

Abstract
Cholesterol has been linked to the pathogenesis of sporadic Alzheimer’s disease (AD) as a risk factor increasing β-amyloid (Aβ) and oxidative stress levels. Caffeine has anti-oxidant properties and has been demonstrated to reduce Aβ levels in transgenic mouse models of familial AD. However, the effects of caffeine on cholesterol-induced sporadic AD pathology have not been determined. In the present study, we determined the effects of caffeine on Aβ levels, tau phosphorylation, oxidative stress generation, and caffeine-target receptors in rabbits fed a 2% cholesterol-enriched diet, a model system for sporadic AD. Our results showed that the cholesterol-enriched diet increased levels of Aβ, tau phosphorylation as well as oxidative stress measured as increased levels of reactive oxygen species, isoprostanes, glutathione depletion, and increased levels of endoplasmic reticulum (ER) stress marker proteins. Additionally, the cholesterol-enriched diet reduced levels of adenosine A1 (A1R) but not ryanodine (RyR) or adenosine A2A (A2AR) receptors. Caffeine, administered at 0.5 mg and 30 mg/day in the drinking water, reduced cholesterol-induced increase in Aβ, phosphorylated tau and oxidative stress levels, and reversed cholesterol-induced decrease in A1R levels. Our results suggest that even very low doses of caffeine might protect against sporadic AD-like pathology.
Keywords: Adenosine receptors, Alzheimer’s disease, β-Amyloid, Caffeine, Cholesterol, Endoplasmic reticulum stress, Oxidative stress, Tau
Introduction
Accumulation of β-amyloid (Aβ) peptide, hyperphosphorylation of tau protein, and increased oxidative stress play important roles in the pathogenesis of Alzheimer’s disease (AD). However, the causes of the vast majority of late-onset sporadic forms of AD remain ill-defined. Nevertheless, a few factors have been identified which increase the risk for incidence and severity of late-onset AD.
The most widely studied risk factor for sporadic AD is apolipoprotein E (ApoE). People who carry one or two copies of the ApoE ε4 allele carry an increased risk of developing AD, however the ε4 allele is not necessary or sufficient to cause AD. ApoE is tightly linked to cholesterol transport as it carries cholesterol from the blood into the brain and shuttles cholesterol from astrocytes to neurons; presence of the ApoE ε4 allele is associated with increased plasma cholesterol levels. Because of the ApoE-cholesterol connection, cholesterol homeostasis has been considered for the last decade as a potential risk factor for AD. Indeed, a recent epidemiological study on a large cohort of men and women demonstrated that high cholesterol levels during mid-life was associated with a 66% increase in risk of developing AD late in life and even borderline high levels of cholesterol were associated with increased risk of vascular dementia. These findings suggest that even modest increase in plasma cholesterol levels at mid-age may determine our degree of risk for developing AD at late age. Recently, two genome-wide association studies established clusterin as a new genetic risk factor for late-onset sporadic AD and clusterin, like ApoE, is an abundantly expressed lipoprotein in brain. All together, these studies suggest that cholesterol metabolism is linked to the pathogenesis of AD.
Accumulation of Aβ and hyperphosphorylated tau in the brain is suggested to play a key role in the neurodegenerative processes that occur in AD by triggering neuronal damage. In addition to Aβ and phosphorylated tau, oxidative stress is an important hallmark of AD. Furthermore, oxidative stress may be the earliest change that occurs in the pathogenesis of AD. Hypercholesterolemia has been demonstrated to cause oxidative stress by increasing the generation of reactive oxygen species (ROS). Subsequent to oxidant stress, F2 isoprostane (iP) levels increase and glutathione (GSH) levels decrease. Oxidative stress can also activate the endoplasmic reticulum (ER) stress response, and sustained ER stress can lead to further oxidative damage and cell death. Based on the potential induction of oxidative damage by hypercholesterolemia and the possible contribution of oxidative stress in the pathogenesis of AD, antioxidant therapy may represent an efficient strategy to control oxidative damage and protect against AD.
The discovery that rabbits fed a high cholesterol diet exhibit cerebral Aβ plaques suggests the cholesterol-fed rabbit as a model system for sporadic AD. We have also shown that, in addition to increasing Aβ, cholesterol-enriched diets increase tau phosphorylation and oxidative stress in rabbit brains. Caffeine, the most widely used psychoactive drug, has antioxidant properties and has been shown to reduce brain levels of Aβ in transgenic mouse models for early-onset familial AD. In the present study, we determined the effects of very low and high doses of caffeine on AD-like pathology in the cholesterol-fed rabbit model system for late onset sporadic AD. We further determined the effect of caffeine on cellular mechanisms that lead to increased Aβ production, tau phosphorylation and oxidative stress as well as on caffeine-target receptors.
Materials and Methods
Animals and Treatments
New Zealand white male rabbits (1.5-2 years of age and weighing 3-4 kg, Charles River Laboratories International, Inc., Wilmington, MA) were assigned randomly to the following groups (n=6 each). Group 1 rabbits were fed normal chow. Group 2 rabbits were fed a 2% cholesterol-enriched diet (Harlan Teklad Global Diets, Madison, WI). Group 3 rabbits were fed normal chow + 0.5 mg of caffeine/day. Group 4 rabbits were fed normal chow + 30 mg of caffeine/day. Group 5 rabbits were fed a 2% cholesterol-enriched diet + 0.5 mg of caffeine/day. Group 6 rabbits were fed a 2% cholesterol-enriched diet + 30 mg of caffeine/day. All rabbits were maintained on these respective diets and caffeine treatments for 12 weeks after which time rabbits were euthanized. Caffeine was prepared fresh daily and was administered in the drinking water. The very low (0.5 mg/day) and higher (30 mg/day) doses of caffeine were chosen based on findings that the basal metabolic rate in animals is proportional to the 3/4 power of body mass. Accordingly, the metabolic rate for a 3-4 kg rabbit is about twice that of a 70 kg human and thus a 30 mg caffeine/day in a rabbit is roughly equivalent to a human ingesting 60 mg of caffeine. For reference, a 5 ounce cup of instant coffee contains 60 mg of caffeine and the 0.5 mg dose of caffeine corresponds to about 20 ounces of decaffeinated coffee. At necropsy, animals were perfused with Dulbecco’s phosphate-buffered saline at 37 °C and the brains were promptly removed and cut to yield two symmetrical hemispheres. One hemisphere was used for immunohistochemistry and the hippocampus from the other hemisphere was used for ROS, isoprostane, glutathione (GSH), ELISA and Western blot analyses. All animal procedures were carried out in accordance with the U.S. Public Health Service Policy on the Humane Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of North Dakota.
Quantification of Aβ Levels by ELISA
Aβ40 and 42 levels were quantified in the hippocampus of all animals using an ELISA kit from Biosource as per the manufacturer’s protocol and as described by us previously. The values of Aβ levels obtained by ELISA were normalized with the amount of protein in the samples. The values were expressed as mean ± standard error. The changes in the levels of Aβ were considered to be significant at p<0.05. Levels of Aβ40 and Aβ42 were expressed as pg/mg of protein.
Western Blot Analyses
Hippocampi from all animals were homogenized using tissue protein extraction reagent (T-PER) containing protease and phosphatase inhibitors (Thermo Scientific, Rockford, IL). Protein concentrations were determined by BCA protein assay (Thermo Scientific, Rockford, IL). For Western blot analyses, proteins (10 µg) were separated by SDS-PAGE (10% and 12.5% gels), followed by transfer to a polyvinylidene difluoride membrane (Millipore, Temecula, CA), and were incubated overnight at 4°C with antibodies to amyloid-β precursor protein (AβPP, 1:100, Millipore, Temecula, CA), β-secretase (BACE1) enzyme that initiates the cleavage of AβPP to yield Aβ (1:100, Millipore, Temecula, CA), insulin degrading enzyme (IDE) that degrades Aβ in the brain (1:100, Chemicon International, Temecula, CA), total tau (1:200, Calbiochem, La Jolla, CA), phosphorylated tau (CP13 and PHF-1, 1:500; gift from Dr. Peter Davis, Albert Einstein College of Medicine), GSK-3β and pTyr216GSK- 3β (1:250, BD Biosciences, San Jose, CA) that phosphorylate tau, the protein markers of ER stress gadd153 (1:100, Abcam, Cambridge, MA), glucose regulated proteins grp78 and grp94 (grp78, 1:100 Assay Designs, Ann Arbor, MI and grp94, 1:100, Affinity Bioreagents, Golden, CO), calreticulin (1:100, Affinity Bioreagents, Golden, CO), and to the caffeine target receptors RyR (1:100, Abcam, Cambridge, MA), A2AR (1:100, Upstate, Temecula, CA), and A1R (1:500, Abcam, Cambridge, MA). β-actin was used as a gel-loading control. The blots were developed with enhanced chemiluminiscence (Immun-star HRP chemiluminiscent kit, Biorad, Herculus, CA). The results were quantified by densitometry and represented as total integrated densitometric values.
Reactive Oxygen Species Assay
ROS generation was determined using 2’-7’-dichlorofluorescin-diacetate (DCFH-DA) as well as fluorometric detection of H2O2. For the DCFH-DA assay, protein samples (25 μg) from hippocampus of all animals were diluted in PBS and incubated with 5.0 μM DCFH-DA (Sigma, St. Louis, MO) in the dark for 15 minutes at 37°C. Fluorescence was measured every 15 min for 1 hr with excitation and emission wavelengths of 488 nm and 525 nm respectively, using a Spectramax Gemini-EM microplate reader (Molecular Probes, Sunnyvale, CA). Values were expressed as percent increase in fluorescence compared to controls.
H2O2 was measured using the horseradish peroxidase (HRP)-linked fluorometric assay (Amplex Ultra Red; Invitrogen, Carlsbad, CA). Hippocampus (20 μg) of the control and treated rabbits was added to a 96-well plate containing 100 μl of reaction buffer containing 0.1 U/ml HRP, 50 μM Amplex UltraRed. Resorufin fluorescence was followed by a Spectramax Gemini-EM (Molecular Probes, Sunnyvale, CA) with excitation 530-560 nm and emission at 590 nm.
Glutathione Depletion
Glutathione (gamma-glutamyl-cysteinyl-glycine; GSH) plays an important role in antioxidant defense in animal cells. Increased levels of oxidative stress lead to the accumulation of oxidized glutathione (GSSG) and a subsequent decrease in the ratio of reduced glutathione (GSH) to GSSG. A luminescent based GSH-Glo Assay (Promega Corporation, Madison, WI) was used for quantification of glutathione in rabbit hippocampus according to the manufacturer’s recommendation.
Isoprostane Assay
The F2-isoprostane 8-Iso-prostaglandin F2α (8-iso-PGF2α) is produced by lipid peroxidation and is a marker of oxidative stress. 8-iso-PGF2α levels were quantified in the hippocampus of all rabbits using the isoprostane oxidative stress assay kit B (Biomol International, Plymouth Meeting, PA). Briefly, tissue samples frozen on liquid nitrogen were powdered and homogenized with an excess volume of 2N sodium hydroxide (NaOH) (10 μg to 1 mg of tissue per 1.0 mL of 2N NaOH). Samples in 2N NaOH were covered and heated at 45°C for 2 hrs to ensure hydrolysis. After hydrolysis, the samples were cooled and treated with an equal volume of 2N hydrochloric acid (HCl) to neutralize the NaOH. The neutralized samples were centrifuged at 3,000 rpm in a microcentrifuge. The pH of the samples was adjusted to be in the range of 6-8. The samples were further diluted with the “direct 8-iso sample dilute” provided with the kit and the assay was carried out following the manufacturer’s protocol. The color developed in the standards and samples was read on a SpectraMax Plus microplate reader (Molecular Devices, Sunnyvale, CA) at 405 nm. The measured optical density was used to calculate the concentration of 8-iso PGF-α.
Plasma Cholesterol Levels
Total plasma cholesterol in rabbits was measured in blood collected from an ear vein after overnight fasting immediately before euthanasia. The measurements were carried out using a Flex reagent cartridge and the Dimension clinical chemistry system (Dade Behring Inc). Total brain cholesterol was shown previously to be unchanged in cholesterol-fed rabbits and therefore was not measured here.
Statistical Analysis
Data were analyzed for statistical significance using one-way analysis of variance (ANOVA) followed by Newman-Keuls Multiple Comparison Test with GraphPad Prism software 4.01. All values in each group were expressed as mean value ± SEM. All group comparisons were considered significant at p < 0.05.
Results
Caffeine decreases cholesterol-enriched diet-induced increase in Aβ production and accumulation
Total amounts of guanidine-solubilized Aβ were significantly increased (p<0.001) in the hippocampus of cholesterol-fed rabbits in comparison to control rabbits; 2-fold increases were observed for Aβ40 (Figure 1a) and 3-fold increases were observed for Aβ42 levels (Figure 1b). Caffeine produced statistically significant reductions in cholesterol-induced increases in both Aβ40 (Figure 1a) and Aβ42 (Figure 1b). For both Aβ40 and Aβ42, the lower dose of caffeine (0.5 mg/day) was more effective than the 30 mg/day dose in reducing Aβ40 and Aβ42 to levels similar to control levels.
Fig. 1.
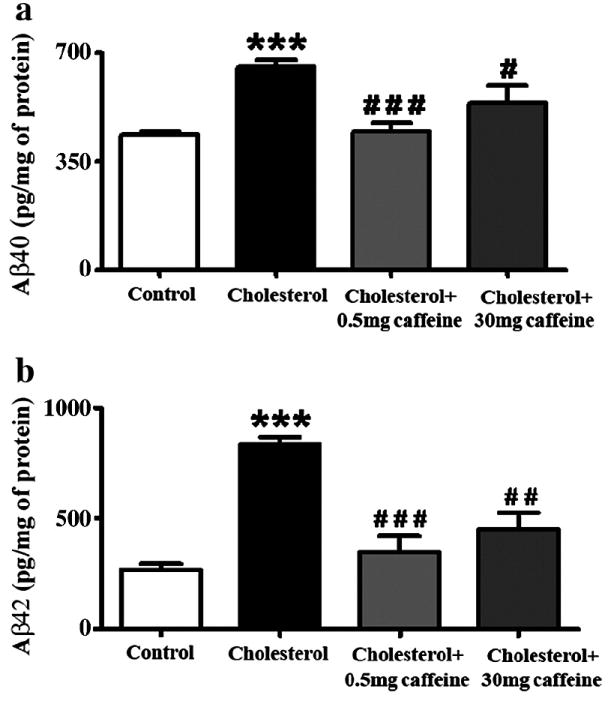
Caffeine reduces cholesterol-enriched diet-induced increase in Aβ40 (a) and Aβ42 (b) levels in rabbit hippocampus. Caffeine was administered at 0.5 mg and 30 mg/day in drinking water to rabbits fed normal chow or a 2% cholesterol-enriched diet for 12 weeks. The amount of Aβ measured by ELISA was normalized with the protein concentration in the samples and expressed as pg/mg of protein. Values expressed are mean value ± SEM from three different experiments. ***p<0.001 versus controls, #p<0.05, # #p<0.01, # # #p<0.001 versus rabbits fed with the 2% cholesterol-enriched diet (cholesterol).
The increased levels of Aβ induced by the cholesterol-enriched diet was accompanied by increased levels of AβPP and BACE1, and by decreased levels of IDE (Figure 2a,b). Caffeine at both doses (0.5 and 30 mg) produced statistically significant (p<0.05) reduction in the cholesterol-induced increases in AβPP. Caffeine only at the high dose (30 mg) produced statistically significant (p<0.001) reductions in the cholesterol-induced increases in BACE1. Caffeine only at the low dose (0.5 mg) produced statistically significant (p<0.01) elevation in the levels of IDE that were decreased by the cholesterol-enriched diet. These results suggest that increased levels of Aβ result from increased processing of AβPP by BACE1 and/or reduced degradation of Aβ by IDE.
Fig. 2.
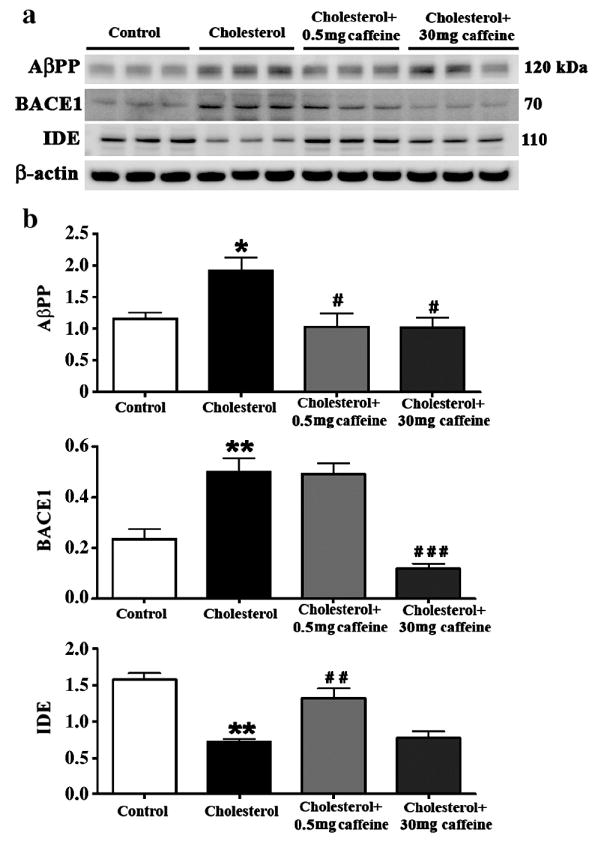
Caffeine dose-dependently regulates Aβ production and degradation in hippocampus from cholesterol-fed rabbits. Representative Western blots (a) and densitometric analysis (b) showing that while both 0.5 and 30 mg/day caffeine reduce the cholesterol-enriched diet-induced increase in AβPP, caffeine reduces BACE1 at 30 mg/day and increases IDE levels at 0.5 mg/day. Values are expressed as mean value ± SEM from three different experiments. *p<0.05, **p<0.01 compared to controls, #p<0.05, # #p<0.01, # # #p<0.001 compared to rabbits fed with 2% cholesterol diet.
Caffeine reduces cholesterol-induced increase in phosphorylation of tau
Because hyperphosphorylation of tau protein is a cardinal hallmark of AD neuropathology and we have showed previously that high cholesterol diet can increase levels of hyperphosphorylated tau in rabbit brain, we next tested the extent to which caffeine decreases cholesterol-induced increase in phosphorylated tau. The cholesterol-enriched diet caused 10-fold increase in levels of phosphorylated tau as determined with PHF-1 and CP13 (Figure 3a,b) antibodies that detected tau phosphorylated at Ser396/404 and Ser202, respectively. Caffeine at 0.5 mg/day did not, but caffeine at 30 mg/day did produce statistically significant (p<0.05) reductions in the cholesterol-induced increases in phosphorylated tau. Mechanistically, the enzyme GSK-3β is involved in the phosphorylation of tau. Accordingly, we next determined the extent to which cholesterol increases levels of the active form of GSK-3β (pTyr216 GSK-3β) and caffeine is able to attenuate pTyr216 GSK-3β. The cholesterol-enriched diet increased by about 2-fold (p<0.05) levels of pTyr216 GSK-3β (Figure 4a,b). Caffeine at both doses (0.5 and 30 mg/day) significantly (p<0.01) reduced levels of pTyr216 GSK-3β to levels similar to those found in rabbits fed control diet.
Fig. 3.
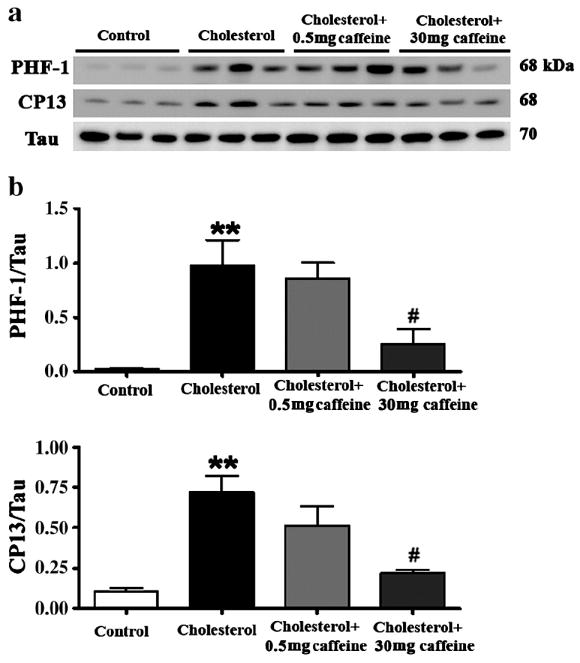
Caffeine regulates tau phosphorylation in cholesterol-fed rabbits in a dose-dependent manner. Western blots (a) and densitometric analyses (b) show that caffeine at 30 mg/day but not at 0.5 mg/day reduces tau phosphorylation in hippocampus from rabbits fed a 2% cholesterol-enriched diet. Tau phosphorylation is detected by PHF-1 and CP13 antibodies. **p<0.01 versus controls, #p<0.05 versus rabbits fed with 2% cholesterol diet.
Fig.4.
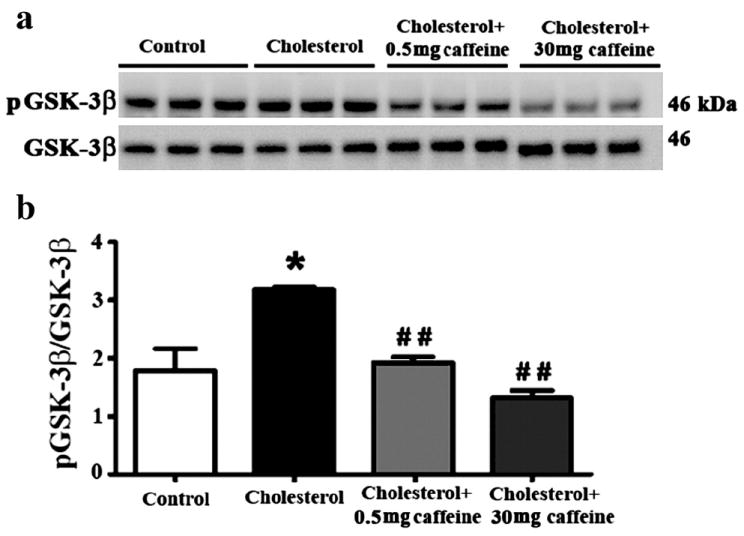
Caffeine, at 0.5 and 30 mg/day reduces levels of p–GSK-3β (a,b) enzyme that is involved in tau phosphorylation. Although it reduced p–GSK-3β levels, 0.5 mg/day caffeine didn’t reduce tau phosphorylation, suggesting that reduction of p–GSK-3β is not sufficient to prevent tau phosphorylation in cholesterol-fed rabbits. *p<0.05 versus controls, # #p<0.01 versus rabbits fed with 2% cholesterol diet.
Caffeine attenuates cholesterol-induced increase in ROS and 8-Iso-PGF2α levels, and reduces glutathione depletion
We next determined levels of ROS, the isoprostane 8-iso-PGF-2α and glutathione depletion because all have been implicated in the neuropathogenesis of AD. The cholesterol-enriched diet significantly increased levels of ROS as demonstrated with both DCFH-DA andfluorometric detection of H2O2 (p<0.01, Figure 5a and b) and 8-iso-PGF-2α (p<0.001, Figure 5c), and significantly (p<0.05) reduced levels of glutathione (Table II) in rabbit hippocampus. While caffeine at 0.5 mg/day did not significantly affect cholesterol-induced changes in ROS and glutathione levels, it did significantly (p<0.001) reduce ROS generation (Figure 5a and b) and reverse glutathione depletion (Table II) at 30 mg/day. Caffeine at doses of 0.5 and 30 mg/day reduced significantly (p<0.001) cholesterol-induced increases in isoprostane levels (Figure 5c).
Fig. 5.
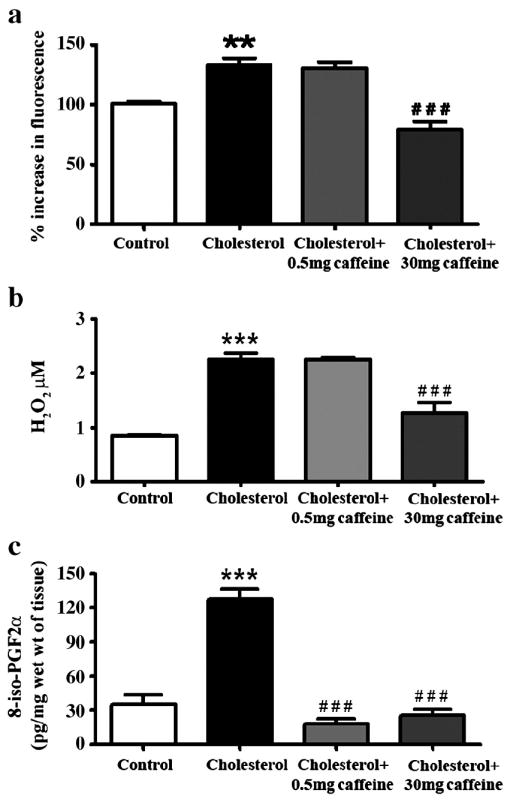
Caffeine dose-dependently reduces reactive oxygen species (ROS) generation, glutathione depletion and isoprostane formation. ROS generation was measured with increased fluorescence with the DCFH-DA assay as well as H2O2 production, and isoprostane level is detected with formation of 8-iso-PGF2α. Caffeine reduces the cholesterol-enriched diet-induced increase in ROS generation (a, b) at 30 mg/day and reduced isoprostane formation at both 0.5 and 30 mg/day (c). **p<0.01, ***p<0.001 versus controls, # # #p<0.001 versus rabbits fed with 2% cholesterol-enriched diet.
Table II.
2% cholesterol-enriched diet for 12 weeks reduces glutathione (GSH)/oxidized glutathione (GSSG) ratio in rabbit hippocampus. Caffeine administered in the drinking water reduced the decrease in the GSH/GSSG ratio.
| Treatment | GSH (μM/mg wet wt of tissue) | GSSG (μM/mg wet wt of tissue) | GSH/GSSG Ratio |
|---|---|---|---|
| Control | 0.267 ± 0.005 | 0.148 ± 0.004 | 1.84 ± 0.045 |
| Cholesterol | 0.220 ± 0.019 | 0.195 ± 0.017 | 1.14 ± 0.205 ** |
| Cholesterol+0.5mg caffeine | 0.233 ± 0.007 | 0.173 ± 0.005 | 1.36 ± 0.047 |
| Cholesterol+30mg caffeine | 0.263 ± 0.008 | 0.161 ± 0.009 | 1.70 ± 0.123 # # |
p<0.01 compared to control rabbits
p<0.01 compared to cholesterol treated rabbits
Caffeine protects against cholesterol-induced ER stress
Oxidative stress and ER stress are tightly linked; ER stress can intiate ROS production and ROS can cause ER stress. Accordingly, we tested next the extent to which the 2% cholesterol-enriched diet alters levels of the ER chaperone proteins calreticulin, grp78 and grp94 as well as the ability of caffeine to protect against cholesterol-induced changes in levels of ER stress-related proteins (Figure 6a,b). The cholesterol-enriched diet significantly increased levels of the ER chaperone proteins calreticulin (p<0.05), grp78 (p<0.01) and grp94 (p<0.05) in rabbit hippocampus (Figure 6a,b). Additionally, levels of gadd153, a transcription factor that sensitizes cells to ER stress through mechanisms that involve increased oxidative stress, were significantly (p<0.05) increased in hippocampus of cholesterol-fed rabbits. Caffeine administered at 0.5 mg/day significantly reduced the levels of calreticulin (p<0.05) and grp78 (p<0.01) but not the levels of grp94 and gadd153. At 30 mg/day, caffeine significantly reduced cholesterol-induced increase in calreticulin (p<0.05), grp78 (p<0.01), grp94 (p<0.05) and gadd153 (p<0.05). These data suggest that caffeine at 30 mg/day can protect cells against cholesterol-induced changes to a wide spectrum of markers for ER stress (Figure 6a,b).
Fig. 6.
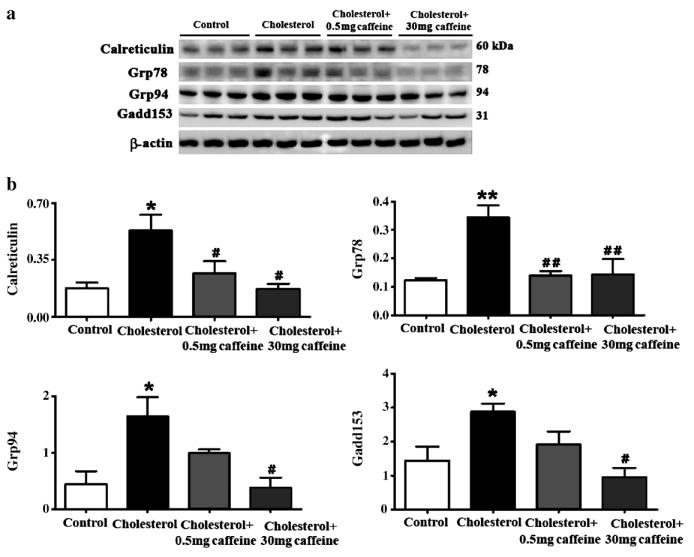
Caffeine dose-dependently reduces endoplasmic reticulum (ER) stress induced by cholesterol-enriched diet in rabbit hippocampus. Representative Western blots (a) and densitometric analysis (b) showing increased levels of the ER-resident proteins calreticulin, grp78 and grp94 and of the ER stress marker protein gadd153. Caffeine administered at 30 mg/day of caffeine/day in drinking water decreased the cholesterol-induced increase in calreticulin, grp78, grpP94, and gadd153 levels. At 0.5 mg/day, caffeine only reduces grp78 and calreticulin levels. *p<0.05, **p<0.01 versus controls, #p<0.05, # #p<0.01 versus rabbits fed with 2% cholesterol-enriched diet.
Caffeine restores adenosine A1R levels and does not affect cholesterol levels
Caffeine exerts its pharmacological effects by acting on various signaling systems and target receptors including adenosine A1R, adenosine A2AR and RyR. We determined levels of these receptors and found that the cholesterol-enriched diets did not affect A2AR or RyR levels, but did significantly (p<0.01) reduce protein levels of A1R (Figure 7a,b). While caffeine at both doses tested (0.5 and 30 mg/day) did not alter levels of A2AR or RyR, caffeine at 0.5 and 30 mg/day afforded significant protection against cholesterol-induced decreases in A1R levels (Figure 7a,b). Thus, A1R may be important in the protective action of caffeine against the effects of the cholesterol-enriched diet.
Fig. 7.
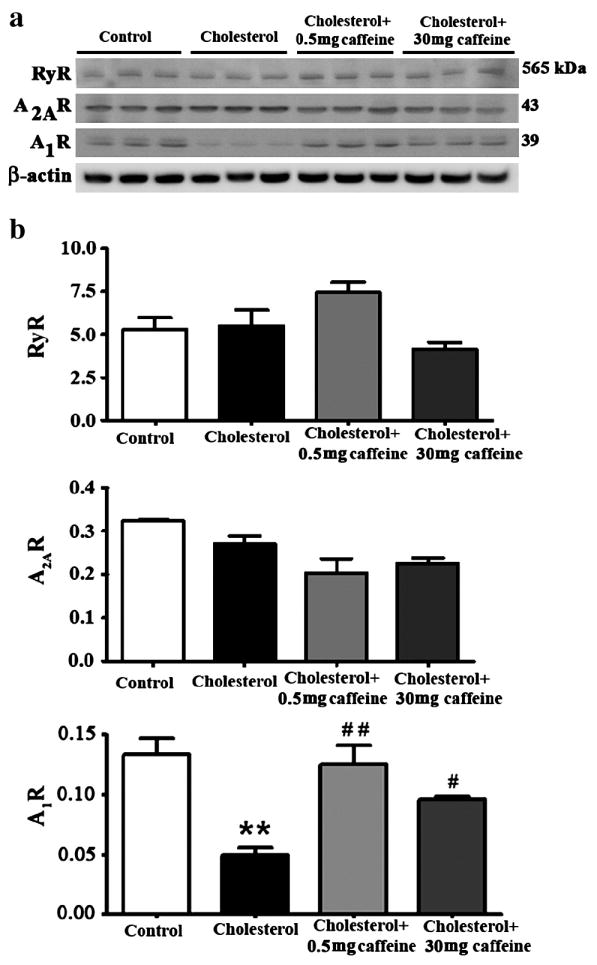
Caffeine restores adenosine A1 receptor (A1R) levels. Representative Western blots (a) and densitometric analysis (b) showing that the 2% cholesterol-enriched diet doesn’t affect levels of ryanodine receptor (RyR) or adenosine 2A receptor (A2AR) but reduces and adenosine A1 receptor (A1R) levels. Caffeine, at 0.5 mg/day > 30 mg/day, restores A1R levels. **p<0.01 versus controls, #p<0.05, # #p<0.01 versus rabbits fed with 2% cholesterol diet.
In order to determine the extent to which caffeine effects may involve changes in cholesterol levels, plasma cholesterol levels were measured (Table 1). The cholesterol-enriched diet markedly increased plasma cholesterol levels from 64.0 ± 7.41 mg/dl to 658 ± 101.5 mg/dl. Caffeine administration did not significantly affect plasma cholesterol levels in either control or cholesterol-fed rabbits. These results suggest that the effects of caffeine on Aβ, tau, and oxidative stress did not involve changes in plasma cholesterol levels. In addition, we found that caffeine administration to control rabbits fed a normal chow did not alter levels of Aβ or its metabolism, tau phosphorylation, ROS generation, isoprostanes, glutathione depletion, ER stress marker proteins, or caffeine-target receptors (data not shown).
Table I.
Caffeine administered in the drinking water at 0.5 mg/day and 30 mg/day didn’t affect total plasma cholesterol levels (mg/dl) in rabbits fed with normal chow or a 2% cholesterol-enriched diet for 12 weeks.
| Treatment | Total plasma cholesterol (mg/dl) |
|---|---|
| Control (n=6) | 64.0 ± 7.41 |
| 2% cholesterol (n=6) | 658.3 ± 101.5 *** |
| 2% Cholesterol + 0.5mg caffeine (n=6) | 593.6 ± 124.6 |
| 2% cholesterol + 30mg caffeine (n=6) | 742.8 ± 92.43 |
| 0.5mg caffeine (n=6) | 84.20 ± 4.97 |
| 30mg caffeine (n=6) | 87.71 ±12.55 |
Values are expressed as mean ± SEM.
p<0.001 versus control group.
Discussion
In the present study, we demonstrate that a 2% cholesterol-enriched diet increased Aβ levels, tau phosphorylation and oxidative stress in rabbit hippocampus. Oxidative stress involved ROS generation, glutathione depletion, increased isoprostane levels, and induction of ER stress. ER stress was evidenced by disturbances in the levels of the ER-resident chaperones calreticulin, grp78 and grp94, and by the increase in levels of gadd153. Increased levels of gadd153 is a specific event to ER stress that plays a key role in generating or exacerbating oxidative damage and ultimately causes cell death (see for review). Caffeine, administered in the drinking water, reduced the cholesterol-enriched diet-induced increase in Aβ, tau phosphorylation, and oxidative stress. The protective effects of caffeine were dose-related and associated with the regulation of A1R but not cholesterol levels. These results indicate that cholesterol-enriched diet is a pro-oxidant that can trigger AD-like pathology and that caffeine possibly through its anti-oxidant properties can protect against AD pathology.
We have shown previously that cholesterol-enriched diets increase Aβ and phosphorylated tau levels in rabbit brain. In the present study we show that 2% cholesterol-enriched diets also increase oxidative stress. Oxidative stress results from the overproduction of ROS and/or from deficiency of antioxidant enzymes. The brain is very rich in polyunsaturated fatty acids that can be attacked by ROS causing lipid peroxidation and the formation of the prostaglandin-like compounds F2-isoprostanes. Glutathione can play an important role in removing lipid peroxides by scavenging free radicals and other types of ROS. Increased levels of oxidative stress lead to the depletion of glutathione levels, a phenomenon that can be assessed by a decrease in the ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG). We demonstrate in the present study that a cholesterol-enriched diet increases ROS generation, isoprostane levels and glutathione depletion in addition to increasing Aβ and phosphorylated tau levels. Although it is generally accepted that Aβ accumulation precedes and causes tau hyperphosphorylation, it remains unclear whether oxidative stress or Aβ is generated first and which causes cell dysfunction and death in AD. Certainly, much has been investigated and documented about the ability of Aβ to cause neuronal cell dysfunction and death. Similarly, oxidative stress contributes to the pathogenesis of a number of diseases including AD; cognitive impairment correlates with plasma antioxidant capacity in AD patients and decreases in GSH levels and GSH/GSSG ratios have been noted in patients with AD and mild cognitive impairment (MCI) patients. Thus, oxidative stress might represent a marker of AD at the MCI stage.
High cholesterol-enriched diets cause Aβ accumulation and oxidative stress as we demonstrate in the present study. Aβ peptide can also cause cholesterol accumulation and oxidative stress, which in turn can alter cholesterol metabolism. Indeed, exposure of neurons to Aβ induced oxidative stress and cholesterol accumulation, and oxidative stress was associated with cholesterol accumulation in the brains of AD patients. These results suggest that disturbances in cholesterol metabolism, oxidative stress and Aβ accumulation are intrinsically related. However, whether Aβ or oxidative stress is generated first in the cholesterol-fed rabbit model is still to be determined. In the present study, we demonstrate that caffeine reduced both Aβ accumulation and oxidative stress in a dose-related manner. Whether reduction in oxidative stress prevented Aβ accumulation or reduction in Aβ levels precluded oxidative stress in cholesterol-fed rabbits treated with caffeine is a question that has to be answered in future studies. Caffeine prevented Aβ accumulation through two mechanisms, reducing the production and increasing the degradation of this peptide. First, caffeine reduced levels of the precursor protein of Aβ, AβPP, and of the enzyme BACE1 that initiates the cleavage of AβPP to Aβ. Reduction in both AβPP and BACE1 ultimately leads to a decrease in Aβ production. Second, caffeine increased protein levels of IDE, an enzyme that contributes to the degradation of the generated Aβ in the brain. Interestingly, these effects of caffeine were dose-related with the low dose (0.5 mg/day) increasing IDE levels and the higher dose (30 mg/day) reducing BACE1 levels. Both doses were similarly efficient in reducing AβPP levels. Reduction in Aβ levels with caffeine is associated with reduction in oxidative stress as demonstrated by reduction in ROS generation, glutathione depletion, isoprostane production, and ER stress protein marker levels. Reduction in ROS and glutathione depletion do not appear to be the major mechanism by which caffeine would reduce Aβ accumulation because the 0.5 mg dose did not affect ROS and glutathione status but reduced Aβ levels. Reduction of isoprostane levels achieved with both the 0.5 and 30 mg/day caffeine doses, suggests that the inhibitory effects of caffeine on isoprostanes may have played a more prominent role in reducing Aβ accumulation.
We further demonstrated that cholesterol-enriched diets alter levels of ER-associated proteins calreticulin, grp78, grp94 and gadd153. Excess cholesterol has been shown to induce disturbances in ER homeostasis in macrophages. We have also shown that feeding rabbits a 1% cholesterol-enriched diet for 6 months caused ER stress in cerebral cortex, but not in hippocampus. Here, rabbits fed a 2% cholesterol-enriched diet over 12 weeks exhibited disturbed levels of ER chaperones in hippocampus. Calreticulin, an ER calcium-binding protein, functions as a molecular chaperone for the β-amyloid precursor protein and the chaperones grp94 and grp78 protect cells to cope with ER stress. In the brain of AD subjects, grp78 expression is increased in surviving neurons and grp94 expression is increased in the parietal cortex. Our results show that the cholesterol-enriched diet increased calreticulin, grp78 and grp94 levels. Caffeine reduced calreticulin and grp78 levels at 0.5 and 30 mg/day, and reduced grp94 levels at 30 but not 0.5 mg/day. Intense or sustained stress in the ER activates the transcription factor gadd153, which in turn can trigger the generation of ROS. Gadd153-induced ROS can foster the production of BACE1, the rate limiting enzyme in AβPP processing to yield Aβ, thus leading to increased Aβ levels. These results strongly suggest that ER stress-induced gadd153 activation can trigger oxidative stress thereby increasing BACE1 levels and potentially resulting in Aβ overproduction. Caffeine, at 30 mg/day, may have prevented ROS generation and subsequent increases in Aβ production by reducing gadd153 levels. Caffeine has been shown to protect membranes from oxidative stress by its antioxidant ability and to inhibit lipid peroxidation induced by ROS. In addition to reducing Aβ and oxidative stress, caffeine dose-dependently reduced tau phosphorylation. Reduction of phosphorylation of GSK-3β with 0.5 mg/day caffeine did not appear to be sufficient to reduce tau phosphorylation. To our knowledge, our results are the first to show that caffeine regulates tau phosphorylation.
The mechanisms by which caffeine reduces Aβ accumulation, tau phosphorylation and oxidative stress in the cholesterol-fed rabbits appear to be multiple. Caffeine is a multifaceted drug that belongs to the family of methylxanthines. At low concentrations caffeine can sensitize RyR, leading to increased Aβ production. Caffeine can also block adenosine A1 and A2A receptors and can inhibit cAMP phosphodiesterase activity at higher doses. We demonstrated that caffeine does not affect RyR or A2AR but significantly increases A1R protein levels. Caffeine, an antagonist of A1R, has been shown to up-regulate these receptors after chronic exposure and as a consequence to reduce ischemic cell damage in gerbil hippocampus. In human neuroblastoma SH-SY5Y cells, A1R agonists increased the production of amyloid precursor protein. A1R are found in the hippocampus and a significant reduction of these receptors was observed in hippocampus and striatum of patients with AD pathology. Of course, we cannot discount a possible involvement of A2A receptors because they too are located in the hippocampus, antagonists of A2A receptors are neuroprotective, and levels of A2A receptors were not decreased in the cholesterol-fed rabbits. The extent to which caffeine protected against cholesterol-enriched diet-induced oxidative damage by mechanisms involving A1R and A2AR levels remains to be tested using specific antagonists and agonists of these sites. Currently the mechanisms by which cholesterol-enriched diets cause pathological hallmarks of AD in brain are not well understood. Plasma cholesterol very poorly enters into the brain when the blood brain barrier (BBB) is intact. Additionally, the brain makes all the cholesterol it needs through de novo synthesis. Thus, under normal conditions brain levels of cholesterol are maintained independently of plasma cholesterol. Our recent published work has shown that cholesterol levels are unchanged in the rabbit brain following ingestion of diets enriched in cholesterol. We have indeed demonstrated that feeding rabbits a 1 or 2% cholesterol-enriched diet dramatically increased plasma cholesterol concentrations but didn’t change brain levels of cholesterol (free or cholesteryl ester) [21,22]. Thus, it is unlikely that the protective effects of caffeine emanate from reducing cholesterol entry into the brain. Additionally, we showed that caffeine administration did not affect basal levels of cholesterol or the cholesterol-enriched diets-induced increases in plasma cholesterol levels. Of importance, we have recently reported that cholesterol-enriched diet compromises the integrity of the BBB in rabbit and that caffeine protects rabbit BBB from the deleterious effects of cholesterol. It is possible that the protective effects of caffeine we report here are due, at least in part, to preventing inflammatory processes and oxidative stress secondary to disruption of BBB integrity following hypercholesterolemia.
In summary, our results demonstrate that cholesterol-enriched diets cause oxidative damage in addition to increasing Aβ and tau phosphorylation levels in rabbit hippocampus. These data strengthen the link between oxidative stress, cholesterol accumulation and sporadic AD pathology. Caffeine has been shown to protect against AD pathology in animal models for familial late-onset AD. In the present study, we show that caffeine also protects against Aβ accumulation and oxidative damage triggered by high cholesterol diets. We have also shown for the first time that caffeine regulates tau phosphorylation and upregulates A1R levels in hippocampus of cholesterol-fed rabbits. Our data adds insight on the relevance of the cholesterolfed rabbit as a model system for sporadic AD exhibiting Aβ accumulation, increased tau phosphorylation and oxidative stress. Our data also demonstrate the usefulness of caffeine as an agent that can protect against the deleterious effects of cholesterol-enriched diets and high plasma cholesterol. Results of this study further provide cellular mechanisms by which caffeine exerts its protective effects and suggests that upregulation of A1R levels is a potential target mechanism for the beneficial effects of caffeine in cholesterol-enriched diets-induced sporadic AD pathology.
Acknowledgments
This work was supported by a Centers of Biomedical Research Excellence grant (2P20-RR017699-08) from the National Center for Research Resources, a division of the National Institutes of Health).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.