

Abstract
Parkinson disease (PD) is a common disorder that leads to motor and cognitive disability. We performed a genome-wide association study (GWAS) with 2000 PD and 1986 control Caucasian subjects from NeuroGenetics Research Consortium.1–5 We confirmed SNCA2,6–8 and MAPT3,7–9; replicated GAK9 (PPankratz+NGRC=3.2×10−9); and detected a novel association with HLA (PNGRC=2.9×10−8) which replicated in two datasets (PMeta-analysis=1.9×10−10). We designate the new PD genes PARK17 (GAK) and PARK18 (HLA). PD-HLA association was uniform across genetic and environmental risk strata, and strong in sporadic (P=5.5×10−10) and late-onset (P=2.4×10−8) PD. The association peak was at rs3129882, a non-coding variant in HLA-DRA. Two studies suggested rs3129882 influences expression of HLA-DR and HLA-DQ.10,11 PD brains exhibit up-regulation of DR antigens and presence of DR-positive reactive microglia.12 Moreover, non-steroidal anti-inflammatory drugs (NSAID) reduce PD risk.4,13 The genetic association with HLA coalesces the evidence for involvement of the immune system and offers new targets for drug development and pharmacogenetics.
Late-onset sporadic PD was long believed to be environmental with no genetic component.14,15 The initial samples that eventually coalesced to form the NGRC dataset was the basis for the first of a series of family studies that revealed the genetic component in PD.1,16 In the last decade, mutations in several genes were identified as causes of early-onset and Mendelian forms of PD, and polymorphisms in the SNCA (chromosome 4q21) and MAPT (17q21.1) regions were established as risk factors for common non-Mendelian PD.2,3,6–9,17 PD risk is also associated, inversely, with cigarette smoking, caffeinated-coffee consumption and NSAID use.4,13,18 Common late-onset PD is currently thought to result from an interplay of genetic susceptibility and environmental exposures, and data in support of this notion is beginning to emerge.5
A GWAS conducted in Japan identified two novel PD loci.19 Five GWAS have been performed in Caucasians, most confirmed the known associations with SNCA and MAPT, but none identified any new genes that reached genome-wide significance.7–9,20,21 We investigated whether the genetic component in PD in Caucasians was due to the genes that have already been identified. Using NGRC data, we estimated heritability before and after excluding the known pathogenic and susceptibility loci. Heritability of PD declined from 0.6 (P<0.0001) to 0.4 (P=0.01), but was still significant, suggesting additional unidentified genes exist.22
We performed a GWAS with 2,000 PD patients, 1,986 control subjects, and 811,597 single nucleotide polymorphisms (SNPs) (Supplementary Table 1). Subjects were recruited from the NGRC clinics in Oregon, Washington, New York and Georgia, using uniform criteria for diagnosis,23 subject selection, data collection and DNA preparation.2–5 25% of PD diagnoses change within the initial 5.4 years;24 therefore follow-up substantially reduces heterogeneity. Mean disease duration at enrollment was 8 years, eliminating most early misdiagnoses, and with an additional mean follow-up of 4 years, we were able to exclude another 47 misdiagnoses before GWAS. Controls were selected by the same investigators and from the same geographic regions as patients. At enrolment, they were on average 12 years older than the patients’ onset age, which increased power by reducing the likelihood that controls were at risk but too young to have developed symptoms. We used the Illumina HumanOmni1-Quad_v1-0_B genotyping array and achieved call rate of 99.92% and reproducibility rate of ≥99.99%. Association analyses were performed using PLINK V1.07.25 We adjusted all analyses for four covariates: age to avoid survival bias, sex because PD affects more men than women, and two principal components (PC1, PC2) which marked significant genetic substructure among Caucasian Americans of European descent (Supplementary Figure 1). Our GWAS confirmed the known PD-susceptibility regions at SNCA (P=3.4×10−11) and MAPT (P=1.3×10−6), as we had previously reported.2,3
We uncovered a novel genetic association with PD in the HLA region (chromosome 6p21.3). The peak significance was at the rs3129882 polymorphism in intron 1 of HLA-DRA gene. The association was genome-wide significance even after adjusting for four covariates (sex, age, PC1, PC2), P=2.9×10−8 (Table 1, Fig. 1 & 2 and Supplementary Fig. 2). 107 HLA SNPs reached P<10−3 for association with PD. We replicated association of rs3129882 in two independent data sets8,9 (Table 1, ORMeta-analysis=1.26, PMeta-analysis=1.9×10−10). The risk allele was the same in all datasets and was in Hardy-Weinberg equilibrium (HWE) in cases and in controls which in addition to the visual inspection of intensity plots supports no major problems with genotyping. Stratified analysis by family history, age at onset, gender and environmental exposures revealed ubiquitous associations across strata, and tests of heterogeneity across strata were not significant (Supplementary Table 2). Associations were particularly strong for sporadic PD (P=5.5×10−10), late-onset PD (P=2.4×10−8), and men (P=1.1×10−7). Most cases of PD are sporadic, late-onset and affect men more than women. There was no evidence for gene-environment or gene-gene interaction between rs219882 and smoking (P=0.42), coffee (P=0.55), NSAIDs (P=0.65), SNCA rs356220 (P=0.78) or MAPT rs199533 (P=0.24).
Table 1.
Association results for PD susceptibility genes*
| Locus | CH | SNP | Base-pair position | Minor/Major allele | Study | N Case | N Control | MAF Case | MAF Control | HWE P-value | Association P-value‡ | OR (95% CI) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PARK18 | HLA-DRA | 6p | rs3129882 | 32,517,508 | G/A | NGRC | 2000 | 1986 | 0.46 | 0.40 | 1.00 | 2.9×10−8 | 1.31 (1.19–1.44) |
| Replication 19 | 843 | 856 | 0.45 | 0.41 | 0.57 | 0.01 (5×10−3) | 1.21 (1.04–1.30) | ||||||
| Replication 28 | 604 | 612 | 0.44 | 0.40 | 0.06 | 0.06 (0.03) | 1.17 (1.00–1.37) | ||||||
| Replication 1+2 | 1447 | 1468 | 0.45 | 0.40 | 0.09 | 1.1×10−3 | 1.18 (1.06–1.31) | ||||||
| Meta-analysis | 3447 | 3454 | 0.45 | 0.40 | 0.27 | 1.9×10−10 | 1.26 (1.17–1.35) | ||||||
| PARK17 | GAK9 | 4p | rs11248051 | 848,332 | T/C | NGRC | 2000 | 1986 | 0.12 | 0.09 | 1.00 | 3.1×10−4 | 1.32 (1.14–1.54) |
| Meta analysis | 2843 | 2842 | 0.12 | 0.09 | 0.42 | 3.2×10−9 | 1.46 (1.29–1.65) | ||||||
| Known genes | SNCA | 4q | rs356220 | 90,860,363 | T/C | NGRC | 2000 | 1986 | 0.44 | 0.36 | 0.38 | 3.4×10−11 | 1.38 (1.25–1.52) |
| MAPT | 17q | rs199533 | 42,184,098 | T/C | NGRC | 2000 | 1986 | 0.17 | 0.22 | 0.05 | 1.3×10−6 | 0.74 (0.66–0.84) | |
| Other 7,19† | BST1 | 4p | rs4698412 | 15,346,446 | A/G | NGRC | 2000 | 1986 | 0.58± | 0.55 | 0.01 | 0.01 | 1.13 (1.03–1.24) |
| LRRK2 | 12q | rs2708453 | 38,764,919 | T/G | NGRC | 2000 | 1986 | 0.17 | 0.15 | 0.86 | 0.03 | 1.15 (1.01–1.31) | |
CH: Chromosome. SNP: Single nucleotide polymorphism. Minor/Major allele refers to the relative frequency of the two alleles. N: number of individuals. MAF: Minor allele frequency. HWE: Hardy Weinberg Equilibrium. OR: Odds ratio. CI: confidence interval
NGRC results were consistently adjusted for four covariates: PC1 and PC2 (for the substructure within Americans of European origin), sex and age.
All P values are for two-sided tests. The only case where a single (no-multiple testing) one-sided hypothesis test was performed was for replication of rs3129882; the one-sided P values are in parentheses.
PARK16 is not included in this table because we did not confirm it; our results were in opposite direction as the original report.7 (see Supplementary Table 4b)
BST1 minor allele (freq<0.5) in the Japanese was the major allele in Caucasians (freq>0.5). We used the same allele as reported for the Japanese to keep the risk allele consistent.
Figure 1. Genome-wide association P-values.
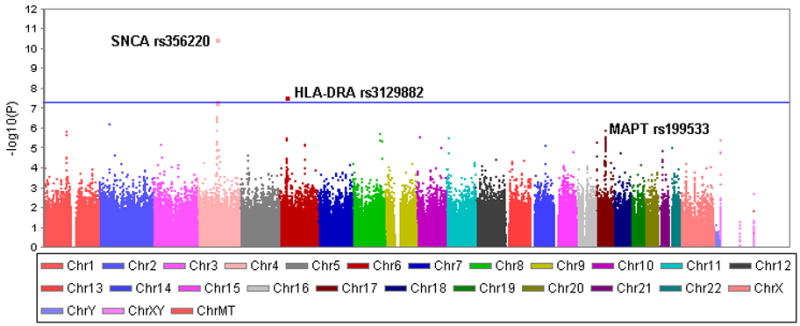
The Manhattan plot shows the P-values for association of 811,597 SNPs with PD. SNPs that surpass genome-wide significance (P<5×10−8) were on chromosomes 4 (SNCA region) and 6 (HLA-DRA). SNCA was known to be associed with PD, the HLA association is novel. The other known PD-associated region is on chromosme 17 (MAPT region) which replicated at P=1.3×10−6.
Figure 2. Signals of PD-association within HLA.

A one megabase (MB) region was defined centered on the association peak, rs3129882 in intron 1 of HLA-DRA, spanning from base pair (BP) position 32,017,508 to 33,017,508. The top panel shows all SNPs in this region plotted according to the significance of their association with PD and color-coded according to their linkage disequilibrium (r2) with the most significant SNP, rs3129882. Note that rs3129882 is not strongly correlated (defined as r2≥ 0.8)with any other HLA variant. The LocusZoom software used here calculates r2 using the HapMap CEU data. SNPs shown in grey were on the Illumina OMNI chip but not on HAPMAP thus r2 was not calculated with this method; however, using the Haploview software and the NGRC data to estimate r2, none of the variants was strongly correlated with rs3129882 (r2<0.6, Supplementary Fig. 6). The bottom panel shows the genes in the region, including the closely linked polymorphic HLA-DRB and DQB loci, and DRA, DRB5, and DRQA2 whose expression is correlated with the variation at rs3129882.
To explore for other true signals that may not have reached GW significance, we performed in silico replication and Meta-analysis of the most significant (P<10−5) SNPs in NGRC, using a publically available dataset from dbGaP (CIDR: Genome Wide Association Study in Familial Parkinson Disease) which has been published by Pankratz et al9. SNPs that replicated were in HLA (six SNPs in addition to rs3129882), SNCA and MAPT regions (Supplementary Table 3). One SNP (rs2046571) in the Hyaluronan synthase 2 (HAS2) gene region on chromosome 8 was marginally significant in CIDR, but did not reach genome-wide significance in Meta-analysis (3.6×10−7).
We used NGRC to replicate findings of previous PD GWAS (Supplementary Table 4). We confirmed the association of PD with cyclin G-associated kinase (GAK, 4p16, Supplementary Figure 3), as suggested by Pankratz et al.9 (rs11248051 PNGRC=3.1×10−4, ORNGRC+Pankratz =1.46, PNGRC+Pankratz =3.2×10−9). Satake et al.19 reported PARK16 (1q32), BST1 (4p15) and common variants (not rare mutations that cause Mendelian-PD) in LRRK2 (12q12) as new PD risk factors in the Japanese, and the accompanying GWAS by Simon-Sanchez et al.7 suggested these associations extend to Caucasians. We did not replicate PARK16 (ORs were in opposite direction from Satake results, P=0.03–0.15). Two of the six reported BST1 SNPs reached P<0.05 in our data. LRRK2 variants that were reported by Simone-Sanchez et al. were not significant in NGRC (P=0.57 & 0.69); however, three LRRK2 SNPs from the Satake’s study yielded P<0.05 in NGRC, and several other LRRK2 SNPs reached P~10−3. In NGRC, 1% of sporadic and 3% of familial PD have rare LRRK2 mutations.26 Since LRRK2 mutations are known to cause Mendelian-PD, association between common LRRK2 variants and non-Mendelian PD warrants follow up.
SNCA, MAPT, GAK and HLA each have a modest effect on PD risk, but when considered together, the cumulative effect can be substantial (Supplementary Fig. 4). To explore the combined effects of the four genes, we classified the subjects by the total number of risk alleles that they carry (0 to 8). Compared to subjects who had one or no risk allele, the risk of PD was doubled for individuals who had four risk alleles (OR=2.49, 95%CI=1.79–3.47, P=6.5×10−8), and was five-fold higher for individuals who had six or more risk alleles (OR=4.95, 95%CI=3.20–7.64, P=5.5×10−13). Thus our data support the long held notion that PD risk is due to cumulative effects of risk factors with modest individual effect.
A persistent problem in genetic association studies is inconsistent reproducibility which often arises from hidden genetic variation, i.e., population substructure. We investigated population structure in depth, using Genomic Control (λ)27 and principal component analysis (PCA),28 augmented with self-reported ethnic and geographic origin (Supplementary Fig 1). Genomic inflation factor was λ=1.03 (Supplementary Fig 2). When compared to HAPMAP reference samples, NGRC clustered well with Caucasians. However, within NGRC, which is a typical mixed Caucasian European-American population, we found evidence for significant genetic diversity (PC1, P=2.9×10−5, Eigen-value=3.97; PC2, P=0.007, Eigen-value=1.26). Using self reported data on ancestry, we determined that the primary clusters correlate with Ashkenazi-Jewish and non-Jewish ancestry, and that the residual diversity in the larger non-Ashkenazi population correlates with the European country from which the subjects’ ancestors had immigrated to the US (Supplemental Fig 1 a-d). Demonstrating the existence of a significant substructure within Americans of European descent, although not surprising, has not been reported before, and may be a major reason for inconsistent findings in genetic association studies. The frequency of rs3129882 risk allele varied significantly in controls (P=0.0007), from 0.36 in Washington to 0.46 in New York. Similarly, we observed a frequency gradient across Europe, low in subjects with Northern-European ancestry and high in Southern-Europeans, particularly Italians (Supplementary Table 2). According to the US census, 14.4% of New Yorkers are Italian vs. 5.6% nationwide, which may explain the high allele frequency in NY. Within each subpopulation (US state, or original country) patients had a higher frequency of the HLA risk allele than controls, which supports association of HLA with PD. However, because of the variable allele frequency, if the population substructure within European Americans is not taken into account, other studies may find no difference or even an inverse association of this allele with disease depending on the mixed origins of their cases and controls. Future studies of HLA and PD will therefore require careful attention to ethnic and geographic origin of the American subjects of European descent.
To assure that the association with HLA was not confounded by population substructure, we corrected all analyses for the two significant PC that marked Jewish ancestry and European country of origin. Additionally, we confirmed that the association of PD with rs3129882 was present in genetically-defined core subsamples of both Jewish (0.04≤PC1≤0.055 and 0.001≤PC2≤0.013) and non-Jewish (−0.0075≤PC1≤0.0025 & −0.005≤PC2≤0.003) clusters (Supplementary Table 2, Supplementary Fig 1c). Lower-order PCs were not significant (PC3, P=0.32, Eigen-value=1.11) indicating that the bulk of genetic diversity within NGRC has been identified and accounted for. We investigated and ruled out the concern that the association of HLA with PD was driven by the association of HLA with PCs (Supplementary Fig. 5, 6, Supplementary Table 5).
The HLA variant that displays the strongest statistical association with PD, rs3129882, is a non-coding polymorphism in intron-1 of HLA-DRA gene (Fig. 2). The protein chains encoded by the closely linked HLA-DRA and HLA-DRB form the class II HLA-DR antigens, which are expressed by antigen presenting cells including microglia in brain, and interact with T-cell receptors. HLA-DRB chains are highly variable and have been associated with numerous disorders. HLA-DRA on the other hand is practically monomorphic and therefore has not been investigated for disease-association. The conventional explanation for our finding is that PD is associated with a classical polymorphic HLA antigen and that rs3129882 is a proxy. Alternatively, PD-association with an intronic DRA variant may represent involvement of regulatory elements, which would be in line with PD-specific over-expression of DR antigens in substantia nigra.29 We used University of Chicago’s expression quantitative trait loci (eQTL) data repository (http://eqtl.uchicago.edu/cgi-bin/gbrowse/eqtl/) to identify potential transcriptional variance associated with rs3129882. Two eQTL analyses, one using microarrays and the other RNA-seq, had identified rs3129882 as a cis-acting regulatory variation that correlated significantly (P=10−7 to 10−4) with expression levels of HLA-DRA, HLA-DQA2, and HLA-DRB5.10,11 One study reports the correlation as exon QTL which suggests the variant also affects alternative splicing expression.10,11
The evidence for genetic association with HLA, particularly when obtained from a hypothesis-free GWAS, lends strong and independent support to the involvement of neuro-inflammation12 and humoral immunity30 in PD pathogenesis. Studies have shown elevated DR expression in the brain29 and cerebrospinal fluid31 of PD patients. Sustained presence of reactive DR-positive microglia has been observed in substantia nigra of PD patients,29 as well as animals32 and humans33 affected with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced parkinsonism. It is postulated that the chronic immune activation and neuro-inflammation is in response to an initial trigger, possibly related to alpha-synuclein accumulation, and produce neurotoxins and oxidative damage that could kill neurons. From a therapeutic perspective, vaccination aimed at neutralizing neuro-immune dysfunction was recently shown to attenuate neurodegeneration in a PD model;34 furthermore, NSAID use is associated with reduced risk of developing PD in humans.4,13 The novel association with HLA highlights the involvement of an important biological pathway in the etiology of PD and a drug target that will stimulate research towards new therapies.
Methods
Human Subjects
Study was approved by Human Subject Committees at participating institutions. Patients and control subjects were recruited from eight NGRC-affiliated neurology clinics in Oregon, Washington, Georgia and New York. Methods were standardized across NGRC. Patients were diagnosed using the modified UK Brain Bank criteria.23 Controls were community volunteers and patient spouses; 353 (ages 67–90; mean 86.3±4.1 years) were evaluated by neurologists in Oregon and were free of neurodegenerative disease, the remaining 1633 (ages 21–90, mean 67.0±12.0 years) self-reported as neurologically healthy. GWAS subjects met seven criteria: (1) Self-reported Caucasian, non-Hispanic, of European origin. (2) Patients with current diagnosis of PD, excluding those whose initial PD diagnosis changed during the ~12 years of follow up. (3) DNA extracted from whole blood, unamplified, concentration ≥ 50ng/μl. (4) Age at blood draw ≥21 years. (5) Known gender. (6) Known age at onset (one missing). (7) No blood-relation to other subjects. We had data on smoking, coffee, and NSAIDs on ~3,000 subjects and on country of ancestral origin for 2,080 subjects. 112 subjects reported as Ashkenazi Jewish (129 were Jewish according to PCA).
Genotyping
To reduce plate effects, DNA samples were randomized on genotyping plates by case-control status, recruitment site, control subjects who were healthy at age ≥85 years, DNA extraction method, and DNA storage time. Samples were genotyped at the Johns Hopkins Center for Inherited Disease Research (CIDR). Data was released for 4,013 study samples (99.5% of attempted samples). Study samples, 90 duplicates, and 170 HapMap controls (151 CEU; 12 YRI, 3 JPT, 4 CHB) were genotyped using Illumina HumanOmni1-Quad_v1-0_B BeadChips (Illumina, San Diego, CA, USA) and the Illumina Infinium II assay protocol. Genotype cluster definitions for each SNP were determined using Illumina BeadStudio Genotyping Module version 3.3.7 and the combined intensity data from all released samples. Genotypes were not called if the quality threshold (Gencall score) was <0.15. Genotype data was released for 1,012,895 SNPs (99.65% of attempted). SNP assay failure criteria were: call rate <85%, cluster separation <0.2, >1 HapMap replicate error, >3% (autosomal) or >4% (X) difference in call rate between genders, >0.3% male AB frequency (X), or >8.8% (autosomal) or >13.2% (XY) difference in AB frequency. Y chromosome and mitochondrial SNPs were manually reviewed and clusters adjusted or genotypes dropped as appropriate. The mean non-Y SNP call rate and mean sample call rate were both 99.9% for the released dataset. Study duplicate reproducibility was ≥99.99% (Supplementary Table 1).
Statistical Analysis
Sex was determined by estimating X chromosome homozygosity and compared to self-reported gender; there was no discrepancy. We identified and excluded 1 patient and 3 controls who were inadvertently enrolled twice, and 13 cases and 10 controls for cryptic relatedness (PI-HAT>0.15). The final N for analysis was 2,000 patients and 1,986 controls. SNPs were excluded if MAF<0.01, call-rate<99%, HWE<10−6, MAF difference in males vs. females >0.15, or missing rate in PD vs. control P<10−5. 811,597 SNPs passed quality-control; with mean call rate of 99.92%. PCA was conducted with HelixTree (www.goldenhelix.com) using a pruned subset of 104,064 SNPs. Pruning was carried-out using PLINK with autosomal SNPs (MAF≥0.05, call rate ≥95%). We used a 50-SNP sliding-window that shifted 5 SNPs with each move and recursively removed SNPs with r2≥0.2, followed by a second round using a 138 SNP sliding-window, resulting in 104,064 SNPs.
Association was tested under an additive model using logistic regression in PLINK V1.07. The analyses were adjusted for sex, age, PC1 and PC2. Linkage disequilibrium (LD) was assessed using Haploview V4.135 and LocusZoom (http://csg.sph.umich.edu/locuszoom/). For replication of NGRC results, we chose datasets that (a) were published by peer review, (b) had genotyped rs3129882, and (c) were Caucasian. Among four publically-available GWAS only “CIDR: Genome Wide Association Study in Familial Parkinson Disease (PD)” by Pankratz et al.9 met our criteria. Two other GWAS have been published;7,8 one was available to us and was used as second replication.8 We applied the same sample and SNP quality-control filters to replication data sets as we did for NGRC, hence our results may vary slightly from their published results. Each replication dataset was individually tested using the tests and adjustments used by authors in their original report. Breslow-Day test-statistics was used to test heterogeneity across three datasets (P=0.6). We performed individual level Meta-analysis using Cochran-Mantel- Haenszel (CMH) test statistics accounting for study and gender. Chi-square test was used to test differences in MAF across disease, ethnic, and geographic strata.
To replicate previously reported GWAS findings, we used genotyped data when available and imputed SNPs that were not present on the array. We used PLINK for imputation, and included SNPs that had call rate ≥95% (information content metric value was >0.8). Meta-analysis was performed using CMH when individual level data was available, otherwise, we used aggregate (OR) data (PLINK was used). We used Breslow-Day test-statistics to test between-study heterogeneity for individual-level data and Q-test for aggregate-level data.
We used logistic regression to test combined effects of four loci (SNCA, MAPT, GAK, HLA). Subjects were classified by the total number of risk alleles (n) that they possessed (minimum=0, maximum=8); carriers of 0 or 1 risk allele were combined due to small numbers and set as the reference for comparison; carriers of 6 or more alleles were also combined due to small numbers at the extremes. Tests were performed comparing subjects with ‘n’ risk alleles to the reference group.
Access to material
Data are publically available via dbGaP (http://www.ncbi.nlm.nih.gov/gap).
Supplementary Material
Acknowledgments
We would like to acknowledge the Parkinson patients, their families and healthy volunteers who participated in this study. We thank Drs. Todd L. Edwards, Jeffery M. Vance, Eden R. Martin, Jonathan L. Haines, and Margaret A. Pericak-Vance for sharing their GWAS data with us; Drs. Richard H. Myers, James F. Gusella, Tatiana Foroud and Nathan Pankratz for making their data public via dbGaP; and Jacob Degner for assistance with the eQTL data repository website at University of Chicago. We acknowledge Ms. Marcia Adams, Ms. Michelle Zilka and the staff of CIDR for excellent genotyping service, Mr. Michael Palumbo, Mr. C. Steven Carmack and the staff of the Computational Biology and Statistics Core of Wadsworth Center for computing support, and Drs. Christophe Lambert and Greta Linse Peterson for developing the randomized plate layout. The project was supported by Award Number R01NS36960 from the National Institute of Neurological Disorders and Stroke. Additional support was provided by an award from The Michael J. Fox Foundation for Parkinson’s Research Edmond J. Safra Global Genetics Consortia initiative, Merit Review Award from the Department of Veterans Affairs (1I01BX000531), National Institutes of Aging (P30AG08017), National Institute of Mental Health (R21MH087336), Office of Research & Development, Clinical Sciences Research & Development Service, Department of Veteran Affairs, The Intramural Research Program of the NIH at National Library of Medicine, and the Close to the Cure Foundation. Genotyping services were provided by the Center for Inherited Disease Research (CIDR), which is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268200782096C. The study of Edwards et al. used for replication was funded by NIH grants AG027944 and NS039764. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Authors have no competing financial interest.
Author Contributions
HP established and directs the NGRC in collaboration with CPZ, SAF and JN. The GWAS was designed by and funded through HP. Subjects were ascertained, diagnosed, and characterized by NGRC investigators AS, AG, JR, SAF, JN and CPZ. DNA and phenotype preparations, data base operations, and final subject selection for GWAS were carried out by JM, DY, DMK, and VIK under the supervision of HP and CPZ. KFD was in charge of GWAS genotyping and genotyping quality control. THH performed all statistical analyses with critical feedback from AT, JP, EP, and HP. VIK and RC contributed to bioinformatics and graphic presentations. WKS provided an independent GWAS dataset for replication. AL uncovered the regulatory function of rs3129882 using bioinformatics. HP, THH and AT wrote the paper. All authors participated in reviewing results and assisting with manuscript preparation.
References
- 1.Payami H, Larsen K, Bernard S, Nutt J. Increased risk of Parkinson’s disease in parents and siblings of patients. Ann Neurol. 1994;36:659–661. doi: 10.1002/ana.410360417. [DOI] [PubMed] [Google Scholar]
- 2.Kay DM, et al. Genetic association between alpha-synuclein and idiopathic Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1222–30. doi: 10.1002/ajmg.b.30758. [DOI] [PubMed] [Google Scholar]
- 3.Zabetian CP, et al. Association analysis of MAPT H1 haplotype and subhaplotypes in Parkinson’s disease. Ann Neurol. 2007;62:137–144. doi: 10.1002/ana.21157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Powers K, et al. Combined effects of smoking, coffee and NSAIDs on Parkinson’s disease risk. Mov Disord. 2008;23:88–95. doi: 10.1002/mds.21782. [DOI] [PubMed] [Google Scholar]
- 5.McCulloch CC, et al. Exploring gene-environment interactions in Parkinson’s disease. Hum Genet. 2008;123:257–65. doi: 10.1007/s00439-008-0466-z. [DOI] [PubMed] [Google Scholar]
- 6.Maraganore DM, et al. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296:661–70. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- 7.Simon-Sanchez J, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–12. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edwards TL, et al. Genome-Wide Association Study Confirms SNPs in SNCA and the MAPT Region as Common Risk Factors for Parkinson Disease. Ann Hum Genet. 2010;74:97–109. doi: 10.1111/j.1469-1809.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pankratz N, et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stranger BE, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–24. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montgomery SB, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–7. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGeer PL, McGeer EG. Glial reactions in Parkinson’s disease. Mov Disord. 2008;23:474–83. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- 13.Chen H, et al. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann Neurol. 2005;58:963–7. doi: 10.1002/ana.20682. [DOI] [PubMed] [Google Scholar]
- 14.Ward CD, et al. Parkinson’s disease in 65 pairs of twins and in a set of quadruplets. Neurology. 1983;33:815–24. doi: 10.1212/wnl.33.7.815. [DOI] [PubMed] [Google Scholar]
- 15.Tanner C, et al. Parkinson disease in twins: An Etiologic study. JAMA. 1999;281:341–346. doi: 10.1001/jama.281.4.341. [DOI] [PubMed] [Google Scholar]
- 16.Thacker EL, Ascherio A. Familial aggregation of Parkinson’s disease: A meta-analysis. Mov Disord. 2008;23:1174–83. doi: 10.1002/mds.22067. [DOI] [PubMed] [Google Scholar]
- 17.Mata IF, et al. A SNCA Variant Associated with Parkinson’s Disease and Plasma α-Synuclein Level. Archives of Neurology. 2010 doi: 10.1001/archneurol.2010.279. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernan MA, Takkouche B, Caamano-Isorna F, Gestal-Otero JJ. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann Neurol. 2002;52:276–84. doi: 10.1002/ana.10277. [DOI] [PubMed] [Google Scholar]
- 19.Satake W, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–7. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 20.Fung HC, et al. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006;5:911–6. doi: 10.1016/S1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- 21.Maraganore DM, et al. High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet. 2005;77:685–93. doi: 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamza TH, Payami H. The heritability of risk and age at onset of Parkinson’s disease after accounting for known genetic risk factors. J Hum Genet. 2010;55:241–3. doi: 10.1038/jhg.2010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibb W, Lees A. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1988;51:745–752. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125:861–70. doi: 10.1093/brain/awf080. [DOI] [PubMed] [Google Scholar]
- 25.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kay DM, et al. Parkinson’s disease and LRRK2: frequency of a common mutation in U.S. movement disorder clinics. Mov Disord. 2006;21:519–23. doi: 10.1002/mds.20751. [DOI] [PubMed] [Google Scholar]
- 27.Devlin B, Roeder K, Wasserman L. Genomic control, a new approach to genetic-based association studies. Theor Popul Biol. 2001;60:155–66. doi: 10.1006/tpbi.2001.1542. [DOI] [PubMed] [Google Scholar]
- 28.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 29.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–91. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 30.Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM. A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain. 2005;128:2665–74. doi: 10.1093/brain/awh625. [DOI] [PubMed] [Google Scholar]
- 31.Fiszer U, Mix E, Fredrikson S, Kostulas V, Link H. Parkinson’s disease and immunological abnormalities: increase of HLA-DR expression on monocytes in cerebrospinal fluid and of CD45RO+ T cells in peripheral blood. Acta Neurol Scand. 1994;90:160–6. doi: 10.1111/j.1600-0404.1994.tb02699.x. [DOI] [PubMed] [Google Scholar]
- 32.McGeer PL, Schwab C, Parent A, Doudet D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann Neurol. 2003;54:599–604. doi: 10.1002/ana.10728. [DOI] [PubMed] [Google Scholar]
- 33.Langston JW, et al. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 34.Reynolds AD, et al. Regulatory T cells attenuate th17 cell-mediated nigrastriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J Immunol. 2010;184:2261–2271. doi: 10.4049/jimmunol.0901852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.