

Abstract
β-arrestins are scaffolding proteins implicated as negative regulators of TLR4 signaling in macrophages and fibroblasts. Unexpectedly, we found that β-arrestin-1 (β-arr-1) and −2 knockout (KO) mice are protected from TLR4-mediated endotoxic shock and lethality. To identify the potential mechanisms involved, we examined the plasma levels of inflammatory cytokines/chemokines in the wild type (WT) and β-arr-1 and −2 KO mice after lipopolysaccharide (LPS, a TLR4 ligand) injection. Consistent with lethality, LPS-induced inflammatory cytokine levels in the plasma were markedly decreased in both β-arr-1 and −2 KO, compared to WT mice. To further explore the cellular mechanisms, we obtained splenocytes (separated into CD11b+ and CD11b−populations) from WT, β-arr-1 and −2 KO mice and examined the effect of LPS on cytokine production. Similar to the in vivo observations, LPS-induced inflammatory cytokines were significantly blocked in both splenocyte populations from the β-arr-2 KO compared to the WT mice. This effect in the β-arr-1 KO mice, however, was restricted to the CD11b− splenocytes. Our studies further indicate that regulation of cytokine production by β-arrestins is likely independent of MAPK and IκBα-NFκB pathways. Our results, however, suggest that LPS-induced chromatin modification is dependent on β-arrestin levels and may be the underlying mechanistic basis for regulation of cytokine levels by β-arrestins in vivo. Taken together, these results indicate that β-arr-1 and −2 mediate LPS-induced cytokine secretion in a cell-type specific manner and that both β-arrestins have overlapping but non-redundant roles in regulating inflammatory cytokine production and endotoxic shock in mice.
Introduction
Toll-like receptors (TLRs) are pattern recognition receptors (PRRs) identified for their role in mediating signaling from the pathogen-associated molecular patterns (PAMPs) from microbial products (Beutler, 2004; Beutler, 2005; Beutler, 2009). TLRs are now implicated in a number of acute and chronic inflammatory diseases including endotoxemia and endotoxic shock (Sabroe et al., 2008; Salomao et al., 2008). Of particular importance, activation of Toll-like receptor-4 (TLR4) (stimulated by lipopolysaccharide (LPS) from gram-negative bacteria) is proposed as one of the primary mechanisms in the pathophysiology of endotoxemic shock and lethality in animals and humans (Armstrong et al., 2004; Daubeuf et al., 2007; Leon et al., 2008; Lorenz et al., 2002; O’Brien et al., 2005; Salomao et al., 2008; Spiller et al., 2008). Endotoxemia and lethality in turn are mediated by production of pro- and anti-inflammatory cytokines/chemokines, the balance of which subsequently determines the disease outcome. LPS stimulation of TLR4 causes an increase in Inflammatory cytokines/chemokines production by mediating cell signaling pathways including that of MAPK and NFκB pathways (Miggin and O’Neill, 2006; O’Neill, 2008), (Symons et al., 2006), (Hayden and Ghosh, 2004; Sun and Ley, 2008).
MAPK signaling typically involves the ERK, JNK and p38 MAPK pathways. These kinases further phosphorylate a number of proteins including transcription factors leading to changes in gene expression (Symons et al., 2006). NFκB pathway is regulated by activation of IκB kinase (IKK) complex, which phosphorylates cytosolic IκB proteins (e.g. IκBα, a well-known member). Under unstimulated conditions IκBα binds to NFκB transcription factors (p50/p65) and retain them in the cytosol. Upon phosphorylation by IKK complex, IκBα proteins undergo ubiquitination and proteosome-dependent degradation resulting in the release of NFκB transcription factors (Hayden and Ghosh, 2004; Sun and Ley, 2008). These transcription factors then translocate into the nucleus and evoke expression of cytokines and chemokines. In addition to MAPK and NFκB signaling pathways, recent studies have clearly demonstrated an important role for chromatin modification, especially histone acetylation in the regulation of cytokine gene expression (Kuo and Allis, 1998). In this regard, histone acetyl transferase (HAT) and histone deacetylase (HDAC) activities have been shown to play a crucial role in cytokine expression and inhibition of these enzymes have profound effects on LPS-induced cytokine production ((Barnes et al., 2005; Choi et al., 2009a; Choi et al., 2009b; Choi et al., 2008; Leoni et al., 2005)).
Arrestins are scaffolding proteins originally discovered for their role in G-protein coupled receptor desensitization. There are four members of the arrestin family (Arrestin-1 to −4), of which arrestin-1 and −4 are restricted to the eyes and arrestin-2 and −3 are ubiquitously distributed. Arrestin-2 (also called β-arrestin-1) and Arrestin-3 (also called β-arrestin-2) are best known for their role in the desensitization and trafficking ofβ-adrenergic receptor (DeWire et al., 2007). Subsequent studies have demonstrated a much broader role for β-arrestins in cell signaling. Although individual knockout of β-arrestin-1 and −2 are viable with some unique phenotypes, homozygous double knockout of β-arrestin-1 and −2 is embryonically lethal, suggesting that at least one of the β-arrestins need to be present for proper embryonic development. Studies have also shown that depending on the cellular context and receptor examined, β-arrestins either mediate or inhibit receptor signaling. This is also the case for TLR4 signaling where β-arrestins’ role is dependent on the cell type (Fan et al., 2007; Parameswaran et al., 2006; Wang et al., 2006). While these studies were primarily done in cell culture and in vitro models, the precise role of β-arrestins in TLR4 signaling in vivo is not clearly understood. Using both in vivo and ex vivo model systems, we demonstrate here that both β-arrestins are positive regulators of TLR4-induced inflammatory cytokine production and endotoxemia in mice and therefore suggest that β-arrestins may be potential drug targets in the treatment of TLR4-mediated inflammatory diseases.
Materials and Methods
Animals
β-arrestin-1 and −2 KO mice have been backcrossed to C57BL6 background for more than 10 generations and have been described previously (Rajagopal et al., 2006). They were kindly provided by Dr. Robert Lefkowitz (Duke University). Wild type control C57BL6 mice were purchased from Jackson Labs. Animals were housed four to five mice per cage at 22–24°C in rooms with 50% humidity and a 12-h light–dark cycle. All animals were given mouse chow and water ad libitum. Purchased mice were housed for at least 1 week before experimental use, and age- and sex-matched animals (~2–3 month old males) were employed as described. All animal procedures were approved by the Michigan State University Institutional Animal Care and Use Committee and conformed to NIH guidelines.
Survival study
Male mice (~2–3 month old) were injected with LPS (E. Coli 0111:B4 from Sigma) intraperitoneally (20 μg/g body weight) and survival of mice monitored for 48 hours (Greten et al., 2007).
Cytokine assays
A mouse 23-plex multiplex-based assay was used to determine the indicated cytokine/chemokine plasma concentrations as per the manufacturer’s instructions (Bio-Rad) via Luminex 100 technology as previously described (Appledorn et al., 2008). For ELISA assays, IL1β, IL-6, IL-10 and TNFα ELISA kits were obtained from eBiosciences and performed per manufacture’s instructions. ELISA assays for IL12, IL5 and IFNγ were performed as described previously (Navuluri et al., 2006; Parvataneni et al., 2009).
Quantitative RT-PCR
To determine relative levels of a specific RNA transcript, tissues were snap-frozen in liquid nitrogen and RNA was harvested using TRIzol reagent (Invitrogen) according to manufacturer’s protocol and as described before (Appledorn et al., 2008; Loniewski et al., 2008). Synthesis of cDNA was performed with reverse transcriptase (RT) with the total RNA using the superscript II kit with Oligo-dt (12–18) primers as recommended by the manufacturer (Invitrogen). cDNA was amplified by PCR in a final reaction volume of 25 μl using SYBR Green Supermix (Invitrogen) with 10 pmol of each primer (Qiagen quantitect primers for COX2 and 18s rRNA primers from IDT). Real-time PCR was performed in MX3000P (Stratagene) thermocycler. RNA free samples (negative control) did not produce any amplicons. Melting curve and gel analysis were used to verify single products of the appropriate base-pair size. Data were analysed with MX3000P software using 18s rRNA for normalization.
Splenocyte preparations
Mouse splenocytes were obtained and cultured as previously described (Parvataneni et al., 2009). Isolation of CD11b+ and CD11b− populations was performed using CD11b antibody (Miltenyi Biotec) and by positive selection (for CD11b+) using MACS column (Miltenyi Biotec) according to manufacturer’s instructions. The negatively selected splenocytes by this method were designated CD11b− cells.
Flow cytometric analysis of Cell surface TLR4 expression
CD11b+ splenocytes were obtained as described above. For bone marrow cells, bone marrows were flushed from tibiae and femurs of wild type, β-arrestin-1 and −2 KO mice and CD11b+ cells were then separated as described for the splenocytes using the MACS column. Cells were then rinsed with staining buffer (1% FBS and 0.09% sodium azide in PBS) and stained with the following antibodies: anti-CD11b-FITC, anti-TLR4-PE, anti-CD3-PerCp-Cy5.5, anti-CD19-PE-Cy7. TLR4 expression was analyzed using LSRII (BD Biosciences) and Flowjo software (Tree Star).
Western blotting
Splenocytes were lysed in lysis buffer (20 mM Tris-HCl (pH 7.4), 1 mM EDTA, 150 mM NaCl) containing 1% Triton X-100 with protease inhibitors. Cell lysates were then centrifuged at maximum speed (13,000 × g) for 10 min at 4°C and the protein concentration of the supernatant determined using Bradford method. Western blotting for p-ERK1/2, ERK2, IκBα, pJNK1/2, JNK1/2, pP38, and tubulin were performed as previously described (Loniewski et al., 2008; Loniewski et al., 2007; Patial et al., 2010). Briefly, equivalent concentrations of protein samples were run on polyacrylamide gels and transferred to nitrocellulose membranes. Blots were then probed with primary and fluorescent secondary antibodies as described (Patial et al., 2010). Blots were scanned and bands were quantified using Li-COR Odyssey scanner. For data analysis, p-ERK1/2 bands were normalized to ERK2, and IκBα levels to tubulin/actin for quantitation.
HAT and HDAC activities
HAT and HDAC activities were assayed using nuclear extracts of the spleen obtained from mice injected with LPS as indicated. Nuclear extracts from the spleen were obtained as described before (Blackwell et al., 1997). HAT activity was performed using the HAT activity colorimetric assay kit from BioVision Research Product (California) following manufacturer’s assay protocol. HDAC activity was performed using HDAC activity assay kit from Caymen, as per manufacturer’s protocol.
Statistics
All data in the figures and text are expressed as mean±SE and data analyzed using GraphPad Prism Software. Each “N” represents individual mouse. The Student’s t-test was used to compare mean values between two experimental groups and Analysis of Variance (with Bonferroni Post-test) for more than two groups. Differences in the survival were determined using the log-rank test. P Values <0.05 were considered significant.
Results and Discussion
LPS-induced mortality is inhibited in β-arrestin-1 and −2 knockout male mice
To determine the role of β-arrestins in LPS-induced endotoxic shock, we injected wild type,β-arrestin-1 (β-arr-1) and −2 knockout mice with LPS (20 μg/g body weight intraperitoneally) and monitored the survival of the three groups of mice for 48 hours. As shown in Fig 1, both β-arrestin knockout mice survived significantly better than the wild type mice. Only 10% of the wild type mice survived by 48 hours compared to 58% of β-arr-1 and 50% of β-arr-2 knockout mice. While the median survival of the wild type mice was 34 hours, median survival of the β-arr-2 KO mice was 47.5 hours and was longer than 48 hours for β-arr-1 KO mice. The surprisingly better survival of β-arr-2 KO male mice is in contrast to the findings of Wang et al who showed that mortality in female β-arr-2 KO mice is significantly quicker than the wild type mice in a Galactosamine-D (Gal-D) sensitized model of endotoxic shock (Wang et al., 2006). Our studies using the Gal-D-sensitized model of endotoxic lethality in males, exhibited the same phenotype as that of our LPS model, i.e. both β-arrestin-1 and −2 knockout had significantly lower mortality rates compared to wild type mice (data not shown). Thus, it is possible that the differences in the study results may be due to the different genders of mice used. Ample evidence indicates that male and female rodents respond quite differently with respect to inflammatory agents (Card et al., 2006; Marriott et al., 2006; Marriott and Huet-Hudson, 2006; Rettew et al., 2009; Rettew et al., 2008). Studies by Rettew et al (Rettew et al., 2009; Rettew et al., 2008) especially have shown that while testosterone reduces the cell surface expression of TLR4 in mouse macrophages, estrogen augments this effect. In addition, removal of endogenous estrogen decreases the plasma levels of inflammatory cytokines in LPS-injected mice. Whether the levels of estrogen are different between the female wild type and β-arrestin KO mice is not known and if the effects observed by Wang et al (Wang et al., 2006) may be attributed to the differences in the levels of TLR4 is also not clear. Interestingly, however, cell surface expression of TLR4 in male wild type and β-arrestin-1 and −2 knockout mice were similar in our studies, as determined by flow cytometry (Fig 2). These results suggest that at least in the males, β-arrestins do not regulate basal cell surface expression of TLR4. Taken together, the difference between our studies and that of Wang et al suggest that the role of β-arrestins in modulating specific inflammatory responses may differ between males and females. Furthermore, our results clearly demonstrate that β-arrestin-1 and −2 are important modulators of endotoxic lethality in male mice.
Figure 1. Endotoxin-induced mortality in wild type, β-arrestin-1 and −2 knockout male mice.

Wild type, β-arrestin-1 and −2 knockout mice were injected intraperitoneally with LPS (20 μg/g body weight) and survival monitored for 48 hours. Survival curves were analyzed using Kaplan-Meier analysis with Graphpad PRISM software. P value = 0.05 for β-arrestin-1 KO mice compared to wild type; P value<0.05 for β-arrestin-2 KO mice compared to wild type. N=10 mice for wild type and β-arrestin-2 KO each and N=12 mice for β-arrestin-1 KO mice. *P<0.05 compared to wild type by Fisher Exact Test.
Figure 2. TLR4 expression in CD11b+ splenocytes (A) and bone marrow cells (B) from wild type, β-arrestin-1 and −2 KO mice.
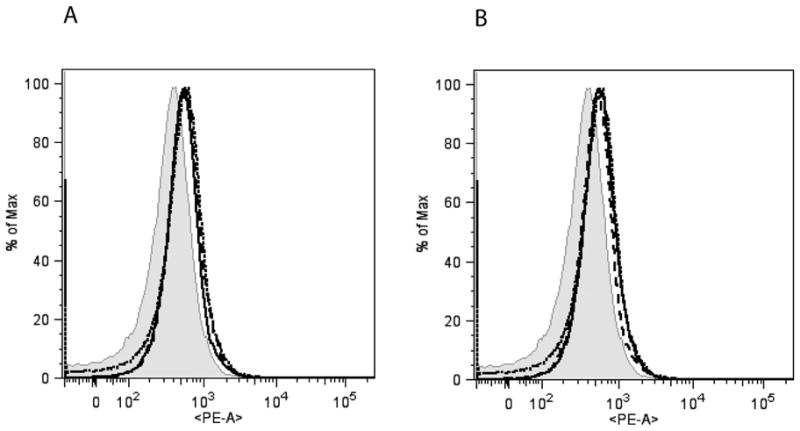
Cell surface expression of TLR4 in CD11b+ splenocytes and bone marrow cells was determined by Flow-cytometry as described in the Methods. A representative experiment (showing the mean fluorescence intensity) from two such experiments is shown. Wild type=dotted line; β-arrestin-1 KO=dashed line and β-arrestin-2 KO=solid line; Isotype control (wild type)=shaded.
β-arrestins positively regulate LPS-induced inflammation in vivo
Endotoxic shock in mice is mediated by a complex array of pro- and anti-inflammatory cytokines and chemokines. Levels of these cytokines/chemokines increase in the plasma with in hours after LPS injection and are strongly associated with mortality in mice. Based on the survival results, we hypothesized that β-arrestins either mediate pro-inflammatory cytokines/chemokines production or inhibit anti-inflammatory cytokines such as IL-10. To test this hypothesis, we injected wild type, β-arr-1 and −2 KO mice with LPS (20 μg/g body weight, intraperitoneally) and determined the plasma levels of various cytokines and chemokines (using 23-plex multiplex cytokine assay) at 1, 3, 6 and 12 hours after injection. Levels of these cytokines/chemokines were also determined in uninjected as well as PBS injected mice to estimate basal/control levels. We did not find any difference in the basal levels of the cytokines between the genotypes (data not shown). Our results however, indicate distinct as well as overlapping roles for β-arrestins in LPS-induced cytokine regulation (Fig 3). Plasma levels of IL-1β, IL12p40, IFNγ, IL-2, IL-3, IL-4, and IL-5 were significantly inhibited in both β-arrestin knockout mice compared to the wild types at the corresponding time points after LPS injection (Fig 3A). Some cytokines (IL1α, IL-13, IL-17, Eotaxin and KC) however, were inhibited only in the β-arrestin-2 knockout mice (Fig 3B). Importantly, levels of some cytokines/chemokines (IL-9, IL-10, IL-6, IL-12p70, G-CSF, GM-CSF, MCP-1, MIP1α and RANTES) were similar between wild types and β-arrestin-1 & −2 knockout mice suggesting selective regulation of inflammatory response by β-arrestins (Fig 3D). Interestingly, as shown by Wang et al (Wang et al., 2006) (for TNFα), LPS-induced TNFα (at 1h) and MIP1β (at 3h) levels were significantly elevated in the β-arrestin-2 knockout mice compared to the wild types (Fig 3C). Given that many pro-inflammatory cytokines are inhibited in the β-arrestin-2 KO mice, the pathophysiological consequence of the negative regulation of TNFα and MIP1β in the current model is not clear. We initially reasoned that the enhanced TNFα in the β-arrestin-2 KO mice might render them more susceptible for LPS/D-Gal-mediated shock (Lehmann et al., 1987), as was shown by Wang et al (Wang et al., 2006). As indicated earlier, however, the survival of the male β-arrestin-2 KO mice was better in the LPS/D-Gal-mediated shock, compared to the wild type (data not shown). Thus the consequence(s) of the initial negative regulation of TNFα and MIP1β remains unclear in this model.
Figure 3. Inflammatory response in wild type, β-arrestin-1 and −2 knockout mice after lipopolysaccharide challenge.


Wild type, β-arrestin-1 and −2 KO mice were injected intraperitoneally with LPS (20 μg/g body weight) or vehicle and mice euthanized and plasma samples collected at the indicated time points. Cytokine/chemokine levels were measured using Biorad’s 23-plex cytokine assay. Cytokine/chemokine levels were undetectable or were not different between uninjected WT, β-arr-1 and −2 KOs (data not shown). Cytokines/chemokines positively regulated by both β-arrestins are shown in (A), cytokines/chemokines regulated only by β-arrestin-2 are shown in (B) and TNFα and MIP1β levels (which are negatively regulated by β-arrestin-2) are shown in (C). Cytokines/chemokines not regulated by β-arrestins in vivo are shown in (D). N=5 mice per genotype for each time point. Data were analyzed by two-way ANOVA with Bonferroni post-test. *p<0.05, **p<0.01, ***p<0.001 compared between β-arrestin-1 KO and wild type mice; #p<0.05, ##p<0.01, ###p<0.001 compared between β-arrestin-2 KO and wild type mice.
Previous studies have demonstrated a strong association between the plasma cytokine levels in sepsis and mortality in animals and humans [reviewed in (Rittirsch et al., 2008)]. In the present study, the better survival of the β-arr-1 and −2 KO mice is strongly associated with the diminished pro-inflammatory cytokine responses observed in the KO mice. In particular, many pro-inflammatory cytokines are significantly attenuated in the β-arrestin-1 and −2 knockout mice in response to LPS injection. Many of these cytokines have also been shown to have significant effects in the pathogenesis of endotoxemia (Greten et al., 2007; Kohler et al., 1993; Wysocka et al., 1995; Zisman et al., 1997). Thus, even though levels of anti-inflammatory cytokine such as IL-10 did not differ between the genotypes, the profound decrease in the pro-inflammatory cytokines (especially IL1β, IFNγ and IL12) is probably sufficient to tip the “balance” towards the anti-inflammatory phenotype observed in the KO mice, and this may be associated with the better survival of these mice.
Regulation of Cyclooxygenase-2 expression by β-arrestins
In addition to the panel of inflammatory cytokines, LPS also induces the expression of cyclooxygenases (COX), enzymes that play a key role in the production of prostaglandins (PGs). Of the various PGs, PGE2 has been shown to be critical in the pathogenesis of endotoxemia and sepsis (reviewed in (Cook, 2005; Funk, 2001). Two isoforms of COX have been identified: COX1 and COX2. COX1 is constitutively expressed and is responsible for basal constitutive prostaglandin synthesis. COX2, however, is induced under various inflammatory states including endotoxemia and importantly, selective inhibition of COX2 has been shown to improve early survival in mouse model of endotoxemia (Reddy et al., 2001). Because deletion of β-arrestins in mice appears to be protective in terms of survival in endotoxemia, we hypothesized that in addition to the regulation of inflammatory cytokine production, prostaglandin production would also be attenuated in the β-arrestin-1 and −2 KO mice. To test this, we determined the levels of ptgs2 (mRNA for COX2) in the spleen of mice injected with LPS for various time points. Similar to the cytokine response, our results demonstrate that the expression of COX2 is markedly inhibited in both β-arrestin-1 and −2 KO mice compared to the wild types (Fig 4), suggesting that this may also contribute to the better survival of the β-arrestin KO mice. Whether the decreased levels of COX2 in the β-arrestin-1 and −2 KO mice is a result of, or a consequence from, diminished cytokine responses is unclear and will be tested in future studies.
Figure 4. Regulation of LPS-induced ptgs2 mRNA by β-arrestins in vivo.
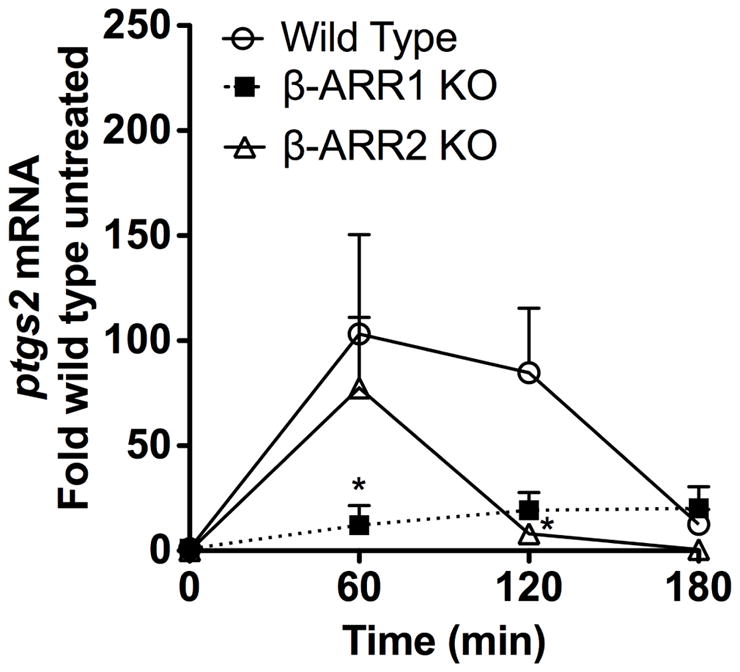
Wild type, β-arrestin-1 and −2 KO mice were injected with LPS as described in Fig 2. Spleens were collected at the indicated time points after LPS-injection or from untreated mice (indicated as 0 time point). Messenger RNA levels for ptgs2 was determined by real-time Q-RT-PCR. N=3 mice per genotype for each time point. Data were analyzed by two-way ANOVA with Bonferroni post-test. *p<0.05 compared between β-arrestin-1 or −2 and wild type mice at the respective time points.
Selective role of β-arrestin-1 and −2 in CD11b+ and CD11b− splenocytes
Our results so far suggest that both β-arrestins have overlapping as well as distinct functions in terms of cytokine production. To further elucidate the cellular and signaling mechanisms by which β-arrestins mediate the effects described above, we focused our studies on the splenocytes. We chose spleen because of its known role in the inflammatory responses observed in endotoxemia and sepsis models (Deriy et al., 2002; Moeniralam et al., 1997; Senaldi et al., 1999). We collected spleens from wild type, β-arrestin-1 and −2 knockout mice and separated the splenocytes into CD11b+ and CD11b− populations (using a MACS column with CD11b antibodies). The negatively selected population in this manner was designated as CD11b− population. On an average the CD11b+ population was about 5–10% of the total splenocyte population and using the MACS column we obtained about 85–90% pure CD11b+ cells as assessed by flow cytometry (data not shown). Furthermore, the CD11b− population was ~95% pure (assessed by flow cytometry). Approximately 50% of the CD11b+ cells were also positive for Gr-1 (marker for granulocytes, monocytes and plasmacytoid dendritic cells). Furthermore, macrophages (F4/80+) constituted ~30% of the CD11b+ population whereas myeloid dendritic cells (CD11c+) constituted ~11% as assessed by flow cytometry (data not shown). Importantly, the CD11b− population contained mainly T- (CD3+, ~42%) and B-lymphocytes (CD19+, ~49%) and insignificant proportion of macrophages (F4/80+, 1%) and myeloid dendritic cells (CD11c+, 0.875%) as assessed by flow cytometry (data not shown). Both CD11b+ and CD11b− populations were treated with LPS for various time points (0, 24, 48 and 72 hours) and the culture supernatants were collected and used for cytokine assay. We examined the levels of IL-1β, IL-12 (p40 and p70), IL-5, IFN-γ, IL-6, IL-10 and TNFα using an ELISA assay. These cytokines were chosen based on the in vivo responses observed in the β-arrestin knockout mice after LPS injection as well as on the known role of these cytokines in endotoxic shock (reviewed in (Rittirsch et al., 2008)). In the wild type CD11b+ splenocytes, LPS treatment caused a significant increase in IL-1β, IL-12 (p40 & 70), IFNγ, IL-6, IL-10 and TNFα (Fig 5 and 6). There was, however, no detectable increase in IL-5 in the CD11b+ splenocytes (data not shown). Although these responses were similar in the β-arr-1 KO cells, levels of IL-1β, IL-12, and IFN-γ were significantly blocked in the β-arr-2 KO splenocytes, similar to in vivo observations (Fig 5). Interestingly, levels of IL-6 and TNFα (but not IL-10) were also modestly inhibited in the β-arrestin-2 knockout splenocytes suggesting that the local production of these two pro-inflammatory cytokines may be potentially regulated by β-arrestin-2 similar to that of IL-1β, IL12 and IFNγ. It should be noted, however, that even though TNFα levels were enhanced in vivo in the β-arrestin-2 KO mice after LPS injection, in vitro treatment of splenocytes resulted in a modest inhibition. Whether this can be attributed as an artifact of in vitro experiment or to the difference in the kinetics of in vivo v/s in vitro production of TNFα is not known. Furthermore, IFNγ secretion was enhanced in the β-arrestin-1 knockout cells (compared to wild types) but the significance of this to the in vivo findings is also not clear. Together, however, these results suggest that β-arrestin-2 but not β-arrestin-1 selectively mediates LPS-induced production of IL-1β, IL-12 and IFNγ in CD11b+ splenocytes. All three of these cytokines have been shown to play a major role in the development of endotoxic shock (Greten et al., 2007; Kohler et al., 1993; Wysocka et al., 1995; Zisman et al., 1997) and thus reduction in these cytokines observed in both in vivo and in vitro experiments in the β-arrestin-2 KO mice may in part explain the resistance of this genotype to endotoxic shock.
Figure 5. Regulation of IL-1β (A), IL-12 (B&C) and IFNγ (D) production by β-arrestins in CD11b+ splenocytes.

CD11b+ splenocytes were obtained as described in the methods. Cells were treated or not with LPS (5 μg/ml) for the indicated time points, supernatants collected and cytokine levels determined by ELISA. The levels (in pg) were then normalized to μg of total cellular protein. N=5 mice per genotype. Data were analyzed by two-way ANOVA with Bonferroni post-test. #p<0.05, ##p<0.01, ###p<0.001 compared between β-arrestin-2 KO and wild type mice.
Figure 6. Regulation of IL-6 (A), IL-10 (B), and TNFα (C) production by β-arrestins in CD11b+ splenocytes.

Cell culture supernatants from LPS-treated (24 hour time-point) CD11b+ splenocytes (experiment as described in Figure 4) were also tested for the levels of IL-6, IL-10 and TNFα by ELISA. N=5 mice per genotype.
In CD11b− splenocytes, LPS treatment significantly increased IL1β, IL12p70, IL-5 and IFNγ, (but not IL12p40) in the wild type mice (Fig 7 and data not shown for IL12p40). In contrast to the results in CD11b+ splenocytes, LPS-induced IL-1β, IL12p70, IFNγ and IL-5 were markedly inhibited in both β-arr-1 and −2 knockout splenocytes (CD11b− population) (Fig 7). Using this in vitro model system, our results reveal that the role of β-arrestin-1 and −2 are exquisitely cell type specific, in that, TLR4 signaling (with respect to cytokine production) is mediated only by β-arrestin-2 in the CD11b+ splenocytes, whereas TLR4 signaling is mediated by both β-arrestins in the CD11b− splenocytes. Further in vivo studies are obviously necessary to distinguish the subpopulation of the splenocytes to determine which subtype of immune cell population is involved in this regulation. Based on our current analysis that, the proportion of macrophages and dendritic cells between the CD11b+ and CD11b− population is markedly different, suggests that the role of β-arrestin-2 may be selective for macrophages/dendrtic cells (predominant in the CD11b+ population). On the contrary, both β-arrestins may be required for TLR4 signaling in lymphocytes (the predominant splenocyte population in the CD11b− cells, which constitute 90–95% of the total splenocyte population).
Figure 7. Regulation of IL-1β (A), IL-12 (B), IL-5 (C) and IFNγ (D) production by β-arrestins in CD11b− splenocytes.

Splenocytes were obtained as described in the methods. Cells were treated with LPS and ELISA and data analysis performed as described in Fig 3. N=5 mice per genotype. Data were analyzed by two-way ANOVA with Bonferroni post-test. *p<0.05, **p<0.01, ***p<0.001 compared between β-arrestin-1 KO and wild type mice; #p<0.05, ##p<0.01, ###p<0.001 compared between β-arrestin-2 KO and wild type mice.
Role of β-arrestins in LPS-induced NFκB and MAPK pathways in the splenocyte populations
To understand the signaling mechanisms mediating the cytokine response in the β-arrestin-1 and −2 KO mice, we first examined the various components of the NFκB and MAPK signaling pathways in the CD11b+ and CD11b− splenocyte populations after 30 and 60 minutes of LPS treatment. To assess the NFκB pathway, we determined the levels of IκBα in total cell lysates. Previous studies have shown thatβ-arrestins negatively regulate signal-induced IκBα degradation in cells in culture (Gao et al., 2004; Wang et al., 2006; Witherow et al., 2004). In this model, however, we did not observe any significant role for β-arr-1 or −2 in IκBα degradation in either CD11b+ or CD11b− splenocytes (Fig 8). These results suggest that the cytokine effects observed in the β-arr-1 and −2 KO splenocytes are independent of IκBα and therefore likely to be independent of the canonical NFκB pathway.
Figure 8. Regulation of IκBα levels by β-arrestins in CD11b+ (A) and CD11b− (B) splenocytes.

Splenocytes were obtained as described in the methods. Cells were then treated with LPS for the indicated time points and IκBα levels determined by Western blotting as described in the methods. IκBα levels were normalized to tubulin/actin for quantitation. N=4 mice per genotype. Quantitation is shown in the top and representative blots in the bottom.
β-arrestins have also been shown to be important regulators of MAPK signaling pathways and depending on the cellular context, β-arrestins mediate or inhibit MAPKs. In particular, previous studies have shown that β-arrestins act as negative regulators as well as positive regulators of TLR4-induced ERK signaling pathway in different cell types (Fan et al., 2007; Parameswaran et al., 2006). Therefore, we examined the role of these MAPK pathways in the splenocyte population to determine if activation of these kinases is impaired in the β-arrestin1/2 KO mice cells. For this, we examined the levels of pERK, pJNK and pP38 in both CD11b+ and CD11b− splenocyte populations. In the CD11b+ cells, LPS treatment increased pERK1/2 levels in the wild type mice and these effects were similar in the β-arrestin-1 and −2 knockout mice (Fig 9). In CD11b− splenocyte population however, there was no ERK activation (data not shown). Furthermore, we did not observe any consistent increase in either phospho-JNK or phospho-p38 after LPS treatment in the wild type splenocytes (both CD11b+ and CD11b−) (data not shown). Taken together, these results indicate that neither β-arr-1 norβ-arr-2 KO mice splenocytes are defective in either IκBα or MAPK signaling. Therefore these results suggest that β-arrestins likely mediate TLR4-induced cytokine production independent of these pathways.
Figure 9. Regulation of ERK1/2 phosphorylation by β-arrestins in CD11b+ splenocytes.

CD11b+ splenocytes were treated with LPS for the indicated time points and lysates were Western blotted for pERK1/2 and ERK2 levels. pERK1/2 levels were normalized with ERK2 for quantitation. N=4 mice per genotype. Quantitation is shown in the top and representative blots in the bottom.
β-arrestins regulate LPS-mediated histone deacetylase and histone acetyl transferase activities in vivo
Recent studies have suggested that β-arrestins not only play a role in cell signaling pathways, but also play an important role in gene expression by regulating histone acetyl transferases (HATs) (Kang et al., 2005). Other studies have shown that in addition to HATs, histone deacetylases also play a crucial role in chromatin modification and gene expression and that the balance of HAT and HDAC might eventually determine the outcome on gene expression. In addition, studies have shown that pharmacological modulation of HATs and HDACs lead to marked changes in inflammatory gene expression (Aung et al., 2006; Barnes et al., 2005; Choi et al., 2009a; Choi et al., 2009b; Choi et al., 2008; Kuo and Allis, 1998; Leoni et al., 2005; Sailhamer et al., 2008; Yu et al., 2002). Because neither IκBα nor MAPK signaling was consistently affected in β-arrestin-1 and −2 KO mice, we reasoned that β-arrestins might modulate chromatin modification and thus mediate production of several cytokines. To test this hypothesis, we obtained nuclear extracts from the spleen of wild type, β-arrestin-1 and −2 KO mice before and after LPS injection and performed HAT and HDAC activities. As shown in Fig 10, LPS injection in wild type mice led to a marked increase (2.5 fold) in the ratio of HAT:HDAC activity compared to the untreated wild type mice. Interestingly, compared to the wild type mice, HAT:HDAC ratio after LPS injection did not change in either β-arrestin-1 or −2 KO mice. These results suggest that LPS-induced increase in HAT:HDAC ratio in vivo, is mediated by β-arrestins.
Figure 10. Regulation of Histone deacetylase and histone acetyl transferase activities by β-arrestins in vivo.
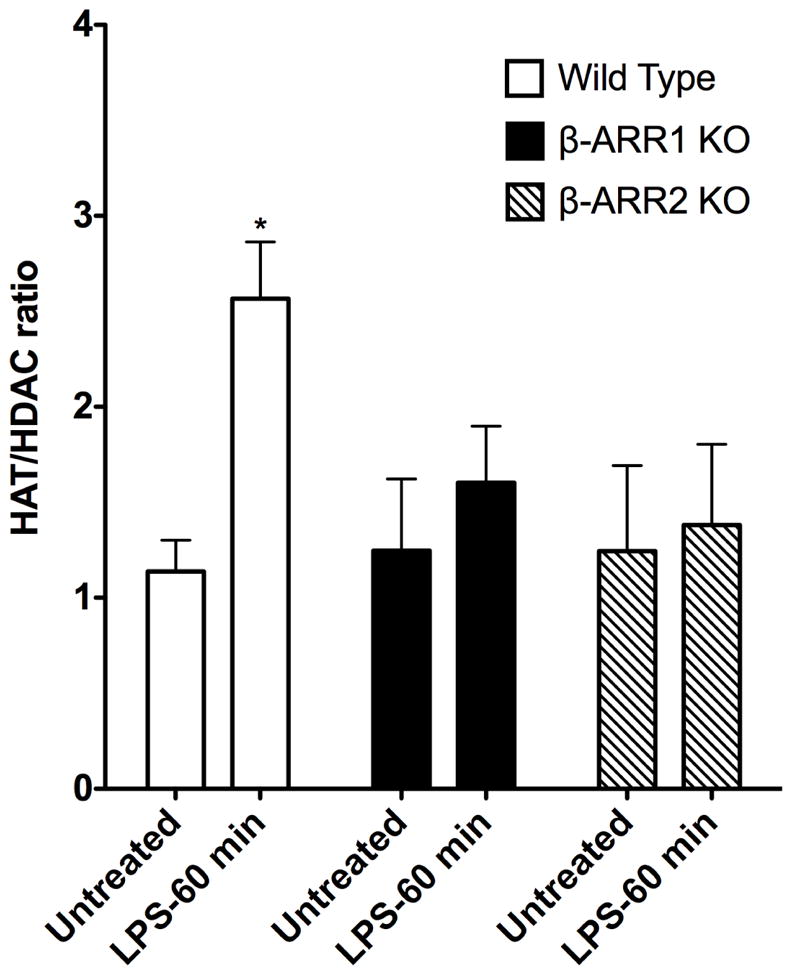
Wild type and β-arrestin KO mice were untreated or injected with LPS (for 60 min) and spleen collected and snap frozen. Nuclear lysates were extracted from the spleen and HAT/HDAC activities were determined as described in the methods. Results are expressed as ratio of HAT:HDAC activities in the untreated and LPS treated groups. N=3 mice each for β-arrestin-1 and −2 KO mice (per time point) and N=4 mice for wild type/time point. Data were analyzed by ANOVA with Bonferroni post-test. *P<0.05 compared between wild type and β-arrestin-2 KO mice at the corresponding time point.
Even though we observed specific differences in cytokine responses between β-arrestin-1 and −2 KO in the CD11b+ cells, we did not see this difference in vivo using this whole spleen model likely because the CD11b+ population constitutes only ~5% of the total cells. In the remaining CD11b− cells (~95%) role of β-arrestin-1 and −2 is similar with respect to TLR4-induced cytokine production. Further in vitro studies are necessary, however, to determine if β-arrestin-2 mediates TLR4 signaling in the CD11b+ splenocytes through other mechanisms or if the mechanism of LPS-induced HAT/HDAC activities selectively utilizes only β-arrestin-2 and not β-arrestin-1 in this population. Based on the results presented here, we postulate that in the absence of β-arrestins, modulation of histone acetylation and chromatin modification by LPS is impaired. Because previous studies have demonstrated the importance of HAT/HDACs in the production of cytokines in response to LPS, we propose that β-arrestins modulate LPS-induced cytokine production and survival at least in part by regulating HAT:HDAC activity ratio.
It is important to note that in the present study we measured the total HAT and HDAC activities and therefore do not demonstrate which HDACs and HATs in particular are being regulated by β-arr-1 and −2. Also, whether β-arrestins directly regulate these enzymes or modulate them through other signaling pathways is not evident from our studies. In future work, we will determine members of HATs/HDACs that are modulated by LPS in vivo and, which HATs/HDACs are being regulated by β-arr-1 and −2, and whether there are cell-type specific differences. It is important to note that in addition to being critical for chromatin modification, HATs and HDACs also have cell signaling function i.e. acetylation/deacetylation of cell signaling proteins (Buchwald et al., 2009; Spange et al., 2009). Therefore, it is also possible that the consequences of HAT/HDAC modulation by β-arrestins may in turn regulate other signaling molecules that might ultimately affect cytokine production. In addition, based on the role of chromatin modification in endotoxin tolerance (reviewed in (Biswas and Lopez-Collazo, 2009), it is possible that the effect of β-arrestins on HAT/HDAC activity might reflect the ability of β-arrestin KO mice to modulate endotoxin tolerance. In this regard, Foster et al (Foster et al., 2007) showed that in LPS-tolerant macrophages, there are two subsets of genes being regulated: Inflammatory genes that are inhibited and, anti-microbial genes that are upregulated or are inducible. Based on its ability to regulate in vivo HAT/HDAC ratio, it would be interesting to test whether LPS-tolerant macrophages from β-arrestin KO mice are able to modify these subsets of genes during endotoxin tolerance. This will be examined in future studies.
Previous studies have shown that β-arrestin functions depend on the extracellular signal and the cellular context. Our studies suggest that in vivo it may depend on the pathogenic stimulus. In this regard, we reported that β-arr-1 and −2 have opposing functions in terms of innate immune response elicited by adenovirus vector (which signals via both TLR-dependent and –independent pathways) (Seregin et al.). We found adenovirus vector-induced cytokine response to be inhibited in the β-arr-1 knockout mice (similar to the present study). However, in contrast to the results presented here, we observed enhanced cytokine levels in response to adenovirus vector in the β-arr-2 knockout mice. Thus the role of β-arrestins, especially β-arresitin-2 in innate immune response may be dependent on the extracellular signal that induces the inflammatory response. This also translates into the possibility that β-arrestins’ role in inflammatory disease may depend on the specific pathogenic aspect of the disease. It remains to be seen whether inhibition of β-arrestins’ functions may used as a strategy to treat inflammatory diseases.
In conclusion, our studies demonstrate that β-arrestin-1 and −2 have overlapping, non-redundant, previously undescribed positive regulatory role in TLR4 signaling in vivo and in vitro. Interestingly, majority of β-arrestins’ functions are not compensated in one knockout by the presence of the other β-arrestin, in this LPS model. Furthermore, β-arrestin-1 and −2 have immune cell type-specific effects on TLR4 signaling, and their effects are largely independent of MAPK and IκBα pathways. Based on our results on HAT/HDAC activities in the spleen, we postulate that β-arrestins may mediate TLR4-induced cytokine production in vivo by modulating LPS-induced chromatin modification or by altering the acetylation status of some signaling proteins that may ultimately affect cytokine production and survival of mice.
Acknowledgments
We wish to thank Dr. Robert J. Lefkowitz (Duke University) for his generosity in providing us with β-arrestin-1 and −2 knockout mice. We also appreciate the Michigan State University Laboratory Animal support facility for their assistance in the humane care and maintenance of the animals utilized in this work. We gratefully acknowledge the support of the National Institutes of Health grants (AR056680, HL095637 and AR055726) to N.P. We would also like to express our gratitude to Dr. Laura McCabe (Department of Physiology, MSU) for critical reading of this manuscript.
References
- Appledorn DM, Patial S, McBride A, Godbehere S, Van Rooijen N, Parameswaran N, Amalfitano A. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J Immunol. 2008;181(3):2134–2144. doi: 10.4049/jimmunol.181.3.2134. [DOI] [PubMed] [Google Scholar]
- Armstrong L, Medford AR, Hunter KJ, Uppington KM, Millar AB. Differential expression of Toll-like receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clin Exp Immunol. 2004;136(2):312–319. doi: 10.1111/j.1365-2249.2004.02433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aung HT, Schroder K, Himes SR, Brion K, van Zuylen W, Trieu A, Suzuki H, Hayashizaki Y, Hume DA, Sweet MJ, Ravasi T. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J. 2006;20(9):1315–1327. doi: 10.1096/fj.05-5360com. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. 2005;25(3):552–563. doi: 10.1183/09031936.05.00117504. [DOI] [PubMed] [Google Scholar]
- Beutler B. Innate immunity: an overview. Mol Immunol. 2004;40(12):845–859. doi: 10.1016/j.molimm.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Beutler B. The Toll-like receptors: analysis by forward genetic methods. Immunogenetics. 2005;57(6):385–392. doi: 10.1007/s00251-005-0011-3. [DOI] [PubMed] [Google Scholar]
- Beutler BA. TLRs and innate immunity. Blood. 2009;113(7):1399–1407. doi: 10.1182/blood-2008-07-019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30(10):475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Blackwell TS, Blackwell TR, Christman JW. Impaired activation of nuclear factor-kappaB in endotoxin-tolerant rats is associated with down-regulation of chemokine gene expression and inhibition of neutrophilic lung inflammation. J Immunol. 1997;158(12):5934–5940. [PubMed] [Google Scholar]
- Buchwald M, Kramer OH, Heinzel T. HDACi--targets beyond chromatin. Cancer Lett. 2009;280(2):160–167. doi: 10.1016/j.canlet.2009.02.028. [DOI] [PubMed] [Google Scholar]
- Card JW, Carey MA, Bradbury JA, DeGraff LM, Morgan DL, Moorman MP, Flake GP, Zeldin DC. Gender differences in murine airway responsiveness and lipopolysaccharide-induced inflammation. J Immunol. 2006;177(1):621–630. doi: 10.4049/jimmunol.177.1.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KC, Jung MG, Lee YH, Yoon JC, Kwon SH, Kang HB, Kim MJ, Cha JH, Kim YJ, Jun WJ, Lee JM, Yoon HG. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009a;69(2):583–592. doi: 10.1158/0008-5472.CAN-08-2442. [DOI] [PubMed] [Google Scholar]
- Choi KC, Lee YH, Jung MG, Kwon SH, Kim MJ, Jun WJ, Lee J, Lee JM, Yoon HG. Gallic acid suppresses lipopolysaccharide-induced nuclear factor-kappaB signaling by preventing RelA acetylation in A549 lung cancer cells. Mol Cancer Res. 2009b;7(12):2011–2021. doi: 10.1158/1541-7786.MCR-09-0239. [DOI] [PubMed] [Google Scholar]
- Choi Y, Park SK, Kim HM, Kang JS, Yoon YD, Han SB, Han JW, Yang JS, Han G. Histone deacetylase inhibitor KBH-A42 inhibits cytokine production in RAW 264.7 macrophage cells and in vivo endotoxemia model. Exp Mol Med. 2008;40(5):574–581. doi: 10.3858/emm.2008.40.5.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook JA. Eicosanoids. Crit Care Med. 2005;33(12 Suppl):S488–491. doi: 10.1097/01.ccm.0000196028.19746.42. [DOI] [PubMed] [Google Scholar]
- Daubeuf B, Mathison J, Spiller S, Hugues S, Herren S, Ferlin W, Kosco-Vilbois M, Wagner H, Kirschning CJ, Ulevitch R, Elson G. TLR4/MD-2 monoclonal antibody therapy affords protection in experimental models of septic shock. J Immunol. 2007;179(9):6107–6114. doi: 10.4049/jimmunol.179.9.6107. [DOI] [PubMed] [Google Scholar]
- Deriy LV, Beno DW, Uhing MR, Jiyamapa-Serna VA, Kimura RE. Splenectomy ablates endotoxin-induced IFNgamma response in rats. Shock. 2002;17(4):312–315. doi: 10.1097/00024382-200204000-00013. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Fan H, Luttrell LM, Tempel GE, Senn JJ, Halushka PV, Cook JA. Beta-arrestins 1 and 2 differentially regulate LPS-induced signaling and pro-inflammatory gene expression. Mol Immunol. 2007;44(12):3092–3099. doi: 10.1016/j.molimm.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447(7147):972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294(5548):1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, Pei G. Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol Cell. 2004;14(3):303–317. doi: 10.1016/s1097-2765(04)00216-3. [DOI] [PubMed] [Google Scholar]
- Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Goktuna SI, Neuenhahn M, Fierer J, Paxian S, Van Rooijen N, Xu Y, O’Cain T, Jaffee BB, Busch DH, Duyster J, Schmid RM, Eckmann L, Karin M. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130(5):918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes & development. 2004;18(18):2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Kang J, Shi Y, Xiang B, Qu B, Su W, Zhu M, Zhang M, Bao G, Wang F, Zhang X, Yang R, Fan F, Chen X, Pei G, Ma L. A nuclear function of beta-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell. 2005;123(5):833–847. doi: 10.1016/j.cell.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Kohler J, Heumann D, Garotta G, LeRoy D, Bailat S, Barras C, Baumgartner JD, Glauser MP. IFN-gamma involvement in the severity of gram-negative infections in mice. J Immunol. 1993;151(2):916–921. [PubMed] [Google Scholar]
- Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20(8):615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Lehmann V, Freudenberg MA, Galanos C. Lethal toxicity of lipopolysaccharide and tumor necrosis factor in normal and D-galactosamine-treated mice. J Exp Med. 1987;165(3):657–663. doi: 10.1084/jem.165.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon CG, Tory R, Jia J, Sivak O, Wasan KM. Discovery and development of toll-like receptor 4 (TLR4) antagonists: a new paradigm for treating sepsis and other diseases. Pharm Res. 2008;25(8):1751–1761. doi: 10.1007/s11095-008-9571-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni F, Fossati G, Lewis EC, Lee JK, Porro G, Pagani P, Modena D, Moras ML, Pozzi P, Reznikov LL, Siegmund B, Fantuzzi G, Dinarello CA, Mascagni P. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med. 2005;11(1–12):1–15. doi: 10.2119/2006-00005.Dinarello. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loniewski K, Shi Y, Pestka J, Parameswaran N. Toll-like receptors differentially regulate GPCR kinases and arrestins in primary macrophages. Mol Immunol. 2008;45(8):2312–2322. doi: 10.1016/j.molimm.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Loniewski KJ, Patial S, Parameswaran N. Sensitivity of TLR4- and −7-induced NF kappa B1 p105-TPL2-ERK pathway to TNF-receptor-associated-factor-6 revealed by RNAi in mouse macrophages. Mol Immunol. 2007;44(15):3715–3723. doi: 10.1016/j.molimm.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Lorenz E, Mira JP, Frees KL, Schwartz DA. Relevance of mutations in the TLR4 receptor in patients with gram-negative septic shock. Arch Intern Med. 2002;162(9):1028–1032. doi: 10.1001/archinte.162.9.1028. [DOI] [PubMed] [Google Scholar]
- Marriott I, Bost KL, Huet-Hudson YM. Sexual dimorphism in expression of receptors for bacterial lipopolysaccharides in murine macrophages: a possible mechanism for gender-based differences in endotoxic shock susceptibility. J Reprod Immunol. 2006;71(1):12–27. doi: 10.1016/j.jri.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Marriott I, Huet-Hudson YM. Sexual dimorphism in innate immune responses to infectious organisms. Immunol Res. 2006;34(3):177–192. doi: 10.1385/IR:34:3:177. [DOI] [PubMed] [Google Scholar]
- Miggin SM, O’Neill LA. New insights into the regulation of TLR signaling. J Leukoc Biol. 2006;80(2):220–226. doi: 10.1189/jlb.1105672. [DOI] [PubMed] [Google Scholar]
- Moeniralam HS, Bemelman WA, Endert E, Koopmans R, Sauerwein HP, Romijn JA. The decrease in nonsplenic interleukin-6 (IL-6) production after splenectomy indicates the existence of a positive feedback loop of IL-6 production during endotoxemia in dogs. Infect Immun. 1997;65(6):2299–2305. doi: 10.1128/iai.65.6.2299-2305.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navuluri L, Parvataneni S, Hassan H, Birmingham NP, Kelly C, Gangur V. Allergic and anaphylactic response to sesame seeds in mice: identification of Ses i 3 and basic subunit of 11s globulins as allergens. Int Arch Allergy Immunol. 2006;140(3):270–276. doi: 10.1159/000093284. [DOI] [PubMed] [Google Scholar]
- O’Brien GC, Wang JH, Redmond HP. Bacterial lipoprotein induces resistance to Gram-negative sepsis in TLR4-deficient mice via enhanced bacterial clearance. J Immunol. 2005;174(2):1020–1026. doi: 10.4049/jimmunol.174.2.1020. [DOI] [PubMed] [Google Scholar]
- O’Neill LA. ‘Fine tuning’ TLR signaling. Nat Immunol. 2008;9(5):459–461. doi: 10.1038/ni0508-459. [DOI] [PubMed] [Google Scholar]
- Parameswaran N, Pao CS, Leonhard KS, Kang DS, Kratz M, Ley SC, Benovic JL. Arrestin-2 and G protein-coupled receptor kinase 5 interact with NFkappaB1 p105 and negatively regulate lipopolysaccharide-stimulated ERK1/2 activation in macrophages. J Biol Chem. 2006;281(45):34159–34170. doi: 10.1074/jbc.M605376200. [DOI] [PubMed] [Google Scholar]
- Parvataneni S, Birmingham NP, Gonipeta B, Gangur V. Dominant, non-MHC genetic control of food allergy in an adjuvant-free mouse model. Int J Immunogenet. 2009;36(5):261–267. doi: 10.1111/j.1744-313X.2009.00860.x. [DOI] [PubMed] [Google Scholar]
- Patial S, Luo J, Porter KJ, Benovic JL, Parameswaran N. G-protein-coupled-receptor kinases mediate TNFalpha-induced NF-kappaB signalling via direct interaction with and phosphorylation of IkappaBalpha. Biochem J. 2010;425(1):169–178. doi: 10.1042/BJ20090908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, Coffman TM, Rockman HA, Lefkowitz RJ. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci U S A. 2006;103(44):16284–16289. doi: 10.1073/pnas.0607583103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy RC, Chen GH, Tateda K, Tsai WC, Phare SM, Mancuso P, Peters-Golden M, Standiford TJ. Selective inhibition of COX-2 improves early survival in murine endotoxemia but not in bacterial peritonitis. Am J Physiol Lung Cell Mol Physiol. 2001;281(3):L537–543. doi: 10.1152/ajplung.2001.281.3.L537. [DOI] [PubMed] [Google Scholar]
- Rettew JA, Huet YM, Marriott I. Estrogens augment cell surface TLR4 expression on murine macrophages and regulate sepsis susceptibility in vivo. Endocrinology. 2009 doi: 10.1210/en.2009-0098. [DOI] [PubMed] [Google Scholar]
- Rettew JA, Huet-Hudson YM, Marriott I. Testosterone reduces macrophage expression in the mouse of toll-like receptor 4, a trigger for inflammation and innate immunity. Biol Reprod. 2008;78(3):432–437. doi: 10.1095/biolreprod.107.063545. [DOI] [PubMed] [Google Scholar]
- Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8(10):776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabroe I, Parker LC, Dower SK, Whyte MK. The role of TLR activation in inflammation. J Pathol. 2008;214(2):126–135. doi: 10.1002/path.2264. [DOI] [PubMed] [Google Scholar]
- Sailhamer EA, Li Y, Smith EJ, Shuja F, Shults C, Liu B, Soupir C, deMoya M, Velmahos G, Alam HB. Acetylation: a novel method for modulation of the immune response following trauma/hemorrhage and inflammatory second hit in animals and humans. Surgery. 2008;144(2):204–216. doi: 10.1016/j.surg.2008.03.034. [DOI] [PubMed] [Google Scholar]
- Salomao R, Martins PS, Brunialti MK, Fernandes Mda L, Martos LS, Mendes ME, Gomes NE, Rigato O. TLR signaling pathway in patients with sepsis. Shock. 2008;30(Suppl 1):73–77. doi: 10.1097/SHK.0b013e318181af2a. [DOI] [PubMed] [Google Scholar]
- Senaldi G, Shaklee CL, Guo J, Martin L, Boone T, Mak TW, Ulich TR. Protection against the mortality associated with disease models mediated by TNF and IFN-gamma in mice lacking IFN regulatory factor-1. J Immunol. 1999;163(12):6820–6826. [PubMed] [Google Scholar]
- Seregin SS, Appledorn DM, Patial S, Bujold M, Nance W, Godbehere S, Parameswaran N, Amalfitano A. beta-Arrestins modulate Adenovirus-vector-induced innate immune responses: differential regulation by beta-arrestin-1 and beta-arrestin-2. Virus Res. 147(1):123–134. doi: 10.1016/j.virusres.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41(1):185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- Spiller S, Elson G, Ferstl R, Dreher S, Mueller T, Freudenberg M, Daubeuf B, Wagner H, Kirschning CJ. TLR4-induced IFN-gamma production increases TLR2 sensitivity and drives Gram-negative sepsis in mice. J Exp Med. 2008;205(8):1747–1754. doi: 10.1084/jem.20071990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SC, Ley SC. New insights into NF-kappaB regulation and function. Trends Immunol. 2008;29(10):469–478. doi: 10.1016/j.it.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons A, Beinke S, Ley SC. MAP kinase kinase kinases and innate immunity. Trends Immunol. 2006;27(1):40–48. doi: 10.1016/j.it.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tang Y, Teng L, Wu Y, Zhao X, Pei G. Association of beta-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2006;7(2):139–147. doi: 10.1038/ni1294. [DOI] [PubMed] [Google Scholar]
- Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ. beta-Arrestin inhibits NF-kappaB activity by means of its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc Natl Acad Sci U S A. 2004;101(23):8603–8607. doi: 10.1073/pnas.0402851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka M, Kubin M, Vieira LQ, Ozmen L, Garotta G, Scott P, Trinchieri G. Interleukin-12 is required for interferon-gamma production and lethality in lipopolysaccharide-induced shock in mice. Eur J Immunol. 1995;25(3):672–676. doi: 10.1002/eji.1830250307. [DOI] [PubMed] [Google Scholar]
- Yu Z, Zhang W, Kone BC. Histone deacetylases augment cytokine induction of the iNOS gene. J Am Soc Nephrol. 2002;13(8):2009–2017. doi: 10.1097/01.asn.0000024253.59665.f1. [DOI] [PubMed] [Google Scholar]
- Zisman DA, Kunkel SL, Strieter RM, Gauldie J, Tsai WC, Bramson J, Wilkowski JM, Bucknell KA, Standiford TJ. Anti-interleukin-12 therapy protects mice in lethal endotoxemia but impairs bacterial clearance in murine Escherichia coli peritoneal sepsis. Shock. 1997;8(5):349–356. doi: 10.1097/00024382-199711000-00006. [DOI] [PubMed] [Google Scholar]