

Abstract
There is overwhelming evidence that tyrosine kinases play an important role in cancer development. As a prototype of targeted therapy, tyrosine kinase inhibitors are now successfully applied to cancer treatment. However, as single agents, tyrosine kinase inhibitors have not achieved satisfactory results in the treatment of prostate cancer, principally due to their inability to efficiently kill tumor cells. The authors’ laboratory has been interested in the role of the Src complex in prostate cancer progression, including the induction of androgen independence and metastasis. Previously, the authors reported that Src inhibitors such as saracatinib and PP2 caused G1 growth arrest and diminished invasiveness in prostate cancer cells but rarely apoptosis. Here, they have shown that Src family kinase (SFK) inhibitors can induce a high level of autophagy, which protects treated cells from undergoing apoptosis. Src siRNA knockdown experiments confirmed that autophagy was indeed caused by the lack of Src activity. The SFK inhibitor-induced autophagy is accompanied by the inhibition of the PI3K (type I)/Akt/mTOR signaling pathway. To test whether autophagy blockade could lead to enhanced cell death, pharmacological inhibitors (3-methyladenine and chloroquine) and a genetic inhibitor (siRNA targeting Atg7) were used in combination with SFK inhibitors. The results showed that autophagy inhibition effectively enhanced cell killing induced by SFK inhibitors. Importantly, the authors showed that a combination of saracatinib with chloroquine in mice significantly reduced prostate cancer (PC3) xenograft growth compared with the control group. Taken together, these data suggest that (1) autophagy serves a protective role in SFK inhibitor-mediated cell killing, and (2) clinically acceptable autophagy modulators may be used beneficially as adjunctive therapeutic agents for SFK inhibitors.
Keywords: Src tyrosine kinase, prostate cancer, autophagy, saracatinib, chloroquine
Introduction
Prostate cancer (PCa) is the most frequently diagnosed cancer in American men and the third leading cause of cancer mortality in the United States.1 Currently, the most common form of treatment for metastatic PCa is androgen ablation therapy via chemo-castration, which unfortunately is not curative, as hormone-refractory or castration-resistant tumors eventually develop.2 Thus, there is a need to develop novel and effective therapy for metastatic PCa.3 Molecularly targeted therapies, especially tyrosine kinase inhibitors, have lately achieved remarkable success in the treatment of certain types of cancers, including chronic myelogenous leukemia and lung adenocarcinoma.4-7 Effective therapeutic strategies for PCa based on tyrosine kinase inhibitors have yet to be developed.8
Src is the most extensively characterized member of the largest family of nonreceptor tyrosine kinases. Although Src is rarely found as a mutated oncogene in human malignancies, elevated expression and activation of Src family kinases (SFKs) have been implicated in a variety of cancers, including glioblastoma, colon, lung, breast, and prostate cancers.9 Among SFK members, Src10,11 and Lyn12 have been functionally linked to prostate carcinogenesis, and Fyn has been found to be overexpressed in PCa.13 Upregulated Src activity correlates with the development of castration-resistant PCa and is involved in promoting cell growth, survival, metastasis, and angiogenesis.14-17 We and others have reported the role of Src tyrosine kinase in the androgen-independent activation of androgen receptor, mediated by neuropeptides, interleukin-8, and epidermal growth factor.18-21 Recently, Src kinase has been reported to physically associate with and directly phosphorylate AR at residue Y534, resulting in the nuclear translocation, stabilization, and enhancement of AR transactivation function under androgen-deficient conditions.21 The studies reported by Park et al.12 and by us22 further demonstrated the role of SFK members in lymph node metastasis. Thus, SFK members represent themselves as important targets for castration-resistant and metastatic PCa.23-25
There are several SFK inhibitors in clinical development—notably, dasatinib, bosutinib, and saracatinib (formerly AZD0530).26 Previously, we demonstrated that saracatinib can inhibit PCa cell proliferation and migration in vitro and lymph node metastasis in an orthotopic nude mouse model.11,22 Flow cytometric analysis of the treated cells revealed significant growth arrest with only marginal apoptosis, a phenomenon also associated with other SFK inhibitors.27-29 In an effort to search for strategies that could enhance cancer cell killing mediated by SFK inhibitors, we looked for possible pro-survival pathways that are activated in response to the drugs. Here we report the induction of pronounced macroautophagy or autophagy by saracatinib.
Autophagy is an evolutionarily conserved process designed to degrade long-lived proteins and organelles to maintain homeostasis.30,31 Under cellular stress conditions, autophagy is rapidly upregulated, providing an alternative source of energy to enable continuous cell survival.32 Excessive or unquenched autophagy, however, can lead to type II programmed cell death (PCD II), which is morphologically distinct from apoptosis and usually caspase independent.32 A hallmark of autophagy is the formation of a double-membrane cytosolic vacuole, the autophagosome, which sequesters cytoplasmic “retired” proteins and organelles and delivers them to the lysosome for degradation.33 Upon induction of autophagy, microtubule-associated protein light chain 3 (LC3) is conjugated to phosphatidylethanolamine for insertion into autophagic membranes, and its eGFP-fusion derivative has been effectively used as a visual marker for autophagosome formation.34
The regulation of autophagy is complex. The PI3K (type I)/Akt pathway is known to inhibit autophagy through the activation of mammalian target of rapamycin (mTOR), which serves as a gatekeeper for autophagy initiation.35,36 AMP kinase (AMPK), sensing cellular AMP/adenosine triphosphate (ATP) ratios, can also inhibit mTOR through activation of tuberous sclerosis 2 (TSC2).37 The role of autophagy in cancer remains unclear.38-40 Defective autophagy may contribute to tumorigenesis, while functional autophagy in response to chemotherapy may lead to chemoresistance of different carcinoma cells.41-43 Accordingly, in the context of SFK inhibitors and PCa, it is not clear whether the induced autophagy contributes to the demise or survival of the treated cells.
In this study, we show that SFK inhibitors such as PP2 and saracatinib effectively induce autophagy in PCa cells, as does siRNA-targeted inhibition of Src expression. These data suggest a role for Src activity in the suppression of autophagy. We also identify Src-induced and autophagy-related signaling pathways, which are affected by SFK inhibitors. Importantly, we demonstrate that inhibition of autophagy using either pharmacological inhibitors or RNA interference of essential autophagy genes promotes cell death induced by Src inhibitors. Notably, the combination of saracatinib with chloroquine (CQ), an inhibitor of autophagy, resulted in 64% tumor growth inhibition and enhanced apoptosis in a xenograft mouse model. Taken together, these findings strongly suggest that inhibition of autophagy may enhance the therapeutic efficacy of SFK inhibitors in the treatment of prostate cancer.
Results and Discussion
Inhibition of Src kinase induces autophagy in prostate cancer cells
Previously, we reported that saracatinib-treated PCa cells were growth arrested but did not undergo extensive apoptosis.11 As autophagy is known to modulate apoptosis, we analyzed the occurrence of autophagy in these cells. PC3 and LNCaP cells were stably transfected to express eGFP-LC3, and they were examined by fluorescent microscopy with or without treatment with the SFK inhibitors, PP2, or saracatinib. Under normal conditions, LC3-I is evenly distributed throughout the cytoplasm. Upon induction of autophagy, a significant fraction of LC3-I undergoes lipidation and is converted into LC3-II (a nonsoluble form),44 which marks autophagosome membranes and is detected as bright puncta in the presence of eGFP-LC3.30 This was clearly seen in cells treated with either 10 μM PP2 or 1 μM saracatinib (Fig. 1A). The fraction of cells that underwent autophagy (as reflected by autophagosome formation) significantly increased after treatment with either PP2 or saracatinib (Fig. 1B). The degree of autophagy induction was comparable to that induced by rapamycin, an mTOR inhibitor and a known autophagy inducer.36 Additional evidence of autophagy induction came from Western blot analysis in the conversion of LC3-I to LC3-II. The ratio of LC3-II to LC3-I increased in a time-dependent manner after PP2 treatment, showing a 19-fold increase at 72 h posttreatment (Fig. 1C). Furthermore, transcriptional upregulation of autophagy genes (i.e., PI3KC3; Atg3, 5, and 7) was also detected (Fig. 1E). To ensure the autophagy induction was indeed due to the inhibition of Src activity, we silenced the expression of Src with specific small interfering RNA (siRNA) targeting Src. Western blotting with a Src antibody showed a distinct reduction in the level of Src protein, which was accompanied by an increase in LC3-II protein levels (Fig. 1D). In addition to LC3, it is reported that p62/SQSTM1, a link between LC3 and ubiquitinated substrates, can be used as a marker for monitoring autophagic flux.45 Concomitant with an increase in the ratio of LC3-II to LC-I is a decrease of p62 protein levels upon Src siRNA knockdown (Fig. 1D). These data indicate that inhibition of Src activity leads to autophagy induction. While this is the first time such a phenomenon has been reported for PCa, the results are consistent with recent reports that dasatinib and imatinib, a Bcr/Abl inhibitor, induce autophagy in glioblastoma and chronic myelogenous leukemia, respectively.29,46
Figure 1.
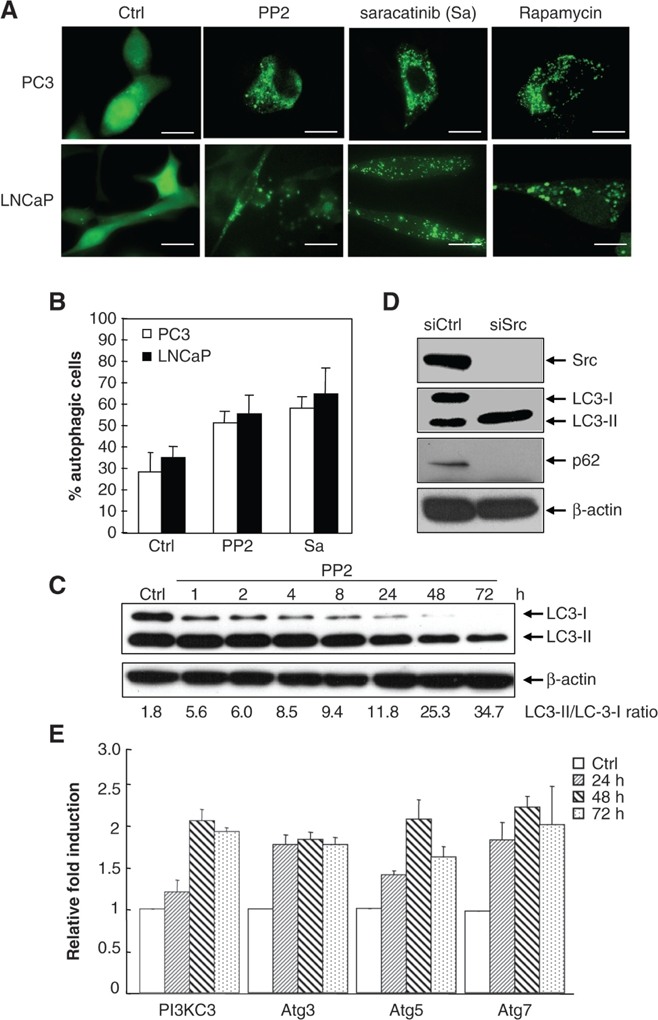
Src inhibitors, PP2 and saracatinib, induce autophagy in prostate cancer cell lines LNCaP and PC3. (A) Representative micrographs of PC3-eGFP-LC3 and LNCaP-eGFP-LC3 cells showing GFP-LC3 localization. PC3-eGFP-LC3 (upper panel) and LNCaP-eGFP-LC3 (lower panel) stable cell lines expressing eGFP-LC3 were treated with DMSO (vehicle control), 10 μM PP2, or 1 μM saracatinib for 48 h and were then analyzed by fluorescence microscopy. Puncta represent autophagosome formation. PC3-eGFP-LC3 and LNCaP-eGFP-LC3 cells treated with 2 μM rapamycin for 4 h were used as a positive control. Scale bar = 30 μm. (B) Quantification plot of autophagic cell numbers in (A). The percentage of autophagic cells was determined by randomly counting 200 cells under the fluorescent microscope. Data represented as mean ± SE. (C) A time-dependent increase in the ratio of LC3-II/LC3-I under the treatment of 10 μM PP2. PC3 cells were treated with 10 μM PP2, and cell lysates were harvested at different time points as indicated and analyzed by Western blotting for LC3-I and LC3-II. β-actin was used as the loading control. (D) PC3 cells were collected at 72 h after being transfected with the negative control siRNA or 100 pmol of Src siRNA oligonucleotides. Cell lysates were then subjected to immunoblot analysis with the antibodies indicated. β-actin was detected as loading control. (E) Induction of PI3KC3, Atg3, Atg5, and Atg7 expression in PC3 cells. PI3KC3, Atg3, Atg5, and Atg7 mRNA levels were measured by quantitative real-time PCR in cells treated for 24, 48, and 72 h with 10 μM PP2. The expression levels were compared to the vehicle control DMSO-treated expression levels. Values represent mean ± SE.
Src inhibitors induce autophagy by inhibiting the Akt/mTOR/p70S6K signaling pathway in PC3 cells
Having demonstrated autophagy induction by Src inhibitors, we investigated the potential signaling pathways involved. Because the PI3K (type I)/Akt/mTOR/p70S6K axis is the main regulatory pathway by which autophagy is suppressed and is also activated by Src (Fig. 2C),47-49 we asked whether Src inhibitors may inactivate this pathway, thereby inducing autophagy. For these experiments, parental PC3 cells were treated with 10 μM PP2 or 1 μM saracatinib, and the cell lysates were subjected to immunoblot with antibodies specific for the signaling molecules as well as their phospho-counterparts (activated forms). We first confirmed that PP2 and saracatinib indeed inhibited Src activity by immunoblotting with α-pSrc (Y416), which is specific for the activated form of SFKs. As expected, phosphorylation of SFK was significantly reduced by both PP2 and saracatinib in a dose-dependent manner (Fig. 2A). Reduced phosphorylation of Akt and mTOR was evident after treatment with either inhibitor, although the kinetics of shutdown differed (Fig. 2B). Likewise, the level of phospho-S6 ribosomal protein, a substrate of p70S6K and an indicator of p70S6K activity, also decreased.50 The total amounts of nonphosphorylated proteins remained similar throughout the treatment duration (Fig. 2B). Alternatively, mTOR can be suppressed by the activation of Ras/Raf-1/MEK1/2/Erk1/2, AMPKα/TSC, and JNK pathways, resulting in autophagy induction.35 None of these kinases were activated upon treatment with SFK inhibitors (see Fig. 2B for phospho-AMPKα and unpublished data). These results point to mTOR inhibition by SFK inhibitors as a potential pathway involved in autophagy induction.
Figure 2.
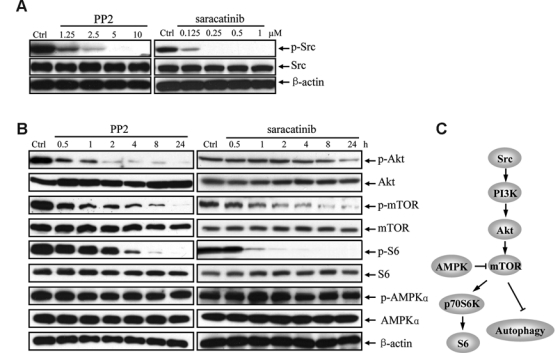
PP2 and saracatinib inhibit mTOR signaling pathway through the PI3K (type I)/Akt pathway. (A) Src phosphorylation in PC3 cells was inhibited in a dose-dependent manner by PP2 and saracatinib following a 30-min treatment. Cell lysates were analyzed by immunoblotting with antibodies indicated. β-actin was detected as loading control. (B) PC3 cells were treated with 10 μM PP2 (left panel)/1 μM saracatinib (right panel) for 0.5, 1, 2, 4, 8, and 24 h. Cell lysates were analyzed by immunoblotting with antibodies as indicated. Controls were treated with vehicle alone. β-actin was detected as loading control. (C) A model depicting Src-mediated signal pathway, involved in the suppression of autophagy.
Pharmacological and genetic inhibitors of autophagy enhance SFK inhibitor-induced apoptosis in PC3 cells
Several studies have demonstrated that autophagy may serve as a protective mechanism in tumor cells and that therapy-induced cell death can be potentiated through autophagy inhibition.31,51 To investigate whether autophagy serves as a survival mechanism in our system, we inhibited autophagy by 3 independent approaches and evaluated its impact on cell killing induced by PP2 or saracatinib. We first tested 2 pharmacological inhibitors, 3-MA and CQ. 3-MA inhibits the catalytic activity of PI3KC3, an enzyme involved in the initiation of autophagosome formation,52 whereas CQ disrupts the function of lysosomes, thus acting at a later stage.51 While 3-MA is primarily a research agent, CQ has been clinically approved as an antimalaria agent. As shown in Figure 3A and B, under the conditions tested, neither 3-MA nor CQ induced any significant level of cell death. They, however, both augment cell killing induced by either PP2 or saracatinib. The increased cell killing is largely due to enhanced apoptosis, as evidenced by a pronounced increase in caspase-3/7 activity within 24 h after combined treatment (Fig. 3A, right panel and 3B, right panel). While 3-MA and CQ each may have other nonspecific effects on cells, the congruent results obtained argue that inhibition of autophagy is the cause of enhanced cell killing induced by SFK inhibitors.
Figure 3.

Inhibition of autophagy enhances PP2 and saracatinib-induced PC3 cell death. (A, B, left and middle panels) PC3 cells were seeded in 96-well plates and cultured overnight. The following day, the cells were treated with 0.1% DMSO as vehicle control, 10 μM CQ or 1 mM 3-MA, 5 μM PP2 or 1 μM saracatinib alone or in combination with 10 μM CQ, or 1 mM 3-MA. After 48 h, cell viability was assessed by MTT assay. Significant difference between PP2/Sa and PP2/Sa plus 3-MA/CQ (P < 0.0005). Values represent mean ± SE. (A, B, right panel) PC3 cells were treated with DMSO (vehicle control), 10 μM CQ, 5 μM PP2, or 1 μM saracatinib alone or in combination with 10 μM CQ. After 24 h, the cultured cells were assayed for caspase-3/7 activity. Significant difference between PP2/Sa and PP2/Sa plus CQ (P < 0.05). Values represent mean ± SE. (C) PC3 cell death was assessed by propidium iodide (PI) staining and flow cytometry analysis after treatment with DMSO (vehicle control), 5 μM PP2, Atg7 siRNA, or 5 μM PP2 plus Atg7 siRNA for 72 h. Fluorescence-activated cell sorting (FACS) analysis shows sub-G1 content. (D, upper panel) Immunoblot for cell lysates from PC3 cells transiently transfected with negative control siRNA or Atg7 siRNA indicating the knockdown of Atg7 in PC3 cells. (D, lower panel) The percentage of sub-G1 content for (3C).
To further confirm that inhibition of autophagy enhances SFK inhibitor-induced cell death, we used siRNA, siAtg7, to knockdown Atg7, a key autophagy gene essential for autophagosome formation.53 Transfection of siAtg7 effectively knocked down the expression of endogenous Atg7 (Fig. 3D, upper panel). Flow cytometric analysis of the siAtg7-treated populations revealed a significant increase of the sub-G1 fraction as compared to the control siRNA-treated samples, when combined with PP2 treatment (Fig. 3C and D). The sub-G1 fraction represents cells with fragmented chromosomes, thus a measure of apoptosis. The results presented in this section, taken together, strongly suggest that autophagy induced by SFK inhibitors protects tumor cells from apoptosis and that autophagy blockade will abrogate the apoptosis resistance associated with SFK inhibitor treatment.
Chloroquine enhances the therapeutic efficacy of saracatinib in a prostate xenograft mouse model
Encouraged by the above in vitro results, we assessed whether treatment with CQ would potentiate the effects of saracatinib in vivo. Nude mice were implanted subcutaneously with 1 × 106 GFP-LC3-PC3 cells and randomly divided into 4 groups with 8 mice in each group: (a) control-treated mice, (b) mice treated with CQ only, (c) mice treated with saracatinib only, and (d) mice treated with saracatinib and CQ. At 14 days, when tumors were palpable, mice were treated daily with vehicle control, 50 mg/kg CQ, 25 mg/kg saracatinib, or the saracatinib/CQ combination. As expected, saracatinib treatment resulted in a decrease in tumor growth by 26% compared to control-treated mice. This decrease was even more marked in the mice treated with saracatinib and CQ, which inhibited tumor growth by 64% (Fig. 4A and B). To determine whether saracatinib effectively inhibited activation of Src and that CQ blocked autophagy in vivo, tumor tissues were harvested, fixed or snap frozen, and prepared for immunohistochemical analysis, TdT-mediated dUTP nick end labeling (TUNEL) staining, or fluorescence analysis. As represented in Figure 4C (upper and middle panel, respectively), saracatinib effectively inhibited Src phosphorylation, and CQ blocked autophagosome fusion with lysosome, resulting in an increased number of autophagosomes. In addition, we further determined the number of apoptotic cells by TUNEL staining of fixed tissues. We observed an at least 2-fold increase in the number of apoptotic tumor cells in the group treated with both saracatinib and CQ (Fig. 4C, lower panel and summary diagram in Fig. 4D). These data confirmed our in vitro results and support our hypothesis that suppression of autophagy sensitizes the killing effect of saracatinib on PCa cells.
Figure 4.
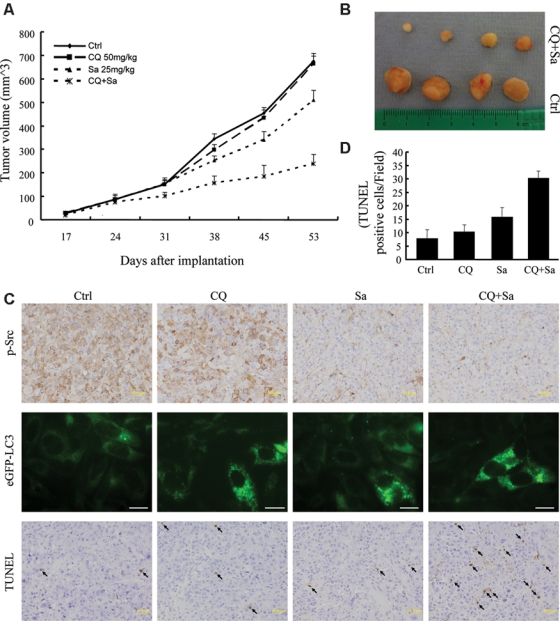
Combination treatment of CQ and saracatinib synergistically inhibits tumor growth in vivo. (A) Mice with PC3-eGFP-LC3 implants were treated with vehicle, 50 mg/kg CQ, 25 mg/kg saracatinib, or CQ plus saracatinib daily. Mice were euthanized after 53 days. Tumor volumes are reported as mean ± SE. (B) Representative tumor samples, including combined treatment of 50 mg/kg CQ with 25 mg/kg saracatinib (upper panel) and vehicle control group (lower panel). (C) Representative micrographs of immunohistochemistry using anti-pSrc antibody (upper panel). Scale bar = 50 μm. Representative fluorescent microscope images of frozen tumor tissue sections for eGFP-LC3 (middle panel). Scale bar = 30 μm. Representative TdT-mediated dUTP nick end labeling (TUNEL) staining of tumor tissues (lower panel). TUNEL-positive cells are indicated by arrows. Scale bar = 50 μm. (D) Tumor sections were analyzed for percentage of apoptotic cells by TUNEL assay. Average numbers of TUNEL-positive cells were counted in 4 randomly selected fields in 3 tumor samples from each group; values represent mean ± SE.
Therapeutic implications
A major conclusion of this study is that SFK inhibitors engender autophagy responses in PCa cells, which are apparently protective, thus lowering the therapeutic efficacy of the inhibitors. We suggest that a combination of autophagy inhibitors such as CQ and SFK inhibitors will provide improved benefits. To our knowledge, this is the first time that autophagy induction is reported for saracatinib and that autophagy blockade is shown to be effective in enhancing cell killing mediated by SFK inhibitors both in vitro and in vivo. While our molecular studies have focused on Src tyrosine kinase, saracatinib inhibits all SFKs, and thus the effects we detected are likely to be a combination of inhibition of Src, Lyn, and Fyn, 3 members reported to be present in PCa. Although saracatinib also inhibits Abl and Arg at a higher concentration,54 these kinases are not activated in PC3 cells11 and thus unlikely to be the targets.
Clinical trials involving SFK inhibitors as an adjunctive therapy for PCa are under way,55 and the inclusion of CQ may be considered. As single agents, SFK or other tyrosine kinase inhibitors have been ineffective in eradicating cancer cells.56-58 The induction of the pro-survival autophagic process in response to tyrosine kinase inhibitors could be one of the underlying reasons for tumor resistance. As illustrated by our study, disrupting autophagy in PCa cells treated with saracatinib, either by pharmacological or genetic inhibitors, resulted in cell death. Our data are in good agreement with the recent report that autophagy blockade potentiates imatinib-induced cell death in chronic myeloid leukemia (CML)46 but are in contrast to the report by Milano et al.,29 where autophagy induction by a combination of dasatinib and temozolomide contributed to cell death in glioma. Perhaps the most striking aspect of our data is the effective inhibition in vivo of PC3 xenograft growth by a combination of saracatinib and CQ. Immunohistochemical studies of the recovered tissues clearly indicated the diminishing signals of activated Src and the increased level of apoptotic cells. An earlier study suggested that CQ promotes cell death by a p53-dependent pathway.59 Since PC3 has a p53-null background, the cause of apoptosis in this case is unlikely due to the intrinsic toxicity of CQ but rather its ability to inhibit autophagic flux. This also suggests that the present protocol should be effective in PCas with either p53+ or p53– status. In addition to CQ, other autophagy inhibitors can potentially be used to enhance the therapeutic efficacy of tyrosine kinase inhibitors. The present study motivates us to develop and characterize other autophagy inhibitors as modulators of targeted therapy.
Materials and Methods
Reagents
Saracatinib was obtained from AstraZeneca International (Alderley Park, UK). 4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) and rapamycin were purchased from Calbiochem (San Diego, CA). Chloroquine (CQ), 3-methyladenine (3-MA), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (Saint Louis, MO). Saracatinib and PP2 were dissolved in DMSO. CQ and 3-MA were dissolved in phosphate-buffered saline (PBS).
Cell culture
Prostate cancer cell lines LNCaP and PC3 were purchased from the American Type Culture Collection (ATCC; Manassas, VA) and maintained in RPMI 1640 containing 10% fetal bovine serum and 1% penicillin/streptomycin/glutamine. LNCaP and PC3 cells stably expressing eGFP-LC3 (LNCaP-eGFP-LC3 and PC3-eGFP-LC3) were generated as described previously60 and selected with 400 μg/mL and 500 μg/mL of G418 (Sigma-Aldrich), respectively.
Fluorescent microscopy
LNCaP-eGFP-LC3 and PC3-eGFP-LC3 were seeded on poly-lysine-coated coverslips and treated with 10 μM PP2 or 1 μM saracatinib for 48 h or 2 μM rapamycin for 4 h, followed by 4% paraformaldehyde fixation and mounting with SlowFade with DAPI (Invitrogen, Carlsbad, CA). eGFP-LC3 images were examined under a 60x lens on an Olympus BX61 motorized reflected fluorescence microscope with FITC filter (excitation, 480 nm; emission, 535 nm) by using SlideBook4.1 software (Intelligent Imaging Innovations, Denver, CO).
Western blotting and antibodies
Western blotting was performed as described previously.61 Proteins were detected by the following antibodies: β-actin (Sigma-Aldrich), p62 (Santa Cruz Biotechnology, Santa Cruz, CA), phospho-Src (Tyr416), Src, LC3, phospho-Akt (Ser473), Akt, phospho-mTOR (Ser2448), mTOR, phospho-S6 (Ser235/236), S6, phospho-AMPKα (Thr172), and AMPKα (Cell Signaling Technology, Beverly, MA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
PC3 cells were seeded in 96-well plates and cultured overnight, followed by treatment with 0.1% DMSO as vehicle control, 10 μM CQ or 1 mM 3-MA, 5 μM PP2 or 1 μM saracatinib alone or in combination with 10 μM CQ, or 1 mM 3-MA for 48 h. The cell viability was then examined by MTT assay (Roche Diagnostic, Mannheim, Germany) according to the manufacturer’s instructions.
Reverse transcription and quantitative real-time PCR (qRT-PCR)
Reverse transcription and qRT-PCR were done as described previously.60 Primer sequences used to amplify PI3KC3, Atg3, Atg5, and Atg7 fragments are listed as follows: PI3KC3 (F) 5′-CTGCGAAGGTATTCTAATCTG-3′ and (R) 5′-GGTCTAAGCGGAATTTATCCT-3′; Atg3 (F) 5′-ATCACAACACAGGTATTACAG-3′ and (R) 5′-TCCTTCATCTTC ATCTTCTTCC3′; Atg5 (F) 5′-GCAAGCCAGACAGGAAAAAG-3′ and (R) 5′-GA CCTTCAGTTGGTCCGGTAA-3′; and Atg7 (F) 5′-CAGGAGA TTCAACCAGAGAC-3′ and (R) 5′-AGATACCATCAATTCCACGG-3′.
Flow cytometry
PC3 cells were transfected with negative control siRNA or Atg7 siRNA and incubated for 72 h. The cells were then treated with 5 μM PP2 or 0.1% DMSO (vehicle control) for an additional 72 h. The DNA content of the cells was determined by propidium iodide (Sigma-Aldrich) staining.60 Cells were analyzed using a Coulter Epics XL flow cytometer (Beckman Coulter, Miami, FL).
RNA interference
PC3 cells were seeded in 60-mm dishes and transiently transfected with 100 pmol negative control siRNA (Ambion, Foster City, CA) or Src siRNA ON-TARGETplus SMARTpool (Thermo Scientific, Bellefonte, PA) using DharmaFECT reagent (Thermo Scientific). Cell lysates were prepared and then analyzed by Western blot as described previously. To inhibit autophagy, PC3 cells were transfected with negative control siRNA or Atg7 siRNA as described above. Cells were then treated with DMSO as vehicle control or 5 μM PP2 for 72 h and analyzed by fluorescence-activated cell sorting (FACS) analysis for sub-G1 content.
Analysis of caspase-3/7 activities
Caspase-3/7 activities were measured by using the Apo-ONE Homogeneous Caspase-3/7 Assay kit (Promega, Madison, WI) according to the manufacturer’s protocol. PC3 cells were plated in triplicate in 96-well cell culture plates (Costar, Corning, Inc., Corning, NY). The following day, the cells were treated with 0.1% DMSO as vehicle control, 10 μM CQ, 5 μM PP2, or 1 μM saracatinib alone or in combination with 10 μM CQ for 24 h. The cells were then incubated at room temperature with a mixture of substrate and reaction buffer for 30 min prior to analysis. Caspase-3/7 activities were measured by using a fluorescence microplate reader (Tecan Systems, San Jose, CA). Results are presented as the mean ± SE.
In vivo tumor biology
Animal housing and experimental conditions were in accordance with the protocol approved by the Institutional Animal Care and Use Committee at the University of California, Davis. Then, 1 × 106 PC3-eGFP-LC3 cells were mixed with an equal volume of Matrigel (Becton Dickinson, San Jose, CA) and injected subcutaneously into the flanks of 5- to 6-week-old male nude mice. Mice were randomly divided into 4 groups and treated with buffer only (control), 50 mg/kg/d CQ, 25 mg/kg/d saracatinib, or a combination of CQ and saracatinib. Saracatinib was resuspended in 0.5% hydroxymethylcellulose and 0.1% Tween-80 and administered orally. CQ was dissolved in PBS and injected intraperitoneally. Mice were treated daily, starting from 17 days after injection of PC3-eGFP-LC3 cells. The size of tumors was measured by a caliper on the fifth day of each week. After 53 days treatment, mice were sacrificed and tumors were harvested. Half of the tumor sample was fixed in 10% neutral buffered formalin, and the other half was snap frozen in liquid nitrogen and stored at –80°C.
Paraffin-embedded tumor tissues were sectioned to 5-μm thickness and mounted on positively charged microscope slides, and 1 mM EDTA (pH 8.0) was used for antigen retrieval. Immunohistochemistry was performed by an UltraVision LP Detection system HRP Polymer and DAB Plus Chromagen (Thermo Scientific) using 1:100 dilution of phospho-Src (Tyr416) antibody (Cell Signaling). Sections were counterstained with hematoxylin (American Master Tech Scientific, Lodi, CA). For TUNEL assay, paraffin-embedded sections were stained by the Apoptag Peroxidase In Situ Apoptosis Detection Kit (Millipore, Temecula, CA) following the manufacturer’s instructions. For eGFP-LC3 images, snap-frozen tumor tissues were embedded in Tissue-Tek OCT compound (Sakura Finetek, Torrance, CA) using chilled isopentane for further section. Then, 7-μm cryostat sections were fixed in 4% paraformaldehyde in PBS for 10 min and analyzed as described previously.
Statistics
Paired Student t test was used for statistics.
Acknowledgments
We thank Dr Mel Campbell for helpful comments and editing, as well as Anthony Martinez for providing reagents and advice to this work.
Footnotes
Dr Christopher P. Evans serves as a consultant to AstraZeneca, which provided saracatinib for this work. The other authors disclosed no potential conflict of interest.
The work was supported by NIH grants (DK52659, CA114575, DK0782243) to HJK.
References
- 1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009;59:225-49 [DOI] [PubMed] [Google Scholar]
- 2. Prezioso D, Galasso R, Di Martino M, Iapicca G, Annunziata E, Iacono F. Actual chemotherapeutical possibilities in hormone-refractory prostate cancer (HRPC) patients. Anticancer Res 2007;27:1095-104 [PubMed] [Google Scholar]
- 3. Johnstone PA, Assikis V, Goodman M, Ward KC, Riffenburgh RH, Master V. Lack of survival benefit of post-operative radiation therapy in prostate cancer patients with positive lymph nodes. Prostate Cancer Prostatic Dis 2007;10:185-88 [DOI] [PubMed] [Google Scholar]
- 4. Golas JM, Lucas J, Etienne C, Golas J, Discafani C, Sridharan L, et al. SKI-606, a Src/Abl inhibitor with in vivo activity in colon tumor xenograft models. Cancer Res 2005;65:5358-64 [DOI] [PubMed] [Google Scholar]
- 5. Finn RS, Dering J, Ginther C, Wilson CA, Glaspy P, Tchekmedyian N, et al. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/“triple-negative” breast cancer cell lines growing in vitro. Breast Cancer Res Treat 2007;105:319-26 [DOI] [PubMed] [Google Scholar]
- 6. Steinberg M. Dasatinib: a tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. Clin Ther 2007;29:2289-308 [DOI] [PubMed] [Google Scholar]
- 7. Sanford M, Scott LJ. Gefitinib: a review of its use in the treatment of locally advanced/metastatic non-small cell lung cancer. Drugs 2009;69:2303-28 [DOI] [PubMed] [Google Scholar]
- 8. Brunton VG, Frame MC. Src and focal adhesion kinase as therapeutic targets in cancer. Curr Opin Pharmacol 2008;8:427-32 [DOI] [PubMed] [Google Scholar]
- 9. Yeatman TJ. A renaissance for SRC. Nat Rev Cancer 2004;4:470-80 [DOI] [PubMed] [Google Scholar]
- 10. Chang YM, Kung HJ, Evans CP. Nonreceptor tyrosine kinases in prostate cancer. Neoplasia 2007;9:90-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene 2008;27:6365-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, Volgin AY, et al. Targeting Src family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res 2008;68:3323-33 [DOI] [PubMed] [Google Scholar]
- 13. Posadas EM, Al-Ahmadie H, Robinson VL, Jagadeeswaran R, Otto K, Kasza KE, et al. FYN is overexpressed in human prostate cancer. BJU Int 2009;103:171-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Trevino JG, Summy JM, Gray MJ, Nilsson MB, Lesslie DP, Baker CH, et al. Expression and activity of SRC regulate interleukin-8 expression in pancreatic adenocarcinoma cells: implications for angiogenesis. Cancer Res 2005;65:7214-22 [DOI] [PubMed] [Google Scholar]
- 15. Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 2006;10:309-19 [DOI] [PubMed] [Google Scholar]
- 16. Kung HJ, Evans CP. Oncogenic activation of androgen receptor. Urol Oncol 2009;27:48-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tatarov O, Mitchell TJ, Seywright M, Leung HY, Brunton VG, Edwards J. Src family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin Cancer Res 2009;15:3540-9 [DOI] [PubMed] [Google Scholar]
- 18. Lee LF, Guan J, Qiu Y, Kung HJ. Neuropeptide-induced androgen independence in prostate cancer cells: roles of nonreceptor tyrosine kinases Etk/Bmx, Src, and focal adhesion kinase. Mol Cell Biol 2001;21:8385-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee LF, Louie MC, Desai SJ, Yang J, Chen HW, Evans CP, et al. Interleukin-8 confers androgen-independent growth and migration of LNCaP: differential effects of tyrosine kinases Src and FAK. Oncogene 2004;23:2197-205 [DOI] [PubMed] [Google Scholar]
- 20. Amorino GP, Deeble PD, Parsons SJ. Neurotensin stimulates mitogenesis of prostate cancer cells through a novel c-Src/Stat5b pathway. Oncogene 2007;26:745-56 [DOI] [PubMed] [Google Scholar]
- 21. DaSilva J, Gioeli D, Weber MJ, Parsons SJ. The neuroendocrine-derived peptide parathyroid hormone-related protein promotes prostate cancer cell growth by stabilizing the androgen receptor. Cancer Res 2009;69:7402-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang JC, Ok J-h, Busby JE, Borowsky AD, Kung H-J, Evans CP. Aberrant activation of androgen receptor in a new neuropeptide-autocrine model of androgen-insensitive prostate cancer. Cancer Res 2009;69:151-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell 2004;6:209-14 [DOI] [PubMed] [Google Scholar]
- 24. Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol 2009;6:587-95 [DOI] [PubMed] [Google Scholar]
- 25. Saad F. Src as a therapeutic target in men with prostate cancer and bone metastases. BJU Int 2009;103:434-40 [DOI] [PubMed] [Google Scholar]
- 26. Finn RS. Targeting Src in breast cancer. Ann Oncol 2008;19:1379-86 [DOI] [PubMed] [Google Scholar]
- 27. Song L, Morris M, Bagui T, Lee FY, Jove R, Haura EB. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res 2006;66:5542-8 [DOI] [PubMed] [Google Scholar]
- 28. Konig H, Copland M, Chu S, Jove R, Holyoake TL, Bhatia R. Effects of dasatinib on SRC kinase activity and downstream intracellular signaling in primitive chronic myelogenous leukemia hematopoietic cells. Cancer Res 2008;68:9624-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Milano V, Piao Y, LaFortune T, de Groot J. Dasatinib-induced autophagy is enhanced in combination with temozolomide in glioma. Mol Cancer Ther 2009;8:394-406 [DOI] [PubMed] [Google Scholar]
- 30. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008;451:1069-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meijer AJ, Codogno P. Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci 2009;46:210-40 [DOI] [PubMed] [Google Scholar]
- 32. Gozuacik D, Kimchi A. Autophagy and cell death. Curr Top Dev Biol 2007;78:217-45 [DOI] [PubMed] [Google Scholar]
- 33. Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004;23:2891-906 [DOI] [PubMed] [Google Scholar]
- 34. Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2004;36:2503-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ 2005;12:1509-18 [DOI] [PubMed] [Google Scholar]
- 36. Cao C, Subhawong T, Albert JM, Kim KW, Geng L, Sekhar KR, et al. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res 2006;66:10040-7 [DOI] [PubMed] [Google Scholar]
- 37. Hardie DG. The AMP-activated protein kinase pathway: new players upstream and downstream. J Cell Sci 2004;117:5479-87 [DOI] [PubMed] [Google Scholar]
- 38. Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007;117:326-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007;7:961-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol 2009;3:366-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carew JS, Nawrocki ST, Cleveland JL. Modulating autophagy for therapeutic benefit. Autophagy 2007;3:464-7 [DOI] [PubMed] [Google Scholar]
- 42. Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007;110:313-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Song J, Qu Z, Guo X, Zhao Q, Zhao X, Gao L, et al. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy 2009. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 44. Kabeya Y, Mizushima N, Uero T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J 2000;19:5720-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005;171:603-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, et al. Targeting autophagy potentiates tyrosine kinase inhibitor induced cell death in Philadelphia chromosome positive cells, including primary CML stem cells. J Clin Invest 2009;119:1109-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen HC, Guan JL. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA 1994;91:10148-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol 1994;14:1680-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol 2000;2:249-56 [DOI] [PubMed] [Google Scholar]
- 50. Blommaart EFC, Luiken JJFP, Blommaart PJE, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem 1995;270:2320-6 [DOI] [PubMed] [Google Scholar]
- 51. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007;8:741-52 [DOI] [PubMed] [Google Scholar]
- 52. Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 1982;79:1889-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tanida I, Mizushima N, Kiyooka M, Ohsumi M, Ueno T, Ohsumi Y, et al. Apg7p/Cvt2p: a novel protein-activating enzyme essential for autophagy. Mol Biol Cell 1999;10:1367-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hannon RA, Clack G, Rimmer M, Swaisland A, Lockton JA, Finkelman RD, et al. Effects of the Src kinase inhibitor saracatinib (AZD0530) on bone turnover in healthy men: a randomized, double-blind, placebo-controlled, multiple ascending dose phase I trial. J Bone Miner Res 2009. Epub ahead of print [DOI] [PubMed] [Google Scholar]
- 55. Lara PN, Jr, Longmate J, Evans CP, Quinn DI, Twardowski P, Chatta G, et al. A phase II trial of the Src-kinase inhibitor AZD0530 in patients with advanced castration-resistant prostate cancer: a California Cancer Consortium study. Anticancer Drugs 2009;20:179-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood 2006;107:4532-9 [DOI] [PubMed] [Google Scholar]
- 57. Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia 2007;21:926-35 [DOI] [PubMed] [Google Scholar]
- 58. Kopetz S, Shah AN, Gallick GE. Src continues aging: current and future clinical directions. Clin Cancer Res 2007;13:7232-6 [DOI] [PubMed] [Google Scholar]
- 59. Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest 2008;118:79-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim RH, Coates JM, Bowles TL, McNerney GP, Sutcliffe J, Jung JU, et al. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res 2009;69:700-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Qiu Y, Robinson D, Pretlow TG, Kung HJ. Etk/Bmx, a tyrosine kinase with a pleckstrin-homology domain, is an effector of phosphatidylinositol 3′kinase and is involved in interleukin 6-induced neuroendocrine differentiation of prostate cancer cells. Proc Natl Acad Sci USA 1998;95:3644-9 [DOI] [PMC free article] [PubMed] [Google Scholar]