

Abstract
The M2 proton channel from influenza A virus is an essential protein that mediates transport of protons across the viral envelope. This protein has a single transmembrane helix, which tetramerizes into the active channel. At the heart of the conduction mechanism is the exchange of protons between the His37 imidazole moieties of M2 and waters confined to the M2 bundle interior. Protons are conducted as the total charge of the four His37 side chains passes through 2+ and 3+ with a pKa near 6. A 1.65 Å resolution X-ray structure of the transmembrane protein (residues 25–46), crystallized at pH 6.5, reveals a pore that is lined by alternating layers of sidechains and well-ordered water clusters, which offer a pathway for proton conduction. The His37 residues form a box-like structure, bounded on either side by water clusters with well-ordered oxygen atoms at close distance. The conformation of the protein, which is intermediate between structures previously solved at higher and lower pH, suggests a mechanism by which conformational changes might facilitate asymmetric diffusion through the channel in the presence of a proton gradient. Moreover, protons diffusing through the channel need not be localized to a single His37 imidazole, but instead may be delocalized over the entire His-box and associated water clusters. Thus, the new crystal structure provides a possible unification of the discrete site versus continuum conduction models.
Keywords: ion channels, M2 proton channel, membrane proteins, water clusters, histidine protonation
Water molecules confined at interfaces or in cavities behave differently from those in the bulk. Studies of water clusters have shed light not only on their fundamental properties (1–4) but also on the mechanism employed by various nano-bio systems to fine-tune water and proton transport (5–9). A relevant example from biology is the M2 protein of the influenza A virus (10, 11), which is the target of the influenza drugs amantadine and rimantadine (12–17). Tetrameric M2 (18) transports protons across the viral envelope to acidify the virion interior and trigger uncoating of the viral RNA prior to fusion of the viral envelope with the endosomal bilayer (19). M2 is one of the smallest bona fide channel/transporter proteins (96 residues), capable of pH-dependent activation and highly selective conduction of protons vs. other ions (20–25). A narrow pore leads to the highly conserved His37 and Trp41 residues (16, 17, 26–29), which are respectively responsible for proton selectivity (30) and asymmetry in the magnitude of conductance when the proton gradient is reversed (31). Thus the control of proton diffusion across the membrane relies on the ability of the imidazole moieties of His37 to accept and store protons from water molecules in the pore.
An M2 peptide (residues 22–46), slightly longer than the transmembrane domain (32), associates into a functional four-helix bundle (33). Solid state 15N nuclear magnetic resonance (ssNMR) experiments indicate that the first protons are bound to the His residues in the tetrad (34) with surprisingly high affinity (pKa = 8.2), stabilized via low-barrier H-bonds. The third pKa of His37 is near the unperturbed pKa of His in water (6.2), which is surprising given the proximity of these three charged groups within the hydrophobic region of a bilayer. Thus, M2 is remarkable in its ability to stabilize up to a 3+ charge (near neutral pH) within a subnanometer enclosure well within the low dielectric environment of the phospholipid membrane. How such a condition is stabilized and contributes to proton conduction constitutes a puzzle in membrane biophysics.
Here we present a high-resolution crystallographic structure of a peptide (M2TM′ hereafter) spanning the TM helical region (residues 25–46) of the M2 protein. A similarly short sequence (22–46) was shown to associate into tetramers that faithfully reproduce the salient electrophysiological and pharmacological features of the full-length protein (33). The present structure reveals a proton conduction path composed of alternating layers of sidechains and well-ordered water clusters. Computational results suggest that this structure corresponds to an intermediate protonation state of the channel, in agreement with the ssNMR measurements (34). The relationship between this structure and previous structures recorded at higher and lower pH (16, 17) suggests that the proton transport mechanism involves conformational changes in the protein bundle coupled to proton motion through the pore.
Results
Sidechain-Mediated Water Wires Line the Pore of M2.
M2TM′ was crystallized at pH ∼6.5, giving crystals that diffract to 1.65 Å. As in previous work (16), Gly34 was converted to Ala to assist forming high quality crystals. This mutant has been shown to be functional in electrophysiological studies (35). An equivalent peptide featuring the wild-type sequence gave crystals with a very similar asymmetric unit, but diffraction was weaker (SI Text).
M2TM′ assembles into a nearly symmetrical helical bundle, similar to previous lower-resolution structures (16, 17, 26, 36, 37). However, the significantly greater resolution now shows that the pore is formed by five layers of sidechains and three intercalated water clusters stacked to form a continuous conduction pathway (Fig. 1A). The outermost or “top” layer of side chains is composed by the four Val27 residues, which form a nearly closed Val27 valve (2 Å pore radius), leading to a central pore lined by small residues, Ala30, Ser31, and Ala34 (Gly34 in the wild type). The conduction pathway is next interrupted by the His37 residues, forming what we term a His-box, similar to aromatic boxes, but smaller in cross section due to the smaller size of the imidazole ring (Fig. 1B). There is no direct H-bonding between the imidazoles, but instead they are connected via a highly structured network of water molecules. The His-box needs to expand only slightly (1–2 Å) to allow passage of a water-sized molecule. Below this motif the sidechains of Trp41 form a Trp-basket with the aromatic rings angled by approximately 45° relative to the bundle axis. Lastly, Asp44 and Arg45 line an Asp/Arg box, defining the cytoplasmic end of the channel. The planar faces of the guanidino groups of Arg45 form a 7 to 8 Å box stabilized at the corners by interaction with an oxygen atom of Asp44. Whereas the electron density for the Asp44 residues is very well-defined, the Arg45 sidechains have higher Debye–Waller factors, suggestive of greater conformational mobility.
Fig. 1.

Structure of M2TM′. (A) The backbone of three monomers is drawn in light gray, and the pore-lining side chain groups are shown as sticks. From the N-terminus (viral exterior) to the C-terminus (viral interior), these are: the Val27 valve (blue), Ser31 (dark gray), the His-box (orange), the Trp-basket (purple), and the Asp/Arg-box (light blue). Water molecules belonging to the entry, bridging, and exit clusters are represented by red spheres. Black lines indicate the observed water–protein H-bonds. The uppermost dimer of waters in the entry cluster exists in two nearly equally occupied configurations in the crystal lattice, related by a 90° rotation down the central axis of the channel. Only one of the two orientations is shown in the figure. In the exit cluster the fifth water molecule (showing a high Debye–Waller factor) is drawn transparent and with a larger radius. The wireframe represents the peak region of the diffuse electron density detected right under the Val27 valve. (B) A closer view of the His-box surrounded by the entry and bridging clusters. (C) A closer view of the exit cluster.
Immediately below the Val27 valve is a region of diffuse density, indicative of dynamically or statically disordered solvent (Fig. 1A); beyond this point, the remainder of the pore is filled by three well-ordered water layers that connect the layers of sidechains. Above the His-box is an entry cluster of 6 waters (Fig. 1 A and B) that consists of a tight water dimer (O–O distance 2.5 Å) atop four waters, which in turn are H-bonded to the Nδ of His37 and the backbone carbonyl of residue 34. Connecting the His-box and Trp-basket is the His37/Trp41 bridging cluster of two waters (Fig. 1 A and B), which H-bonds to the Nε of each His37 residue. This dimer is well positioned to mediate a π-cation interaction (38) between charged His37 residues and the electron-rich faces of Trp41 residues. Lastly, the exit cluster (Fig. 1 A and C) consists of four waters that form H-bonds connecting the indole NH of Trp41 to a carboxylate oxygen of Asp44. A fifth, poorly ordered solvent molecule lies below these four waters, displaced toward the interior of the virus. Throughout the structure, the four-fold symmetry is broken only by the water dimer found in the entry cluster and the His37/Trp41 bridging cluster (Fig. 1 A and B). No ions were detected in the structure, although the solvent density around Ala30 and Ser31 and the fifth solvent molecule in the exit cluster are sufficiently diffuse that it would be difficult to unambiguously rule out disordered chloride ions with partial occupancy at these positions (Fig. S1).
In summary, the pore of M2TM′ is populated by a network of water molecules stably H-bonded to the protein, starting below the Val27 valve. The H-bonding network extends through His37, and is broken only at the π-face of Trp41, near the interior of the virus.
pH-Dependent Conformational Transitions.
Comparison of the current structure to previously solved structures (16, 17) suggests potential conformational changes that might facilitate motion of protons through the channel. Fig. 2A compares the structure of the “neutral form” of M2, solved at pH 7.5-8 in the presence of rimantadine (Left) (17), the “intermediate form” of M2TM′ crystallized here at pH 6.5 (Center), and a “low pH” (3+ or 4+) form of the structure crystallized in the presence of amantadine near pH 5 (Right) (16). As the degree of protonation of the His-box increases, the N-terminal half of the TM helix becomes increasingly tilted (relative to the pore axis): ranging from 16° in the neutral form, to 31° in the present structure, to 38° in the low pH structure. This motion progressively constricts the Val27 valve, which is open with an approximate 3 Å radius in the neutral structure, to be essentially fully closed at low pH. The opposite is observed for the C-terminal aspect of the bundle, where both a flip of the rotamer of Trp41 as well as main-chain structural changes cause progressively greater water access as pH is decreased. At high pH, the hydrophobic benzenoid ring of Trp41 blocks the C-terminal end of the channel. By contrast, at pH 6.5, π-cation interactions with the His-box/bridging cluster stabilize the Trp-basket in a conformation that projects the polar indolic NH toward the pore, allowing interactions with Asp44 via the exit water cluster (Fig. 1 A and C). At even lower pH, the bundle takes a conical shape, and the pore increases in diameter near the C-terminal exit from the channel. The conformational changes between the three structures reflect two types of motions: helix bending and rigid helix tilting. In excellent agreement with ssNMR studies of M2 (26, 39) in bilayers, the current pH structure displays a gentle 12° bend near the center of the bilayer. At low pH, the C-terminal end of the helix swivels into a straighter configuration, accentuating the conical shape of the bundle. Also in agreement with ssNMR, the bend involves small changes in ϕ/Ψ angles over a large number of helical residues, rather than a shift to a nonhelical conformation at a given position (26, 39). An analysis of the position-specific Debye–Waller factors (Fig. 2B) also revealed the greatest degree of conformational variability near the regions of the channel expected to undergo large-scale conformational transitions associated with proton entry and release.
Fig. 2.

M2TM helix bundle in different experimental structures, with Val27, His37, Trp41, Asp44, and Arg45 color coded as in Fig. 1. (A) TM portion of the previously reported NMR structure at pH 7.5–8 (17) (Left). The X-ray structure at pH 6.5 presented here (Center). The previously reported X-ray structure at pH 5.3 (16) (Right). The blue and red cylinders in the top row highlight, respectively, the N- and C-terminal portions of the helices and their angle with respect to the bundle axis. In the X-ray structure presented here the C-terminal portion shows the same angle as the NMR structure (17), whereas the N-terminal one closely resembles that of the previous X-ray structure (16). There is a 12° bend between these two helical sections through several residues around Ala34. The Val27 valve constricts with decreasing pH, whereas the Trp-basket opens up. Importantly, the Trp sidechain rotamer is different in the high pH NMR structure (Lower Left) than in the intermediate pH structure reported here (Lower Center). (B) M2TM′ crystal structure with its backbone B-factors represented by color and helix thickness. B-factors for the crystal structure were normalized, (B - 〈B〉)/σ(B), percent ranked, and averaged over the four helices.
Despite large differences in permeating ions and overall structure, a remarkably similar helix bending transition at a conserved Gly residue underlies gating between open and closed conformations in tetrameric K+ and NaK channels (40) and mechanosensitive channels (41). Thus, this motion appears to represent an energetically and structurally facile solution to couple ion-binding to selectivity filters with large-amplitude gating motions.
Assignment of Protonation State.
The characterization of the pKa’s for the multiple titration sites of the tetrad has been the subject of extensive investigation based on either computational (42) or experimental (34) approaches. A crucial insight onto this issue has been provided by ssNMR spectroscopy on M2TM peptides bearing 15N-labeled histidines; these experiments showed that, remarkably, the first two pKa’s of the tetrad are 8.2 and the third 6.3.
In this work M2TM′ was crystallized in a solution buffered at a pH of approximately 6.5, therefore we expect the X-ray structure to be representative of a configuration of the peptide in which two or three His are protonated (34). The conformation of the bundle, which shows hybrid features compared to the previously determined high pH NMR (17) and low pH crystal structures (16) (Fig. 2A), is consistent with the hypothesis of an intermediate value of the total charge on the tetrad. To assign the likely overall charge and tautomeric state of the neutral His residues, the magnetic shielding tensors were calculated [using density functional theory (DFT)] for model systems based on the X-ray structure and comprising the His and Trp tetrads plus the clusters of water molecules H-bonded to the imidazole moieties, focusing on states with a total charge of 2+ and 3+. The spectrum predicted for the 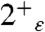 and
and 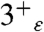 states (in which the neutral His are protonated on the ε nitrogen of the His imidazole) is consistent with the experimental spectrum (34). Therefore these calculations are in agreement with previous studies suggesting that the ε site is the primary protonation site of the neutral His residues in the tetrad. Moreover, small differences in the water/imidazole H-bond distances computed for the
states (in which the neutral His are protonated on the ε nitrogen of the His imidazole) is consistent with the experimental spectrum (34). Therefore these calculations are in agreement with previous studies suggesting that the ε site is the primary protonation site of the neutral His residues in the tetrad. Moreover, small differences in the water/imidazole H-bond distances computed for the 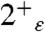 and
and 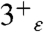 states result in shifts as large as 10 ppm of the NMR peaks for the protonated nitrogen, which is also consistent with the spectra in ref. 34. Thus, the H-bonding between the imidazole and imidazolium species of His37 is likely mediated by a water cluster.
states result in shifts as large as 10 ppm of the NMR peaks for the protonated nitrogen, which is also consistent with the spectra in ref. 34. Thus, the H-bonding between the imidazole and imidazolium species of His37 is likely mediated by a water cluster.
To further assess the set of possible protonation states classical molecular dynamics (MD) simulations were performed. In these simulations the structure of the bundle was held fixed by restraining the backbone atoms to their initial positions, to discover which protonation state would induce a water structure closest to that observed in the crystal. The number of waters and their average position is remarkably sensitive to the total charge of the His-box. In particular, the number of waters within the pocket wedged between the His-box and the Trp-basket correlates very well with the number of protons accepted by the His tetrad (Fig. 3A). The 1+ and 2+ states both have an average of two water molecules in this region, as observed in the bridging dimer (Fig. 3A). Furthermore, the agreement between the calculated and the measured oxygen densities is much better in the 2+ case than in the 1+: Two water molecules are clearly defined near the protonated histidines (Fig. 3B), whereas one of the two is very diffuse in the 1+ case. The overall shape of the calculated water density in the 2+ case is also the one that most closely resembles the experimental density, despite some broadening of the distribution resulting from the difference in temperature (310 K in the simulation as opposed to 100 K in the X-ray diffraction). By contrast, in the 1+ case only one of the two waters is defined, whereas the other is spread across the three neutral histidines. MD simulations, performed without restraints in a hydrated bilayer on the ε-tautomers, also support the hypothesis that the highly charged states such as 3+ and 4+ do not significantly contribute to the X-ray structure. Indeed, the bundle is found to be stable (with a rmsd of less than 1 Å) over the relatively long time interval of ∼80 ns only in the 1+ and 2+ states (Fig. 3C).
Fig. 3.

Spatial distribution of pore waters and structural stability of the bundle. (A) Average number of water molecules localized within the pocket between the His and the Trp tetrads. (B) The water oxygen density from the simulation of the 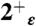 state (light blue shading) is plotted together with the experimental one (red surfaces). The overall structure of the bundle is also presented (gray shading) and the sidechains of some of functionally relevant residues are highlighted as sticks: Val27 (blue), His37 (orange), and Trp41 (green). (C) The rmsd of the heavy atoms from the initial (X-ray) structure is plotted as a function of MD simulation time for the protonation states
state (light blue shading) is plotted together with the experimental one (red surfaces). The overall structure of the bundle is also presented (gray shading) and the sidechains of some of functionally relevant residues are highlighted as sticks: Val27 (blue), His37 (orange), and Trp41 (green). (C) The rmsd of the heavy atoms from the initial (X-ray) structure is plotted as a function of MD simulation time for the protonation states 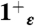 (red),
(red), 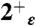 (gold),
(gold), 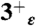 (purple), and 4+ (blue), respectively.
(purple), and 4+ (blue), respectively.
Overall, whereas the calculated magnetic shieldings suggest that 2+ and 3+ states (with the neutral His in the ε-tautomer) are consistent with the NMR spectra (Table S1), the configuration of the water molecules and the stability of the bundle characterized via MD simulations indicate the 2+ as being more consistent with the X-ray structure. Thus, we tentatively assign the structure as representative of the 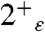 , with the caveat that this conclusion is based on classical MD simulations that do not allow proton delocalization in the water cluster or account for any quantum stabilization of this network.
, with the caveat that this conclusion is based on classical MD simulations that do not allow proton delocalization in the water cluster or account for any quantum stabilization of this network.
Interruption of Water Wires Disrupts M2 Activity.
Among the highly fit transmissible flu viruses the pore-lining residue Gly34 is one of the most conserved, pointing to a particularly important functional role. Hydrophobic mutations at this position are generally nonfunctional, with the exception of G34A (35). The four Ala34 residues are also involved in several interesting properties of the current X-ray structure. First, M2TM′ helices are bent at this position (Fig. 2). Also, their backbone carbonyl groups form H-bonds that stabilize the entry cluster. A small residue at position 34 might assist in hinge formation (43), although all crystallographic and NMR studies suggest that this residue remains in the α-helical region of the Ramachandran map. Moreover, the pore region between Ala34 and His37, where the entry cluster is located, features the highest pore diameter in the M2TM′ structure (Fig. 4A).
Fig. 4.

Effect of mutations of pore-lining residues. (A) The density of water oxygen as a function of the location in the channel pore (z) for the wild type (WT) and 2 mutants (G34A and G34V) is reported in black, red, and blue, respectively. Density profiles were obtained from MD simulations of the 2ε state of each variant. Shaded areas mark peaks conserved in all mutants. The shape of the channel pore is depicted in blue shading along with the sidechains of pore-lining residues. The density in the region adjacent to residue 34 decreases with the increased size of the sidechain and drops to zero for the G34V mutant. (B) Specific activity measurements for M2 wild type, G34A and G34V in Xenopus laevis oocytes. The amplitude of the channel activity (I) was plotted as a function of the immunosignal intensity for each tested oocyte and a straight line was fitted to the data. The slope of each plot represents the relative specific activity for the protein: 6.6 ± 1.5 for M2 and 2.7 ± 0.4 for M2-G34A. M2-G34V showed membrane expression comparable to the wild type, but no detectable channel activity. (C) Comparison of channel activity and proton selectivity between full-length M2, and the G34A and G34V mutants. The channel activity was evoked by rapid exchange of nonactivating solution (pH 8.5) with activating solution (pH 5.5). The reversal voltages were measured in Na+, K+ and NMDG+—based activating solutions and showed no significant differences in the ion selectivity and channel properties between the M2 wild type and the G34A mutant. G34V mutant (marked with *) did not show any pH induced channel activity. Each bar is the mean (± SD) of 5 independent experiments.
To examine the effects of the G34A mutation to the crystal structure and the water density in the pore region, we have employed MD simulations of the M2TM′ structure, using for comparison the WT protein and the nonfunctional G34V mutant (also modeled after the M2TM′ structure). Unrestrained MD simulations in a hydrated lipid bilayer find that the bundle structure is stable in all of the three simulations (Fig. S2); but, marked differences appear in the water distributions. In the WT simulation, the entry cluster is continuously connected to the waters at the N-terminal end, with two to three water molecules between Gly34 residues. In contrast, in G34V only the four water molecules of the entry cluster adjacent to the histidines are present (Fig. 1 A and B); as a result, the water density has a 5 Å gap centered around the Val34 side chains (Fig. 4A). G34A (M2TM′) shows an intermediate behavior between WT and G34V, retaining significant water density through the Ala34 side chains, which is composed of at least one water molecule connecting the entry cluster to the water molecules in the N-terminal section of the pore (Fig. 4A).
Comparison of the MD data with the proton conductance of M2 in oocytes, ion selectivity and surface expression of the WT, G34A and G34V shows a correlation between water density in the channel leading to the His-box and proton conduction. The specific activity of M2-G34A is approximately 2.5-fold lower than wild type (Fig. 4B), whereas M2-G34V does not show any detectable channel activity (Fig. 4B) in spite of normal expression. Consequently the G34V mutation impairs the channel activity by blocking a large fraction the conduction pathway. Proton diffusion through the G34A variant is permitted, but the rate is diminished compared to WT because of the availability of only one water molecule between the Ala34 side chains Thus, a requirement for efficient conduction of protons seems to be the presence of a continuous water density throughout the pore of the channel. Density can be broken only by the His37 side chains themselves, in between which the number of water molecules is the smallest, and negligible in the WT channel (Fig. 4A). The WT protein and the G34A mutant are both equivalently selective for protons, featuring very similar reversal voltages (Vrev) in recording solutions containing different cations (Na+, NMDG+, or K+) (Fig. 4C). It is unlikely that another ion would bind to or penetrate through the doubly protonated His-box.
Discussion
In conjunction with earlier lower-resolution structures, the present high-resolution structure provides a basis for rationalizing the mechanism of proton conduction through the M2 channel. It is important to consider these structures in the context of the pH-dependence of conductance as well as the dynamic nature of the M2 protein. Because the conductance of M2 shows a sigmoidal pH/flux curve, we (10) and others (34, 42) previously suggested that lowering the pH to between 5 and 6 “gates” the channel into an activated state that is competent to conduct protons. However, at high pH other nonselective proton channels such as gramicidin A and LS2 conduct protons with a rate very similar to M2. In fact, the rate of conduction of ions through ion and proton channels is generally proportional to the concentration of the permeant ions in the absence of specific inhibition or activation sites. Proton channels with molecular dimensions similar to M2, such as gramicidin A and LS2 (44), conduct protons with a second order rate constant (k2) of approximately 108 to 109 M-1 sec-1. The corresponding rate of per-channel proton flux (kflux = k2 ∗ [proton]) is 10 to 1,000 in protons/sec at pH 7 to 6, within the same range as that observed for M2 under these pH conditions. Thus, near neutral pH, M2 conducts protons within an order of magnitude or two of that expected for diffusion through a pore, and it is considered a “slow” channel only because the proton concentration is small over its physiological range of pH 7 to 5. What distinguishes M2 from other channels is that its conductance fails to rise linearly with the concentration of the permeant ion, instead leveling below pH 5. Thus, the sigmoidal nature of the curve does not reflect “activation” of the channel, but rather leveling of the rate as the 3+ state is formed.
A second feature that differentiates M2 from other channels is that it undergoes large-scale conformational changes on the same time-scale as the transit of ions through the channel. Solution and ssNMR both show the onset of large-scale conformational transitions occurring on the micro to millisecond timescale as the pH is decreased below 6.5, and these transitions are apparent in both TM peptides as well as longer fragments that incorporate a C-terminal cytoplasmic helix (17). These large-scale conformational changes occur on time scales similar to the opening/closing gating transitions observed in K+ and cation channels (45). However, the latter conduct at much greater rates, due to the considerably higher physiologic concentration of K+ or Na+ versus protons. Thus, a single conformational event can gate a cation channel into a state capable of conducting large numbers of cations, whereas a single proton transits the M2 channel amidst conformational changes.
The structures of the M2 protein solved in different protonation states along with MD calculations (43, 46) suggest a structural mechanism for the conduction of protons down their concentration gradient in biologically relevant conditions when the pH on the outside of the virus (pHout) is lower than pHin (Fig. 5). The channel is proposed to oscillate between distinct states, whose relative stability is modulated by the degree of protonation. In an underprotonated state, protons can diffuse from the outside (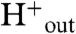 ) into the pore through an open Val27 valve (17); in contrast, the Trp-basket at the opposing end has a relatively small hydrophobic opening, disfavoring proton uptake from the inside. At intermediate protonation the upper part of the M2 bundle contracts. Conversely, the Trp-basket has a larger polar opening, beginning to expose the protonated histidines to the viral interior. This state most closely resembles the crystallographic structure described here. When the highest charge state is reached, the Val27 valve at the outside end nearly completely closes, whereas the Trp-basket opens by several Å (16), making proton release to the viral interior the only viable option to restore a lower charge state in the channel. After one or more protons dissociate from the His-box, the conformational ensemble of M2 will revert back to one with a population of conformers resembling the intermediate pH structure, and even the pH 7.5 structure (17) if the inside pH is still sufficiently high.
) into the pore through an open Val27 valve (17); in contrast, the Trp-basket at the opposing end has a relatively small hydrophobic opening, disfavoring proton uptake from the inside. At intermediate protonation the upper part of the M2 bundle contracts. Conversely, the Trp-basket has a larger polar opening, beginning to expose the protonated histidines to the viral interior. This state most closely resembles the crystallographic structure described here. When the highest charge state is reached, the Val27 valve at the outside end nearly completely closes, whereas the Trp-basket opens by several Å (16), making proton release to the viral interior the only viable option to restore a lower charge state in the channel. After one or more protons dissociate from the His-box, the conformational ensemble of M2 will revert back to one with a population of conformers resembling the intermediate pH structure, and even the pH 7.5 structure (17) if the inside pH is still sufficiently high.
Fig. 5.

M2TM conformational ensemble. (A) Cartoon illustration of the change in the energy landscape as a function of the pH at the N-terminus. Three conformational states of M2TM have been identified by X-ray crystallography and NMR at different pH (16, 17). They are: the OOut, intermediate (INT), and OIn states, depicted in panels B, C, and D, respectively. Each conformation can exist in a variety of protonation states: in this paper we have performed a study of the INT state. As the pH decreases the favored state changes from the high pH OOut state to INT and then to OIn. The OOut and INT states feature different tilt angles of the N-terminal portions of the helices (see also Fig. 2) and rotamers of the Trp41 side chains. The INT and OIn states instead show different conformations of the C-terminal portions.
In this mechanism, the leveling of the rate of conduction at low pH is associated with rate-determining deprotonation of the His-box or conformational changes enabling this event. In the absence of strong buffer catalysis (which is not observed for M2) (47), the rate of deprotonation (koff) of a conjugate acid is defined by its equilibrium dissociation constant (Kdiss = 10-pKa) and the second order rate constant for protonation (kon) according to the equation Kdiss = koff/kon. Given M2’s conducting pKa of approximately 6 and values of kon between 107 to 109 M -1 sec-1 for diffusion through a pore the size of M2, the expected value of koff is 101 to 103 sec-1 in good agreement with the observed maximal rate (47).
The original proposals for conduction have focused on protonation/depronation of discreet His37 residues. However, the well-ordered water clusters seen in the present pore structure suggest that the proton must be delocalized over the His-box and surrounding water clusters. Such a conduction mechanism, which bears anology to the storage of electrons within iron/sulfur clusters, offers a unification of discreet site versus continuum conduction models.
Importantly all three channel conformations (designated OOut for open to the outside, INT for intermediate, and OIn for open to the inside, Fig. 5 B–D) are accessible in the relevant pH range of 6 ± 1, as evidenced by MD calculations and the observation of a mixed conformational state in one crystal structure of M2TM (16, 46). The landscapes in Fig. 5A indicate that the favored conformation in the ensemble changes as a function of protonation state, amidst ongoing, continuous structural interconversion between the individual states in the ensemble. Moreover, although this diagram illustrates only three symmetrical states, motions of the four helices do not necessarily occur in a cooperative all-or-nothing transitions as evidenced by the mixed conformation bundle in a previous crystal structure (16) and conformational heterogeneity seen by ssNMR (39, 43, 48). The presence of a drug molecule in the channel may affect the relative population of the various conformational states, by selectively stabilizing one of them, as suggested by NMR measurements (15, 17).
Methods
Synthesis and Crystallization of the M2TM (wild-type) and M2TM′ (G34A mutant) Peptides.
The 25–46 TM peptide from the Udorn sequence of the M2 protein (designated here M2TM), and the corresponding peptide with the G34A mutation (M2TM′) were synthesized by 9-fluorenylmethoxycarbonyl (Fmoc) chemistry. The N-termini of the sequences were capped with 4-bromobenzoyl group. To make aliquots for crystallization, each peptide was mixed with n-octylglucoside (OG, Sigma Aldrich) in an aqueous:isopropanol (1∶1) stock using ε280peptide = 5853 M-1 cm-1. Both mixtures were evaporated under reduced pressure, and the resulting films were taken up in 5% (w/v) xylitol and mixed with a reservoir solution of 100 mM sodium citrate pH 5.6, 150 mM tri-sodium citrate, 15% v/v isopropanol. M2TM crystals appeared after 5 months, and were grown for additional 2 months. M2TM′ crystals appeared within 2 weeks, and were grown over periods of 2–5 months.
Diffraction Data Collection and Processing.
Data sets were collected from several different crystals of M2TM′, with cryocooling to 100 K during data collection. The M2TM′ crystal that diffracted at the highest resolution (1.65 Å) features the C2221 space group, with cell parameters 48.67, 79.09, and 48.56 Å, respectively. The corresponding highest resolution M2TM crystal diffracted at 2.5 Å and featured the same space group and very similar cell parameters, but degraded before a sufficient dataset could be acquired. Therefore, M2TM diffraction data were only used to test the overall similarity between the M2TM′ and M2TM structures. Table S2 shows the data collection statistics for the two crystals.
Tetramer Model Building, Molecular Replacement, and Refinement of M2TM′.
Initial attempts to determine the M2TM′ structure single- and multiple-wavelength anomalous dispersion were not successful, due to the high disorder associated with the Br atoms’ positions. By comparing with the unit cell dimensions with the previously published 2 Å resolution structure (3BKD; ref. 16), it is evident that the M2TM′ tetramers lie in the ac plane of the crystal, with helices oriented along the b axis (Table S2). One of the α-helices (labeled as the “A” chain) from the previously published structure (3BKD; ref. 16) was used as an initial model for the M2TM′ monomer. An ensemble of tetramer models was generated using as free parameters the three rotation angles around the Cartesian axes, and the radius of the bundle. Only those models with an orientation of Val27 similar to the previous structure were considered for molecular replacement. Model building and iterative refinement were carried out up to Rwork = 19.6% and Rfree = 20.5% (Table S2), with all the residues in the allowed regions (100% favorable) of the Ramachandran plot. Solvent molecules were included only when visible at the 3σ level in an F0–FC map. All atoms were resolved with full occupancy, with the exception of the disordered Br atoms in the bromobenzoyl group (presumably due to radiation damage), and the dimer of water molecules in the entry cluster. The latter was refined in two alternate conformations, with occupancies of 0.6 and 0.4. Further details on the refinement process are provided in SI Text, with statistics in Table S2.
Classical MD Simulations.
The CHARMM27 force field was used for the protein, the lipids, and the Cl- and K+ ions (49), together with the TIP3P force field for water molecules (50). Several systems were simulated (described in SI Text), the largest of which contained 54642 atoms in the simulation box. Three sets of simulations were performed using NAMD 2.7b1 (51) and analyzed using VMD 1.8.7 (52): (i) 15 ns-long trajectories were collected for the 0ε, 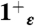 ,
, 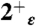 ,
, 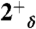 ,
, 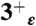 ,
, 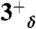 , and 4+ protonation states of the M2TM′ peptide solvated in a water box with the heavy atoms of the peptide backbone kept restrained to their initial position; (ii) 80 ns-long trajectories were collected for the
, and 4+ protonation states of the M2TM′ peptide solvated in a water box with the heavy atoms of the peptide backbone kept restrained to their initial position; (ii) 80 ns-long trajectories were collected for the 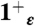 ,
, 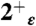 ,
, 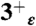 , and 4+ protonation states of the M2TM′ peptide embedded into a 80 × 80 Å2 bilayer of 1-palmitoyl-2-oleoylphosphatidylcholine lipid molecules; and (iii) 15 ns-long trajectories were collected for the
, and 4+ protonation states of the M2TM′ peptide embedded into a 80 × 80 Å2 bilayer of 1-palmitoyl-2-oleoylphosphatidylcholine lipid molecules; and (iii) 15 ns-long trajectories were collected for the 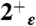 protonation state of the WT, G34A, and G34V peptides in lipid bilayer using the same setup as in (ii).
protonation state of the WT, G34A, and G34V peptides in lipid bilayer using the same setup as in (ii).
Oocyte Electrophysiology Experiments.
Recombinant A/M2-G34A and A/M2-G34V proteins from the Udorn strain of Influenza A virus were prepared and expressed in Xenopus oocytes as described in ref. 53. Channel activity was assayed using a two-micro electrode voltage clamp, and the reversal voltages for the ramp of -60 to +80 mV were measured in Na+ , K+, and NMDG+- based recording solutions as described in ref. 54. Immunofluorescence of living oocytes was performed as described in ref. 53.
Supplementary Material
Acknowledgments.
We thank S. Stayrook and K. Ellis-Guardiola for technical assistance. The research was supported by the National Institutes of Health under Grants AI 74571, AI 20201, and GM56423, as well as by the Human Frontiers Science Program. The computational studies were made possible by the National Science Foundation through TeraGrid resources provided by the Pittsburgh Supercomputing Center, the National Center for Supercomputing Applications, and the National Institute for Computational Sciences (allocation number TG-MCA93S020). W.F.D., R.A.L., M.L.K., and L.H.P. are founders and members of the scientific advisory board of Influmedix (www.influmedix.com).
Footnotes
Conflict of interest statement: R.A.L., L.H.P., W.F.D., and M.L.K. are members of the scientific advisory board of InfluMedix, a company that is developing influenza drugs.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1007071107/-/DCSupplemental.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3LBW).
References
- 1.Headrick JM, et al. Spectral signatures of hydrated proton vibrations in water clusters. Science. 2005;308:1765–1769. doi: 10.1126/science.1113094. [DOI] [PubMed] [Google Scholar]
- 2.Shin J-W, et al. Infrared signature of structures associated with the H+(H2O)n (n = 6 to 27) Science. 2004;304:1137–1140. doi: 10.1126/science.1096466. [DOI] [PubMed] [Google Scholar]
- 3.Robertson WH, et al. Spectroscopic determination of the OH- solvation shell in the OH-(H2O)n clusters. Science. 2003;299:1367–1372. doi: 10.1126/science.1080695. [DOI] [PubMed] [Google Scholar]
- 4.Liu K, Cruzan JD, Saykally RJ. Water clusters. Science. 1996;271:929–933. doi: 10.1126/science.271.5245.59. [DOI] [PubMed] [Google Scholar]
- 5.Harries WEC, et al. The channel architecture of aquaporin 0 at 2.2 Å resolution. Proc Natl Acad Sci USA. 2004;101:14045–14050. doi: 10.1073/pnas.0405274101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tajkhorshid E, et al. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science. 2002;296:525–530. doi: 10.1126/science.1067778. [DOI] [PubMed] [Google Scholar]
- 7.Chakrabarti N, Roux B, Pomes R. Structural determinants of proton blockage in aquaporins. J Mol Biol. 2004;343:493–510. doi: 10.1016/j.jmb.2004.08.036. [DOI] [PubMed] [Google Scholar]
- 8.de Groot BL, Grubmueller H. Water permeation across biological membranes: Mechanism and dynamics of aquaporin-1 and GlpF. Science. 2001;294:2353–2357. doi: 10.1126/science.1066115. [DOI] [PubMed] [Google Scholar]
- 9.Hummer G, Rasaiah JC, Noworyta JP. Water conduction through the hydrophobic channel of a carbon nanotube. Nature. 2001;414:188–190. doi: 10.1038/35102535. [DOI] [PubMed] [Google Scholar]
- 10.Pinto LH, Lamb RA. The M2 proton channels of influenza A and B viruses. J Biol Chem. 2006;281:8997–9000. doi: 10.1074/jbc.R500020200. [DOI] [PubMed] [Google Scholar]
- 11.Pinto LH, et al. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc Natl Acad Sci USA. 1997;94:11301–11306. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang C, Takeuchi K, Pinto LH, Lamb RA. Ion channel activity of influenza A virus M2 protein: Characterization of the amantadine block. J Virol. 1993;67:5585–5594. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tumpey TM, et al. Existing antivirals are effective against influenza viruses with genes from the 1918 pandemic virus. Proc Natl Acad Sci USA. 2002;99:13849–13854. doi: 10.1073/pnas.212519699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jing X, et al. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc Natl Acad Sci USA. 2008;105:10967–10972. doi: 10.1073/pnas.0804958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cady SD, Hong M. Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. Proc Natl Acad Sci USA. 2008;105:1483–1488. doi: 10.1073/pnas.0711500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stouffer AL, et al. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugrue RJ, Hay AJ. Structural characteristics of the M2 protein of influenza A viruses: Evidence that it forms a tetrameric channel. Virology. 1991;180:617–624. doi: 10.1016/0042-6822(91)90075-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhirnov OP. Isolation of matrix protein M1 from influenza viruses by acid-dependent extraction with nonionic detergent. Virology. 1992;186:324–330. doi: 10.1016/0042-6822(92)90090-c. [DOI] [PubMed] [Google Scholar]
- 20.Pinto LH, Holsinger LJ, Lamb RA. Influenza virus M2 protein has ion channel activity. Cell. 1992;69:517–528. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- 21.Chizhmakov IV, et al. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J Physiol-London. 1996;494:329–336. doi: 10.1113/jphysiol.1996.sp021495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin TI, Schroeder C. Definitive assignent of proton selectivity and attoampere unitary current to the M2 ion channel protein of influenza A virus. J Virol. 2001;75:3647–3656. doi: 10.1128/JVI.75.8.3647-3656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, Lamb RA, Pinto LH. Activation of the M2 ion channel of influenza virus: A role for the transmembrane domain histidine residue. Biophys J. 1995;69:1363–1371. doi: 10.1016/S0006-3495(95)80003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vijayvergiya V, et al. Proton conductance of influenza virus M2 protein in planar lipid bilayers. Biophys J. 2004;87:1697–1704. doi: 10.1529/biophysj.104.043018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mould JA, et al. Mechanism for proton conduction of the M2 ion channel of influenza A virus. J Biol Chem. 2000;275:8592–8599. doi: 10.1074/jbc.275.12.8592. [DOI] [PubMed] [Google Scholar]
- 26.Hu J, et al. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from Influenza A virus. Biophys J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cady SD, et al. Structure of the amantidine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pielak RM, Schnell JR, Chou JJ. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc Natl Acad Sci USA. 2009;106:7379–7384. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Witter R, et al. Solid-state 19F NMR spectroscopy reveals that Trp41 participates in the gating mechanism of the M2 proton channel of influenza A virus. J Am Chem Soc. 2008;130:918–924. doi: 10.1021/ja0754305. [DOI] [PubMed] [Google Scholar]
- 30.Venkataraman P, Lamb RA, Pinto LH. Chemical rescue of histidine selectivity filter mutants of the M2 ion channel of influenza A virus. J Biol Chem. 2005;280:21463–21472. doi: 10.1074/jbc.M412406200. [DOI] [PubMed] [Google Scholar]
- 31.Tang Y, Zaitseva F, Lamb RA, Pinto LH. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J Biol Chem. 2002;277:39880–39886. doi: 10.1074/jbc.M206582200. [DOI] [PubMed] [Google Scholar]
- 32.Forrest LR, Tieleman DP, Sansom MSP. Defining the transmembrane helix of M2 protein from influenza A by molecular dynamics simulations in a lipid bilayer. Biophys J. 1999;76:1886–1896. doi: 10.1016/s0006-3495(99)77347-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma C, et al. Identification of the functional core of the influenza A virus A/M2 proton-selective ion channel. Proc Natl Acad Sci USA. 2009;106:12283–12288. doi: 10.1073/pnas.0905726106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu J, et al. Histidines, heart of the hydrogen ion channel from influenza A virus: Toward an understanding of conductance and proton selectivity. Proc Natl Acad Sci USA. 2006;103:6865–6870. doi: 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Balannik V, et al. Functional studies and modeling of pore-lining residue mutants of the influenza A virus M2 ion channel. Biochemistry. 2009;49:696–708. doi: 10.1021/bi901799k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian C, et al. Initial structural and dynamic characterization of the M2 protein transmembrane and amphipathic helices in lipid bilayers. Protein Sci. 2003;12:2597–2605. doi: 10.1110/ps.03168503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinto LH, et al. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc Natl Acad Sci USA. 1997;94:11301–11306. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okada A, Miura T, Takeuchi H. Protonation of histidine and histidine-tryptophan interaction in the activation of the M2 ion channel from influenza A virus. Biochemistry. 2001;40:6053–6060. doi: 10.1021/bi0028441. [DOI] [PubMed] [Google Scholar]
- 39.Cady SD, Luo W, Hu F, Hong M. Structure and function of the influenza A M2 proton channel. Biochemistry. 2009;48:7356–7364. doi: 10.1021/bi9008837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yifrach O, MacKinnon R. Energetics of pore opening in a voltage-gated K+ channel. Cell. 2002;111:231–239. doi: 10.1016/s0092-8674(02)01013-9. [DOI] [PubMed] [Google Scholar]
- 41.Liu Z, Gandhi CS, Rees DC. Structure of a tetrameric MscL in an expanded intermediate state. Nature. 2009;461:120–124. doi: 10.1038/nature08277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen H, Wu Y, Voth GA. Proton transport behavior through the influenza A M2 channel: Insights from molecular simulation. Biophys J. 2007;93:3470–3479. doi: 10.1529/biophysj.107.105742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yi M, Cross TA, Zhou HX. Conformational heterogeneity of the M2 proton channel and a structural model for channel activation. Proc Natl Acad Sci USA. 2009;106:13311–13316. doi: 10.1073/pnas.0906553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Decoursey TE. Voltage-gated proton channels and other proton transfer pathways. Physiol Rev. 2003;83:475–579. doi: 10.1152/physrev.00028.2002. [DOI] [PubMed] [Google Scholar]
- 45.Hille B. Ion Channels of Excitable Membranes. Sunderland, MA: Sinauer; 2001. [Google Scholar]
- 46.Khurana E, et al. Proton gating mechanism of tetrameric membrane-bound M2 protein from influenza A virus: A new perspective from molecular dynamics calculations. Proc Natl Acad Sci USA. 2009;106:1069–1074. doi: 10.1073/pnas.0811720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mould JA, et al. Permeation and activation of the M2 ion channel of influenza A virus. J Biol Chem. 2000;275:31038–31050. doi: 10.1074/jbc.M003663200. [DOI] [PubMed] [Google Scholar]
- 48.Li C, Qin H, Gao FP, Cross TA. Solid-state NMR characterization of conformational plasticity within the transmembrane domain of the influenza A M2 proton channel. Biochim Biophys Acta. 2007;1768:3162–3170. doi: 10.1016/j.bbamem.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.MacKerell AD, Jr, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 50.Jorgensen WL, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 51.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Humphrey W, Dalke A, Schulten K. VMD—Visual Molecular Dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 53.Stouffer AL, et al. The interplay of functional tuning, drug resistance and thermodynamic stability in the evolution of the M2 proton channel from the influenza A virus. Structure. 2008;16:1067–1076. doi: 10.1016/j.str.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma C, et al. Identification of the pore-lining residues of the BM2 ion channel protein of influenza B. virus. J Biol Chem. 2008;283:15921–15931. doi: 10.1074/jbc.M710302200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.