MA)
RESULTS
Sequencing of the exons and splice sites of PTRF gene showed a homozygous mutation in the CGL 178 siblings: c.135delG, resulting in a short protein of p.Lys45AspfsX5 (Fig. 2B). CGL180.3 harbored a homozygous insertion in exon 2 c.481-482insGTGA which is predicted to result in a frame shift making an abnormal protein p.Lys161SerfsX41 (Fig. 2D). CGL 7100 siblings showed compound heterozygous mutations, c.518-521delAAGA, which is predicted to result in frame shift making an abnormal protein of p.Lys173SerfsX101 and c.IVS1+1G>T, which is predicted to retain 143 nucleotides of intron 1, again resulting in the frame shift and an aberrant protein p.Asp158ValfsX49 (Fig. 2F).
The father of the proband from CGL 178 pedigree (CGL 178.1) is a 43-year-old male, who had a heterozygous mutation c.135delG (p.Ile44fsX4). He did not have lipodystrophy but had poorly controlled diabetes with a hemoglobin A1c of 10%. His nonfasting serum cholesterol was 202 mg/dl, triglycerides were 405 mg/dl and HDL-cholesterol was 36 mg/dl. He had a normal CK level. The mother of the proband (CGL 178.2) is 40-year-old female with heterozygous mutation c.135delG (p.Ile44fsX4). She did not have lipodystrophy but did have slightly elevated nonfasting triglycerides of 206 mg/dl and a low HDL-cholesterol level of 37 mg/dl. She did have a normal CK and glucose levels.
The parents and a sibling of the proband from CGL180 pedigree carried the heterozygous mutation c.481-482insGTGA. Phenotypic information on the parents and sibling was not available.
The 42-year-old father (CGL 7100.1) of the probands belonging to CGL 7100 siblings, had the heterozygous mutation c.518-521delAAGA (p.Leu172fsX100). Clinically, he did not have lipodystrophy. He had slightly high triglyceride concentration of 215 mg/dL with normal HDL-cholesterol, glucose, and CK levels. CGL7100.2 is a 35-year-old female and harbors the heterozygous mutation c.IVS1+1G>T (p.Gln157fsX50). She did not have lipodystrophy. She had slightly high serum triglyceride concentration of 200 mg/dL with normal HDL-cholesterol, glucose and CK levels.
DISCUSSION
Genetic as well as phenotypic heterogeneity is well described in patients with CGL. The two major subtypes, CGL1 and CGL2, harbor mutations in AGPAT2 and BSCL2, respectively and have some overlapping and some distinct phenotypic features. For example, CGL2 patients lack any mechanical and metabolically active adipose tissue whereas CGL1 patients lack only metabolically active adipose tissue and have well preserved mechanical adipose tissue [Simha et al., 2003b]. In addition, CGL2 patients have increased prevalence of cardiomyopathy, mental retardation but they tend to have less pronounced lytic lesions in the appendicular skeleton [Agarwal et al., 2003; Van Maldergem et al., 2002]. Patients of either subtype, CGL1 or 2, develop marked metabolic complications such as severe insulin resistance, hypertriglyceridemia, acanthosis nigricans, premature diabetes and hepatic steatosis. On the other hand a single patient, of the subtype, CGL3, with a homozygous null mutation in CAV1 has been reported who had distinctive features compared to the two other major subtypes, i.e., vitamin D resistance, hypocalcemia, reduced bone mineral density, preserved bone marrow fat and short stature [Garg et al., 2008; Kim et al., 2008]. The recent report of a novel subtype of CGL by us [Simha et al., 2008] and the discovery of the fourth CGL locus, PTRF, by Hayashi et al. [2009] prompts the question about the phenotypic heterogeneity of CGL4 compared to the other CGL subtypes.
Only five patients were reported by Hayashi et al [Hayashi et al., 2009]. Our report of an additional five patients adds other interesting features to what had been reported earlier. The main distinguishing feature of CGL4 is concomitant myopathy with weakness, inability to exercise and percussion muscle mounding. Interestingly, all the patients with CGL4 reported so far have had null mutations in PTRF. This prompts the question whether less severe mutations such as missense mutations will be associated with CGL4 or with a less severe phenotype. Heterozygous carriers do not manifest overt lipodystrophy or myopathy, however, we cannot exclude mild metabolic disturbances like hypertriglyceridemia and diabetes amongst them. Similar data were reported previously [Hayashi et al., 2009]. Further studies comparing the phenotype of larger numbers of heterozygotes and normal subjects belonging to CGL4 pedigrees will determine the effect of haploinsufficiency of PTRF.
In contrast to our current patients, patients from Japan had no acanthosis and no metabolic abnormalities. Interestingly, four of our five patients have elevated triglycerides and two of the three patients evaluated for hepatic steatosis have elevated liver fat. Three of the five patients also have acanthosis nigricans in the setting of normal glucose tolerance. Two of them also had hyperinsulinemia and four of them had low leptin and adiponectin levels with normal adiponectin levels being about 7.8 mg/L (1.5–29.4) [Antuna-Puente et al., 2010] Thus, although acanthosis nigricans appears to be mild compared to CGL1 and 2, our data suggest that these patients are also likely to develop severe hyperinsulinemia and metabolic abnormalities.
In addition, three of our five patients had atlantoaxial instability. CGL 7100.3 was discovered to have atlantoaxial instability at a young age but was asymptomatic for some time. CGL 7100.3 and CGL 7100.5 were then involved in a car accident when they were 10 years old and 4 years old, respectively. CGL 7100.3 had continued headaches and an MRI was done for evaluation. The atlantoaxial instability was seen again and due to perceived risk of quadriparesis in future, both patients were operated on to stabilize the cervical spine. Whether atlantoaxial instability poses a neurological risk and needs surgical correction, remains unclear.
Our five patients with PTRF mutations presented with congenital generalized lipodystrophy and myopathy (Table I). Two of the five patients had pyloric stenosis in the first month of life requiring corrective surgery and subsequent fat intolerance. Three of the five patients had normal echocardiograms with normal ventricular size and function. Only one of them had hypertension. None of them had QT prolongation but on exercise testing two of them showed CPVT. CPVT is characterized by stress induced ventricular tachycardia and a mortality of 30% in untreated symptomatic patients [Kontula et al., 2005; Swan et al., 1999]. CPVT has been reported in patients with mutations in ryanodine receptor-2 (RYR2) gene [Priori et al., 2001] and rarely, in calsequestrin-2 (CASQ2) gene [Lahat et al., 2001]. Early initiation of oral β-adrenergic blockers is the treatment of choice [Napolitano et al., 2007]. Our data suggest that mutations in PTRF are also associated with CPVT.
Table I.
Clinical Characteristics of our patients with CGL4
| Characteristic | CGL 178.5 | CGL 178.7 | CGL 180.3 | CGL 7100.3 | CGL 7100.5 |
|---|---|---|---|---|---|
| Age (y)/Gender | 11/M | 1/F | 11/F | 14/F | 8/M |
| Pyloric Stenosis | + | + | − | − | − |
| Atlantoaxial instability | + | n/a | n/a | + | + |
| Acanthosis nigricans | + | − | − | + | + |
| Hepatomegaly | + | + | + | + | + |
| Myopathy | + | + | + | + | + |
| Hypertension | − | n/a | − | + | − |
| Glucose tolerance | normal | n/a | normal | normal | normal |
| Hypertriglyceridemia | + | + | − | + | + |
| Echocardiogram | n/a | n/a | normal | normal | normal |
| catecholaminergic polymorphic ventricular tachycardia (CPVT) | n/a | n/a | n/a | + | + |
M, male; F, female; +, present; −, absent; n/a, data not available
Previously, Rajab et al. [2002] reported two interesting phenotypes of CGL patients from Oman. The patients reported as group A had many of the same characteristics that are well characterized for CGL1 or 2 such as muscle hypertrophy, acanthosis nigricans, raised insulin levels, diabetes mellitus and hypertriglyceridemia [Rajab et al., 2002]. In contrast, 10 Group B CGL patients, age 0–14 years, had hypertrophic pyloric stenosis; and also had cardiomegaly, reduced exercise tolerance and elevated CK levels [Rajab et al., 2002]. Some older patients had arrhythmias and some of them died suddenly. Interestingly, none of them had acanthosis nigricans, diabetes mellitus, raised insulin levels or hypertriglyceridemia. While our paper was under review by the Journal, Rajab et al. [2010] reported mutations in PTRF in eight families with CGL and myopathy. However, detailed phenotypic characteristics were reported for only two patients. One 14-year-old boy from Oman had pyloric stenosis, prolonged QTc interval and ventricular and supraventricular tachyarrhythmias. He had esophageal dysmotility and joint stiffness. He had insulin resistance as assessed by HOMA-IR but no acanthosis nigricans. He did have significant myopathy with elevated CK levels and hypertriglyceridemia. The second patient from the United Kingdom also had prolonged QTc with ventricular and supraventricular tachycardias and died at age 13 from sudden cardiac death due to ventricular fibrillation. She had atlantoaxial dislocation and hepatomegaly. She also had hypertriglyceridemia and insulin resistance but no acanthosis nigricans. Four of the additional nine patients had elevated CK levels and joint contractures. Six of them had evidence of muscle mounding. Eight of them had hypertrophic pyloric stenosis but none had atlantoaxial dislocation.
PTRF is an essential component of caveolae, specialized plasma membrane microdomains appearing as 50–100 nm vesicular invaginations, which function as endocytic carrier vesicles. Caveolae formation requires expression of caveolar proteins, caveolin (CAV) 1, 2, or 3 in various tissues. Mice lacking PTRF have profoundly reduced protein expression of all caveolins and no detectable caveolae [Liu et al., 2008]. It is likely that many manifestations in patients with CGL4 are due to disrupted caveolae formation and diminished expression of CAVs. Interestingly, mutations in CAV3 are associated with autosomal dominant myopathy [Betz et al., 2001; Carbone et al., 2000; Tateyama et al., 2002]; hypertrophic cardiomyopathy [Hayashi et al., 2004], long QT syndrome [Vatta et al., 2006] and abnormal atrio-ventricular conduction [Catteruccia et al., 2009]. Mutations in CAV3 induce an increase in late Na current by affecting Nav1.5 channels present in caveolae and thus may delay cardiac depolarization and increase QT interval [Lehnart et al., 2007; Yarbrough et al., 2002]. It is possible that PTRF mutations induce arrhythmias by a similar mechanism or they may affect other cardiac ion channels that reside in caveolae including pacemaker channel (HCN4) [Barbuti et al., 2007], Ca2+ channel (CAv1.2) [Balijepalli et al., 2006], K+ channels (Kv1.5, Kir6.2/Sur2a) [Balijepalli et al., 2008] and Na+/Ca2+ exchanger (NCX) [Bossuyt et al., 2002; Camors et al., 2006].
In addition, PTRF is expressed mostly in adipocytes and muscle tissue including smooth muscle. There has been evidence of patients showing smooth muscle hypertrophy in the gastrointestinal tract leading to dysmotility, dysphagia and ileus [Rajab et al., 2002]. It is unclear to us whether the observed fat intolerance seen in two of our patients (CGL 178) is due to smooth muscle dysfunction.
In summary, we report five additional CGL patients with novel PTRF mutations who shared some of the same clinical characteristics as previously reported in five patients with from Japan such as CGL, myopathy, percussion muscle mounding, ankle contractures and acromegaloid features. We now add additional clinical features associated with CGL4, such as, acanthosis nigricans, hypertriglyceridemia, hyperinsulinemia, hepatic steatosis, hypertension, atlantoaxial instability, pyloric stenosis, neuromyotonia and CPVT. The heterogeneity even within our own patients illustrates the phenotypic variability within this rare clinical syndrome. The molecular mechanisms by which PTRF mutations cause manifestations in various organ systems and phenotypic variability remain to be elucidated.
Acknowledgments
We thank Dr. Gregory L. Johnson for help in the cardiac evaluation of the affected patients belonging to pedigree CG7100. We thank Lidia Szczepaniak, Ph.D. for interpretation of the liver and muscle MRS studies and Claudia Quittner for her assistance during patient evaluations. We thank Dr. Gulcin Akinci for evaluation of patient belonging to CGL180 pedigree. We also thank Sarah Masood and Crystal Kittisopikul for help with illustrations and mutational screening. This work was supported by the National Institutes of Health grants R01-DK54387, CTSA Grant UL1 RR024982 and Southwest Medical Foundation.
References
- Agarwal AK, Simha V, Oral EA, Moran SA, Gorden P, O’Rahilly S, Zaidi Z, Gurakan F, Arslanian SA, Klar A, Ricker A, White NH, Bindl L, Herbst K, Kennel K, Patel SB, Al-Gazali L, Garg A. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J Clin Endocrinol Metab. 2003;88:4840–4847. doi: 10.1210/jc.2003-030855. [DOI] [PubMed] [Google Scholar]
- Antuna-Puente B, Boutet E, Vigouroux C, Lascols O, Slama L, Caron-Debarle M, Khallouf E, Levy-Marchal C, Capeau J, Bastard JP, Magre J. Higher adiponectin levels in patients with Berardinelli-Seip congenital lipodystrophy due to seipin as compared with 1-acylglycerol-3-phosphate-o-acyltransferase-2 deficiency. J Clin Endocrinol Metab. 95:1463–1468. doi: 10.1210/jc.2009-1824. [DOI] [PubMed] [Google Scholar]
- Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac L-type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proc Natl Acad Sci U S A. 2006;103:7500–7505. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balijepalli RC, Kamp TJ. Caveolae, ion channels and cardiac arrhythmias. Prog Biophys Mol Biol. 2008;98:149–160. doi: 10.1016/j.pbiomolbio.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbuti A, Terragni B, Brioschi C, DiFrancesco D. Localization of f-channels to caveolae mediates specific beta2-adrenergic receptor modulation of rate in sinoatrial myocytes. J Mol Cell Cardiol. 2007;42:71–78. doi: 10.1016/j.yjmcc.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Betz RC, Schoser BG, Kasper D, Ricker K, Ramirez A, Stein V, Torbergsen T, Lee YA, Nothen MM, Wienker TF, Malin JP, Propping P, Reis A, Mortier W, Jentsch TJ, Vorgerd M, Kubisch C. Mutations in CAV3 cause mechanical hyperirritability of skeletal muscle in rippling muscle disease. Nat Genet. 2001;28:218–219. doi: 10.1038/90050. [DOI] [PubMed] [Google Scholar]
- Bossuyt J, Taylor BE, James-Kracke M, Hale CC. The cardiac sodium-calcium exchanger associates with caveolin-3. Ann N Y Acad Sci. 2002;976:197–204. doi: 10.1111/j.1749-6632.2002.tb04741.x. [DOI] [PubMed] [Google Scholar]
- Camors E, Charue D, Trouve P, Monceau V, Loyer X, Russo-Marie F, Charlemagne D. Association of annexin A5 with Na+/Ca2+ exchanger and caveolin-3 in non-failing and failing human heart. J Mol Cell Cardiol. 2006;40:47–55. doi: 10.1016/j.yjmcc.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Carbone I, Bruno C, Sotgia F, Bado M, Broda P, Masetti E, Panella A, Zara F, Bricarelli FD, Cordone G, Lisanti MP, Minetti C. Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology. 2000;54:1373–1376. doi: 10.1212/wnl.54.6.1373. [DOI] [PubMed] [Google Scholar]
- Catteruccia M, Sanna T, Santorelli FM, Tessa A, Di Giacopo R, Sauchelli D, Verbo A, Lo Monaco M, Servidei S. Rippling muscle disease and cardiomyopathy associated with a mutation in the CAV3 gene. Neuromuscul Disord. 2009;19:779–783. doi: 10.1016/j.nmd.2009.08.015. [DOI] [PubMed] [Google Scholar]
- Garg A. Gender differences in the prevalence of metabolic complications in familial partial lipodystrophy (Dunnigan variety) J Clin Endocrinol Metab. 2000;85:1776–1782. doi: 10.1210/jcem.85.5.6605. [DOI] [PubMed] [Google Scholar]
- Garg A, Agarwal AK. Caveolin-1: a new locus for human lipodystrophy. J Clin Endocrinol Metab. 2008;93:1183–1185. doi: 10.1210/jc.2008-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Arimura T, Ueda K, Shibata H, Hohda S, Takahashi M, Hori H, Koga Y, Oka N, Imaizumi T, Yasunami M, Kimura A. Identification and functional analysis of a caveolin-3 mutation associated with familial hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2004;313:178–184. doi: 10.1016/j.bbrc.2003.11.101. [DOI] [PubMed] [Google Scholar]
- Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, Park YE, Nonaka I, Hino-Fukuyo N, Haginoya K, Sugano H, Nishino I. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119:2623–2633. doi: 10.1172/JCI38660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TL, Wilson KE, Heymsfield SB. Dual energy X-Ray absorptiometry body composition reference values from NHANES. PLoS One. 2009;4:e7038. doi: 10.1371/journal.pone.0007038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CA, Delepine M, Boutet E, El Mourabit H, Le Lay S, Meier M, Nemani M, Bridel E, Leite CC, Bertola DR, Semple RK, O’Rahilly S, Dugail I, Capeau J, Lathrop M, Magre J. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93:1129–1134. doi: 10.1210/jc.2007-1328. [DOI] [PubMed] [Google Scholar]
- Kontula K, Laitinen PJ, Lehtonen A, Toivonen L, Viitasalo M, Swan H. Catecholaminergic polymorphic ventricular tachycardia: recent mechanistic insights. Cardiovasc Res. 2005;67:379–387. doi: 10.1016/j.cardiores.2005.04.027. [DOI] [PubMed] [Google Scholar]
- Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, Lancet D, Eldar M. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378–1384. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE, Ackerman MJ, Benson DW, Jr, Brugada R, Clancy CE, Donahue JK, George AL, Jr, Grant AO, Groft SC, January CT, Lathrop DA, Lederer WJ, Makielski JC, Mohler PJ, Moss A, Nerbonne JM, Olson TM, Przywara DA, Towbin JA, Wang LH, Marks AR. Inherited arrhythmias: a National Heart, Lung, and Blood Institute and Office of Rare Diseases workshop consensus report about the diagnosis, phenotyping, molecular mechanisms, and therapeutic approaches for primary cardiomyopathies of gene mutations affecting ion channel function. Circulation. 2007;116:2325–2345. doi: 10.1161/CIRCULATIONAHA.107.711689. [DOI] [PubMed] [Google Scholar]
- Liu L, Brown D, McKee M, Lebrasseur NK, Yang D, Albrecht KH, Ravid K, Pilch PF. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 2008;8:310–317. doi: 10.1016/j.cmet.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano C, Priori SG. Diagnosis and treatment of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:675–678. doi: 10.1016/j.hrthm.2006.12.048. [DOI] [PubMed] [Google Scholar]
- Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- Rajab A, Heathcote K, Joshi S, Jeffery S, Patton M. Heterogeneity for congenital generalized lipodystrophy in seventeen patients from Oman. Am J Med Genet. 2002;110:219–225. doi: 10.1002/ajmg.10437. [DOI] [PubMed] [Google Scholar]
- Rajab A, Straub V, McCann LJ, Seelow D, Varon R, Barresi R, Schulze A, Lucke B, Lutzkendorf S, Karbasiyan M, Bachmann S, Spuler S, Schuelke M. Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS Genet. 2010;6:e1000874. doi: 10.1371/journal.pgen.1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simha V, Agarwal AK, Aronin PA, Iannaccone ST, Garg A. Novel subtype of congenital generalized lipodystrophy associated with muscular weakness and cervical spine instability. Am J Med Genet A. 2008;146A:2318–2326. doi: 10.1002/ajmg.a.32457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simha V, Agarwal AK, Oral EA, Fryns JP, Garg A. Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J Clin Endocrinol Metab. 2003a;88:2821–2824. doi: 10.1210/jc.2002-021575. [DOI] [PubMed] [Google Scholar]
- Simha V, Garg A. Phenotypic heterogeneity in body fat distribution in patients with congenital generalized lipodystrophy caused by mutations in the AGPAT2 or seipin genes. J Clin Endocrinol Metab. 2003b;88:5433–5437. doi: 10.1210/jc.2003-030835. [DOI] [PubMed] [Google Scholar]
- Swan H, Piippo K, Viitasalo M, Heikkila P, Paavonen T, Kainulainen K, Kere J, Keto P, Kontula K, Toivonen L. Arrhythmic disorder mapped to chromosome 1q42–q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J Am Coll Cardiol. 1999;34:2035–2042. doi: 10.1016/s0735-1097(99)00461-1. [DOI] [PubMed] [Google Scholar]
- Tateyama M, Aoki M, Nishino I, Hayashi YK, Sekiguchi S, Shiga Y, Takahashi T, Onodera Y, Haginoya K, Kobayashi K, Iinuma K, Nonaka I, Arahata K, Itoyama Y. Mutation in the caveolin-3 gene causes a peculiar form of distal myopathy. Neurology. 2002;58:323–325. doi: 10.1212/wnl.58.2.323. [DOI] [PubMed] [Google Scholar]
- Van Maldergem L, Magre J, Khallouf TE, Gedde-Dahl T, Jr, Delepine M, Trygstad O, Seemanova E, Stephenson T, Albott CS, Bonnici F, Panz VR, Medina JL, Bogalho P, Huet F, Savasta S, Verloes A, Robert JJ, Loret H, De Kerdanet M, Tubiana-Rufi N, Megarbane A, Maassen J, Polak M, Lacombe D, Kahn CR, Silveira EL, D’Abronzo FH, Grigorescu F, Lathrop M, Capeau J, O’Rahilly S. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet. 2002;39:722–733. doi: 10.1136/jmg.39.10.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- Yarbrough TL, Lu T, Lee HC, Shibata EF. Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude. Circ Res. 2002;90:443–449. doi: 10.1161/hh0402.105177. [DOI] [PubMed] [Google Scholar]