

Abstract
The human epidermal growth factor receptor 2 (HER2) is specifically overexpressed in tumors of several cancers, including an aggressive form of breast cancer. It is therefore a target for both cancer diagnostics and therapy. The 58 amino acid residue Zher2 affibody molecule was previously engineered as a high-affinity binder of HER2. Here we determined the structure of Zher2 in solution and the crystal structure of Zher2 in complex with the HER2 extracellular domain. Zher2 binds to a conformational epitope on HER2 that is distant from those recognized by the therapeutic antibodies trastuzumab and pertuzumab. Its small size and lack of interference may provide Zher2 with advantages for diagnostic use or even for delivery of therapeutic agents to HER2-expressing tumors when trastuzumab or pertuzumab are already employed. Biophysical characterization shows that Zher2 is thermodynamically stable in the folded state yet undergoing conformational interconversion on a submillisecond time scale. The data suggest that it is the HER2-binding conformation that is formed transiently prior to binding. Still, binding is very strong with a dissociation constant KD = 22 pM, and perfect conformational homogeneity is therefore not necessarily required in engineered binding proteins. A comparison of the original Z domain scaffold to free and bound Zher2 structures reveals how high-affinity binding has evolved during selection and affinity maturation and suggests how a compromise between binding surface optimization and stability and dynamics of the unbound state has been reached.
Keywords: protein engineering, molecular recognition, protein–protein interactions, protein conformational dynamics, cancer therapy
The human epidermal growth factor receptor 2 (HER2, ErbB2) is a 185-kDa transmembrane glycoprotein receptor tyrosine kinase involved in the signal transduction pathways leading to cell growth and differentiation (1). In contrast to the other three members of the epidermal growth factor receptor family, HER2 is thought to be an orphan receptor, i.e. lacking a ligand (2). However, HER2 forms heterodimers with any of the related receptors, resulting in receptor activation. Enhanced levels of HER2 have been shown to correlate with one form of aggressive breast cancer, precipitating the development of the HER2-binding monoclonal antibodies (mAb) trastuzumab (3) and pertuzumab (4). Trastuzumab is now established for treating breast cancers shown to be HER2 overexpressing. Pertuzumab binds to a separate site on HER2 (4). It is more efficient at blocking HER2 heterodimerization (4, 5), and it is in clinical trials for its effects on breast cancer and other cancers (6). Trastuzumab conjugated to the microtubule-depolymerizing agent maytansinoid (trastuzumab-DM1) was recently shown to be even more efficacious than native trastuzumab, leading to high response rates in patients who failed trastuzumab therapy (7). These findings agree with previous data indicating that cancer cells do not loose HER2 expression when they become refractory to trastuzumab, but that the biological context needed for efficacy of trastuzumab has been lost (8, 9). Thus, the role of the antibody moiety in trastuzumab-DM1 is primarily to work as a carrier of a cytotoxin. This finding raises interest in evaluating other HER2-binding molecules displaying alternative properties compared to antibodies, including biodistribution. One such molecule is the affibody molecule 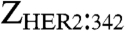 (hereafter Zher2), which binds HER2 with very high affinity [KD = 22 pM (10)].
(hereafter Zher2), which binds HER2 with very high affinity [KD = 22 pM (10)].
Affibody molecules were designed as an alternative to immunoglobulin-based binding proteins. The parental “Z domain” is a small protein of 58 amino acids organized into a three-helix bundle. It was developed as a stabilized variant of the “B domain” of the IgG-binding staphylococcal protein A (11). The original affibody sequence diversity libraries were made by randomizing 13 surface-exposed amino acid residues on the IgG-binding surface of helices 1 and 2 (12). Affibody molecules binding a variety of different target proteins have subsequently been selected from such libraries (13).
The high affinity of Zher2 for its HER2 target is a result of so-called affinity maturation, which is a second round of combinatorial protein engineering and phage display selection (10, 13). Zher2 competes with neither trastuzumab (14) nor pertuzumab (15) for HER2 binding. Zher2 also does not appear to mediate detectable biological effects (16). These properties make it suitable as a tracer when the aim is to study HER2 without affecting its function, even in the presence of therapeutic monoclonal antibodies. Hence, Zher2 allows for molecular imaging of HER2 (15, 17) without interfering with ongoing therapy using either of the two mAbs. Alternatively, it may be used in synergy with any of these, if used as a carrier to direct additional therapeutic agents to their HER2 target.
The potential of the Zher2 affibody molecule for diagnostic and therapeutic use motivates detailed characterization of its structural and biophysical properties. Previous structural studies of affibody molecules (18–20) have revealed a large variability in structure and binding interface, and it is therefore also interesting to learn how the binding surface evolves during affinity maturation. We find that Zher2 adopts the standard three-helix bundle topology, but that the extensive mutagenesis in this case results in two or more similar conformations that interconvert on the submillisecond time scale. This dynamics is apparently not prohibitive of picomolar binding. We argue that it appears as a trade-off in affinity maturation when, for instance, intramolecular hydrogen bonds are sacrificed in order to optimize the HER2 binding surface. The crystal structure of the complex between Zher2 and HER2 allows comparison of the structures of free and bound Zher2 and shows that the binding site on HER2 is distant from the epitopes previously determined for trastuzumab and pertuzumab.
Results
Secondary Structure Content and Stability of Free Zher2.
The circular dichroism (CD) spectrum of Zher2 (Fig. 1A) shows that α-helical secondary structure is dominating. The contribution from the disordered histidine tag to the CD at 222 nm is expected to be minimal (21). If the CD signal at 222 nm originates only from the 58-residue affibody domain, then the mean residue ellipticity MRE222 = -26,100 deg cm2 dmol-1, which corresponds to a helix content of 73% or ca. 42 residues (22). This value compares to 77% (ca. 45 residues) in the parental Z domain (23).
Fig. 1.

Biophysical characterization of the Zher2 affibody molecule. (A) Circular dichroism spectrum at 20 °C. (B) Thermal denaturation monitored by CD at 222 nm. The red line is a fit to a two-state folding model to determine the melting temperature Tm = 66.8 ± 0.2 °C. (C) Chemical denaturation monitored by intrinsic fluorescence at 352 nm. The line is a fit with ΔGunfold = 3.1 ± 0.3 kcal mol-1 and a value of m = -1.2 ± 0.1 kcal mol-1 M-1. (D) 15N HSQC NMR spectrum of Zher2 at 900 MHz and 30 °C. Resonances are colored according to apparent 15N resonance linewidths. These reflect four categories of backbone dynamics, as discussed in the text. n.a., two unassigned resonances; minor, three pairs of resonances from Asn or Gln side chains in unassigned alternate conformations. Many backbone amide resonances are broadened beyond detection by conformational exchange dynamics. (E) 15N R2 relaxation dispersion at 800 MHz (81.1 MHz 15N frequency). Effective R2 rates ( ) of 38 resolved and assigned amide correlations were measured as a function of a constant-time CPMG spin lock field. Twenty-three resonances showed relaxation dispersion (99% confidence level) and were fitted (global R-square = 0.981) to a two-state model with kex = 3574 ± 206 s-1 to determine R2 relaxation rates and Rex. Others, such as Ala54 in the figure, were fitted to a straight line to determine R2. (F) Summary of global fit of all 15N relaxation dispersion profiles. Black bars, R2; red circles, R2 + Rex with estimated standard deviations. α-helical secondary structure is indicated at the top.
) of 38 resolved and assigned amide correlations were measured as a function of a constant-time CPMG spin lock field. Twenty-three resonances showed relaxation dispersion (99% confidence level) and were fitted (global R-square = 0.981) to a two-state model with kex = 3574 ± 206 s-1 to determine R2 relaxation rates and Rex. Others, such as Ala54 in the figure, were fitted to a straight line to determine R2. (F) Summary of global fit of all 15N relaxation dispersion profiles. Black bars, R2; red circles, R2 + Rex with estimated standard deviations. α-helical secondary structure is indicated at the top.
The thermal stability of the present affibody molecule is also good with a melting temperature of 67 °C (Fig. 1B), which is close to the 75 °C of the Z domain at pH 7.2. We measured folding stability at room temperature by monitoring intrinsic protein fluorescence as function of guanidinium chloride denaturant concentration (Fig. 1C). The data can be fitted to a two-state model in which the folded state is stabilized by a free energy ΔGunfold = 3.1 ± 0.3 kcal mol-1. This value is lower than that of the Z domain or the B domain [∼7 kcal mol-1; (24, 25)]. It may be argued that a two-state model of unfolding is inappropriate when there is more then one folded conformation, which we will show is the case here. However, one must consider the folded state(s) as a single thermodynamic state as long as interconverting conformations cannot be distinguished by the experiments, and as long as the two-state model fits the data as in Fig. 1 B and C.
The NMR Spectrum and Dynamics of Free Zher2.
The assigned 15N NMR heteronuclear sequential quantum correlation (HSQC) spectrum of Zher2 at 30 °C is characterized by a large variation in NMR line shapes. These reveal at least four categories of dynamics on different time scales, as indicated by the coloring of resonances in Fig. 1D. Disordered residues at the N terminus are dynamic at the picosecond time scale with sharp NMR resonances. The residues in helix 3, most of helix 2, and in loop regions appear to be well ordered within the folded protein as judged from linewidths and a large number of NOE connectivities (Fig. S1). Resonances from helix 1, and from mutated regions of helix 2, are broadened due to conformational exchange on the microsecond to millisecond time scales. Some of these are too weak to be assigned, and most are in fact broadened beyond detection. Several Asn and Gln side chain resonances are also broadened, and there are at least three sets of unassigned resonances from Gln or Asn amino ( ) groups in alternative slowly exchanging conformations. The same pattern of dynamics is apparent in the 13C HSQC spectrum, where several resonances are undetectable and resonances from methyl groups at the hydrophobic interface of helices 1 and 2 are observable, but broadened.
) groups in alternative slowly exchanging conformations. The same pattern of dynamics is apparent in the 13C HSQC spectrum, where several resonances are undetectable and resonances from methyl groups at the hydrophobic interface of helices 1 and 2 are observable, but broadened.
We used NMR relaxation dispersion experiments (26) to determine the rate of conformational exchange. As expected from the line broadening, many amide 15N resonances show a strong dependence of the R2 relaxation rate on an external CPMG (Carr–Purcell–Meiboom–Gill) field (Fig. 1E). Measured relaxation dispersion profiles could be fit to a two-state model of fast exchange (27) using a single global value of the exchange rate kex = 3574 ± 206 s-1, which is the sum of forward and reverse interconversion rates. Best-fit R2 relaxation rates and dispersion amplitudes confirm that helix 1 and helix 2 are involved in this interconversion, whereas loop regions and helix 3 are less affected (Fig. 1F). The populations of interconverting states cannot be resolved in the fast exchange limit unless there is additional information on chemical shift differences. A global fit of 800- and 600-MHz relaxation data to the full Carver–Richards equation was still attempted to determine both populations and shift differences, but it did not converge.
Resonances from C-terminal residues 59 and 60 appear as doublets in the NMR spectrum (Fig. 1D), indicating that they can adopt two slowly exchanging conformations. These are not part of the parental Z domain and on the DNA level they correspond to a restriction site introduced to enable facile multimerization. Conformational heterogeneity at the C terminus is unlikely to be of relevance for Zher2 stability or binding.
Solution Structure of Free Zher2.
As many as 13 amide resonances in helices 1 and 2 could not be detected and assigned (Fig. 1D), but other resonances in most of the corresponding side chains could still be sequentially assigned based on medium range dαβ(i,i + 3) NOEs in the 3D 13C-NOESY spectrum, and yet others were assigned based on residue type (for instance, Ala12). An ensemble of structures was calculated using simulated annealing (SA) based on distance restraints derived from NOE connectivities, backbone dihedrals derived from NMR chemical shifts, and backbone and side chain hydrogen bonds, which were added at the final stage of refinement (SI Text and Table S1). The structure of most of the molecule, including helix 3, most of helix 2, loop regions, and the hydrophobic core, is well defined (Fig. 2 A and B).
Fig. 2.

Structure and dynamics of the Zher2 affibody molecule. (A) Sequences of the Z domain and Zher2. Gray, randomized positions in affibody libraries; yellow, selected side chains at HER2 interface; cyan, other residues at HER2 interface; black boxes, dynamic nonpolar side chains in core; red boxes, Asn and Gln side chains with alternating conformations. Insets above and below sequences show secondary structure and surface exposure, with darker blue shading reflecting less surface exposure. Residues for which backbone NMR resonances in Zher2 are broadened by dynamics are in gray and ordered regions in orange (matching coloring in C–E). (B) Backbone traces of the Zher2 structure ensemble. The blue/gray colors of residues 1 to 12 at the N terminus show two conformers obtained with unrestrained SA. (C) Zher2 conformational exchange dynamics: orange, no backbone dynamics; gray, backbone dynamics; white spheres, methyl groups at the interface between helices 1 and 2 that undergo conformational dynamics; sticks, dynamic Asn and Gln side chains. (D) The alternative Zher2_alt structure with an intact helix 1. (E) Illustration of how dynamics (as in D) coincides with mutations at the HER2 binding surface (yellow sticks corresponding to yellow shading in A).
However, the backbone conformation of helix 1 and around mutated regions of helix 2 could not be calculated at high precision. Hence, helix 1 appears as two helical fragments connected by a loop at residues 13 and 14 in minimized SA structures, and there are two conformations of residues 1 to 12 (Fig. 2 B and C). Still, chemical shifts and medium range NOE connectivities of all observable resonances in helix 1 suggest that it indeed could be completely helical. To examine this possibility, we calculated a set of alternative Zher2 structures (Zher2_alt) in which an α-helical conformation of residues 6 to 14 was enforced, without removing or changing any experimental restraints. These SA calculations also converged to high-quality structures without violating any of the ca. 100 experimental restraints involving helix 1 (Table S1).
The reason why unbiased SA results in a distorted rather than an intact helix 1 is that the former is slightly favored by the structure calculation force field (conformational plus restraint energies of -499 ± 20 versus -472 ± 24 kcal mol-1, respectively). However, an evaluation of Ramachandran statistics (Table S1) and packing of the hydrophobic core (Fig. 2D) suggest that an intact helix 1 is more favored. We conclude that both conformations are possible. It is not unlikely that the observed dynamics involves interconversion between these two conformations, as discussed below.
X-Ray Crystal Structure of HER2/Zher2.
The complex between the HER2 extracellular domain (HER2ecd) and Zher2 was determined at 2.9-Å resolution (Fig. 3 and Table S2). It shows Zher2 bound to an epitope that includes residues from both domain III and domain IV of HER2. This region is not part of the epitopes in the previously characterized complexes of HER2ecd with Fab fragments of therapeutic antibodies trastuzumab or pertuzumab (Fig. 3A), although it is part of a crystal packing interaction in the pertuzumab-Fab/HER2 complex (4). The conformational arrangement of HER2ecd domains is essentially the same as in previous studies (2, 4, 28). Upon Zher2 binding about 1,250 Å2 of solvent accessible surface (1.4-Å probe radius), of which 58% is nonpolar, is sequestered on each side of the HER2ecd/Zher2 interface, and the shape complementarity statistic is Sc = 0.65. There are 25 residues from HER2ecd and 14 residues from Zher2 that are within 4 Å of the binding partner. These form 11 direct interprotein hydrogen bonds with distances up to 3.3 Å (Fig. 3B and Table S3). These metrics are all consistent with the high-affinity binding.
Fig. 3.

The Zher2 affibody epitope at the junction of domains III and IV on HER2. (A) Crystal structure of the complex between HER2ecd (surface colored by domain) and Zher2 (orange). For reference, Fab fragments of the therapeutic monoclonal antibodies trastuzumab (blue) and pertuzumab (cyan) are shown where they bind to HER2ecd. Zher2 contacts HER2ecd on parts of domains III and IV far from both antibody epitopes. (B) Detailed view of Zher2 binding. The backbone of residues 6 to 39 is colored in orange and side chains that interact with HER2ecd are shown as sticks (helix 3 omitted for clarity). Side chains that have been randomized and selected by phage display are shown with yellow color on carbon and side chains that are not varied, but still interact at the binding surface, are colored in cyan on carbons as in Fig. 2A. Three varied side chains (Asn11, Gln25, and Lys27; not displayed) do not interact with HER2. Some of the observed intermolecular hydrogen bonds (Table S3) are shown as dashed lines.
The conformation of Zher2 in the crystal structure includes a continuous helix 1 as in the alternate Zher2_alt NMR structure, which provides close contacts to HER2 along its entire length. The rms backbone differences between bound-state Zher2 and the Zher2 and Zher2_alt free-state structures are 1.4 and 1.0 Å, respectively, for alpha carbons of residues 6 to 56, and ∼3.0 and 0.9 Å for residues 6 to 13 (in helix 1). There is also mostly good correspondence with side chain conformations in bound-state Zher2 and Zher2_alt, whereas side chain correspondence to the (nonalternate) solution structure is relatively poor for residues in helix 1. The mean temperature factor (B) for Zher2 is the same as that for the entire complex structure, but B values are relatively high at the beginning of helix 1. Zher2 residues preceding helix 1 were not resolved in electron density maps and are absent from the final coordinates.
Discussion
Conformational Dynamics in Free Zher2.
The present HER2-binding affibody molecule is thermodynamically stable and a very strong binder, but it undergoes conformational interconversions on the submillisecond time scale in the free state. There is a clear correlation between sites of mutation and dynamics (Fig. 2 A and C–E). Zher2 has a melting temperature of 67 °C, but it is somewhat less stable to unfolding than the parental Z domain or the B domain of staphylococcal protein A. Still, the conformational dynamics is not likely to be global unfolding, because one would then not expect the localized effects of line broadening observed here. Studies of the B domain show that global protein unfolding begins with helix 1 (29). It is therefore possible that the observed dynamics is due to local unfolding of helix 1.
However, the average helicity observed by CD implies that a major fraction (> 99%) of Zher2 exists as a three-helix bundle at 20 °C. An alternative explanation of the dynamics, which we favor, is therefore that it reflects interconversions between two or more folded conformations of helix 1. It is possible that two of these are the ones shown in Fig. 2 C and D, both of which are consistent with experimental data. Interconversion between only two conformations is supported by the 15N R2 relaxation dispersion data, but the presence of additional conformational states can on the other hand not be ruled out.
The fact that line broadening makes interconverting structures nearly indistinguishable in structure calculations can be explained: NMR resonances that would distinguish between conformations would be expected to experience the largest changes in chemical shift upon interconversion. These are therefore most affected by exchange line broadening, and the critical experimental discriminants (chemical shifts and NOEs) remain undetected as they disappear with line broadening.
Previous structural studies of affibody molecules in complex with target proteins reveal that the three-helix scaffold of the Z domain may remain intact in the complex (19, 20), but that the extensive mutagenesis also can lead to the selection of other very different conformations (18). Affibody molecules may also differ in their folding stability and even be largely disordered while still showing strong binding (30). The present HER2-binding affibody molecule represents yet another variation on these themes: It is thermodynamically stable and a very strong binder, but there is dynamics in mutated regions.
Conformational dynamics in engineered binding proteins will affect binding affinity, though the effect does not have to be large. For instance, if there are two equally populated interconverting states of Zher2 of which one binds the HER2 receptor, the binding affinity is reduced by only 50%, in this case from a theoretical KD = 11 pM to the observed KD = 22 pM. Still, the dynamics leaves room for further optimization of binding affinity, perhaps by optimizing conformational stability as with the ZSPA-1 affibody molecule (31).
A possible reason for destabilization and dynamics in Zher2 is the replacement of Gln9 at the N-terminal end with Met9 and Glu24/Glu25/Asn28 at the C-terminal end with Asn24/Gln25/Arg28. This is because as many as eight hydrogen bonds in the Z domain are lost by these mutations, and only a few alternative hydrogen bonds form (Fig. 4). These replacements can therefore account for destabilization as well as dynamics of helix 1 and 2 and alternative conformations of Asn and Gln side chains at the C-terminal end (Figs. 1D and 2 C and D). Homologous substitutions at positions 9, 24, and 25 could not be identified in initial selections from the full library (32). These were therefore again randomized in the affinity maturation library resulting in selection of Met9 and Asn24 that together with the native Asn6 and the previously selected Arg28 participate in interactions with HER2 (Figs. 3B and 4). Thus, although reinstating original residues might stabilize Zher2, it will probably also affect binding affinity. We therefore suggest that affinity maturation has involved a trade-off between binding surface optimization and folding stability and/or structural homogeneity of unbound molecules in the library. Or in other words, stability has been sacrificed during the selections in order that new side chains can interact with HER2, and with free-state conformational lability and dynamics as by-products.
Fig. 4.

Trade-off between binding affinity and intramolecular hydrogen bonding during affinity maturation of Zher2. (A) Hydrogen bonding (dashed lines) at the C-terminal end of the Z domain (Left), free Zher2 (Middle), and bound Zher2 (Right). Replacements at positions 24, 25, and 28 result in HER2 interactions by Asn24 and Arg28 on the expense of wild-type Z domain hydrogen bonds. (B) Hydrogen bonds formed by Gln9 at the N-terminal end of the Z domain (Left) are lost upon replacement with Met9 in Zher2 (Middle), but both Met9 and Ans6 can instead interact with HER2 (Right).
Bound-State Zher2 Conformation and Interactions.
The complex structure determined using X-ray crystallography is of sufficiently high resolution to permit confident placement of side chains. There is a good correspondence between the residues varied during initial selections and affinity maturation and those found in intimate association with HER2ecd. Of the 13 randomized amino acid positions, 10 are within 4 Å and 6 interact with HER2 via 11 hydrogen bonds (Fig. 3B and Fig. S2). Overall, the bound-state coordinates of Zher2 are clearly more similar to those of the alternate Zher2_alt than to the Zher2 free-state structures, and most differences occur in regions that are dynamic in the free state. This is consistent with a transient population of the bound-state conformation in free Zher2. Furthermore, relatively high temperature factors (B values) are observed for the first part of helix 1, where the two NMR structures differ the most. Hence, some of the flexibility observed with monomeric Zher2 in solution probably remains in complex with HER2. The similarity between bound Zher2 and Zher2_alt extends to almost all side chains at the binding surface. Exceptions are Trp14, for which the conformation is the same as in only 4 of 23 structures of the Zher2_alt ensemble, and Tyr35, which attains a Zher2_alt side chain conformation different from that in the bound state (χ1 = 180° versus 90°). Interactions with HER2 are clearly responsible for the conformations of these side chains in the complex, indicating that some conformational adaptation occurs also at the binding surface.
The high-affinity Zher2 binding is accounted for by the structure. For instance, the binding surface is 50% larger than in any of the four other affibody molecules that have been structurally characterized (18–20). This is because 10 of 13 varied side chains are involved, but also because interactions extend toward the N-terminal end of the molecule (Fig. 3B; e.g., Asn6 and Pro38). The Ile31 side chain, which is native to the Z domain, is centrally located at the Zher2 binding surface as in all other bound-state affibody or Z domain structures. It was suggested that Ile31 mediates some intrinsic property of Z domain binding that is inherited in affibody molecules (19).
The HER2/Zher2 interface also reveals how the evolution of Zher2 into a high-affinity binder has occurred. First, residues Arg10 (or a Lys10), Trp13, Tyr14, Arg28, Arg32, and Tyr35 were all present in two initial binders obtained from a full library with 13 randomized positions (32). These, together with Ile31, Asp36, and Pro38 that were not varied, already constitute the core binding surface (Fig. 3B) that supports dissociation constants of KD≈50 nM and KD≈140 nM, respectively. The maturation library then allowed for optimization around the core HER2 interface. It contained six completely randomized positions and two positions with a more restricted variation. The side chains of Met9, Ala17, Leu18, and Asn24 at four of the six completely randomized positions were selected because they extend the interface and Arg10 was this time present in all selected binders. Presumably also the nonvaried Asn6 became involved at this point. Side chains at three remaining randomized positions that did not participate in binding then “returned” to those present in the original Z domain (Asn11) or as homologous side chains (Gln25 at the position of Glu25, and Lys27 replacing Arg27) to complete the high-affinity binder (10).
Zher2 Recognizes a Previously Unexploited Epitope on the HER2 Receptor.
The Zher2 binding site on HER2 is distant from those of both trastuzumab and pertuzumab. This is consistent with the lack of interference between either trastuzumab or pertuzumab and Zher2. Any antibodies that may have been raised against this epitope would probably not be well studied due to an absence of any effect on HER2 function. Nonetheless, the present structure demonstrates that this and other nominally occult epitopes are available for exploitation for imaging or for targeted delivery of linked chemotherapeutic agents. The relatively small size of Zher2 and the associated reduced potential for steric overlap with therapeutic antibodies make it attractive for use as a molecular imaging tracer during antibody treatment. Indeed, a molecular imaging agent based on Zher2 has been investigated clinically and has been shown to yield images of HER2 overexpressing metastases within hours after injection in patients also undergoing trastuzumab therapy (17).
Materials and Methods
Zher2 with an N-terminal His6 tag (10) used for biophysical and NMR studies was produced in Escherichia coli and purified by immobilized metal ion chromatography followed by size exclusion chromatography (SEC). NMR was performed at 30 °C on a ca. 1 mM uniformly 13C,15N-labeled Zher2 sample in 160 mM NaCl and 16 mM potassium phosphate at pH 6.0 with 0.1% NaN3 and 5% D2O. Structures were calculated using simulated annealing with distance restraints derived from NOEs, backbone dihedral angle restraints derived from chemical shifts, and hydrogen bond restraints derived from hydrogen bonds observed in initially calculated structures.
The 58 amino acid Zher2 peptide used for X-ray crystallography was prepared by solid phase synthesis. HER2ecd was expressed in Chinese hamster ovary cells and purified by affinity chromatography using HERCEPTIN®, diethylaminoethane anion exchange, and SEC. Crystals of the HER2ecd:Zher2 complex formed in sitting drops at 10 mg/mL in 0.1 M NaCl, 5 mM MOPS (pH 7.3) and reservoir containing 15% wt/vol PEG 3350, 0.1 M sodium acetate (pH 5.0), 0.2 M ammonium acetate. Diffraction data extending to 2.9-Å resolution was collected at ESRF beamline ID23-2. The structure was solved by molecular replacement.
Complete descriptions of protein production, biophysical characterization, NMR spectroscopy, and crystallography are in SI Text.
Supplementary Material
Acknowledgments.
We thank Jeremy Murray and MXpress for collecting diffraction data, Clifford Quan for peptide synthesis, and Prof. Vladislav Orekhov at the Swedish NMR Centre at the University of Gothenburg for assistance with NMR experiments. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. The research was in part supported by a grant from the Swedish Research Council (to T.H.).
Footnotes
Conflict of interest statement: L.A. is employed at Affibody AB.
This article is a PNAS Direct Submission.
Data deposition: The HER2/Zher2, Zher2, and Zher2_alt structures have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3MZW, 2KZI, and 2KZJ, respectively).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1005025107/-/DCSupplemental.
References
- 1.Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garrett TP, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell. 2003;11:495–505. doi: 10.1016/s1097-2765(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 3.Cho H-S, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 4.Franklin MC, et al. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5:17–28. doi: 10.1016/s1535-6108(04)00083-2. [DOI] [PubMed] [Google Scholar]
- 5.Scheuer W, et al. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 2009;69:9330–9336. doi: 10.1158/0008-5472.CAN-08-4597. [DOI] [PubMed] [Google Scholar]
- 6.Makhija S, et al. Clinical activity of gemcitabine plus pertuzumab in platinum-resistant ovarian cancer, fallopian tube cancer, or primary peritoneal cancer. J Clin Oncol. 2010;28:1215–1223. doi: 10.1200/JCO.2009.22.3354. [DOI] [PubMed] [Google Scholar]
- 7.Phillips GDL, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68:9280–9290. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 8.Berns K, et al. A functional genetic approach indentifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 9.Junttila TT, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15:429–440. doi: 10.1016/j.ccr.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 10.Orlova A, et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006;66:4339–4348. doi: 10.1158/0008-5472.CAN-05-3521. [DOI] [PubMed] [Google Scholar]
- 11.Nilsson B, et al. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987;1:107–113. doi: 10.1093/protein/1.2.107. [DOI] [PubMed] [Google Scholar]
- 12.Nord K, et al. Binding proteins selected from combinatorial libraries of an α-helical bacterial receptor domain. Nat Biotechnol. 1997;15:772–777. doi: 10.1038/nbt0897-772. [DOI] [PubMed] [Google Scholar]
- 13.Grönwall C, Ståhl S. Engineered affinity proteins—generation and applications. J Biotechnol. 2009;140:254–269. doi: 10.1016/j.jbiotec.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 14.Lee SB, et al. Affibody molecules for in vivo characterization of HER2-positive tumors by near-infrared imaging. Clin Cancer Res. 2008;14:3840–3849. doi: 10.1158/1078-0432.CCR-07-4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tolmachev V, et al. Radionuclide therapy of HER2-positive micro-xenografts using a 177Lu-labeled HER2-specific Affibody molecule. Cancer Res. 2007;67:2773–2782. doi: 10.1158/0008-5472.CAN-06-1630. [DOI] [PubMed] [Google Scholar]
- 16.Ekerljung L, et al. Dimeric HER2-specific affibody molecules inhibit proliferation of the SKBR-3 breast cancer cell line. Biochem Biophys Res Comm. 2008;377:489–494. doi: 10.1016/j.bbrc.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 17.Baum RP, et al. Molecular imaging of HER2-expressing malignant tumors in breast cancer patients using 111In- 68Ga-labeled Affibody molecules. J Nucl Med. 2010;51:892–897. doi: 10.2967/jnumed.109.073239. [DOI] [PubMed] [Google Scholar]
- 18.Hoyer W, Grönwall C, Jonsson A, Ståhl S, Härd T. Stabilization of a β-hairpin in monomeric Alzheimer’s amyloid-β peptide inhibits amyloid formation. Proc Natl Acad Sci USA. 2008;105:5099–5104. doi: 10.1073/pnas.0711731105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lendel C, Dogan J, Härd T. Structural basis for molecular recognition in an affibody:affibody complex. J Mol Biol. 2006;359:1293–1304. doi: 10.1016/j.jmb.2006.04.043. [DOI] [PubMed] [Google Scholar]
- 20.Wahlberg E, et al. An affibody in complex with a target protein: Structure and coupled folding. Proc Natl Acad Sci USA. 2003;100:3185–3190. doi: 10.1073/pnas.0436086100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelly SM, Price NC. The application of circular dichroism to studies of protein folding and unfolding. Biochim Biophys Acta. 1997;1338:161–185. doi: 10.1016/s0167-4838(96)00190-2. [DOI] [PubMed] [Google Scholar]
- 22.Scholtz JM, Quian H, York EJ, Stewart JM, Baldwin RL. Parameters of helix-coil transition theory for alanine-based peptides of varying chain lengths in water. Biopolymers. 1991;31:1463–1470. doi: 10.1002/bip.360311304. [DOI] [PubMed] [Google Scholar]
- 23.Dincbas-Renqvist V, Lendel C, Dogan J, Wahlberg E, Härd T. Thermodynamics of folding, stabilization, and binding in an engineered protein–protein complex. J Am Chem Soc. 2004;126:11220–11230. doi: 10.1021/ja047727y. [DOI] [PubMed] [Google Scholar]
- 24.Bai Y, Dyson HJ, Wright PE. Absence of a stable intermediate on the folding pathway of protein A. Protein Sci. 1997;6:1449–1457. doi: 10.1002/pro.5560060709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cedergren L, Andersson R, Jansson B, Uhlén M, Nilsson B. Mutational analysis of the interaction between staphylococcal protein A and human IgG1. Protein Eng. 1993;6:441–448. doi: 10.1093/protein/6.4.441. [DOI] [PubMed] [Google Scholar]
- 26.Mulder FA, Mittermaier A, Hon B, Dahlquist FW, Kay LE. Studying excited states of proteins by NMR spectroscopy. Nat Struct Biol. 2001;8:932–935. doi: 10.1038/nsb1101-932. [DOI] [PubMed] [Google Scholar]
- 27.Palmer AG, Massi F. Characterization of the dynamics of biomolecules using rotating-frame spin relaxation NMR spectroscopy. Chem Rev. 2006;106:1700–1719. doi: 10.1021/cr0404287. [DOI] [PubMed] [Google Scholar]
- 28.Bostrom J, et al. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science. 2009;323:1610–1614. doi: 10.1126/science.1165480. [DOI] [PubMed] [Google Scholar]
- 29.Bottomley SP, et al. The stability and unfolding of an IgG binding protein based upon the B domain of protein A from Staphylococcus aureus probed by tryptophan substitution and fluorescence spectroscopy. Protein Eng. 1994;7:1463–1470. doi: 10.1093/protein/7.12.1463. [DOI] [PubMed] [Google Scholar]
- 30.Hoyer W, Härd T. Interaction of Alzheimer’s Aβ peptide with an engineered binding protein—thermodynamics and kinetics of coupled folding-binding. J Mol Biol. 2008;378:398–411. doi: 10.1016/j.jmb.2008.02.040. [DOI] [PubMed] [Google Scholar]
- 31.Wahlberg E, Härd T. Conformational stabilization of an engineered binding protein. J Am Chem Soc. 2006;128:7651–7660. doi: 10.1021/ja060933g. [DOI] [PubMed] [Google Scholar]
- 32.Wikman M, et al. Selection and characterization of HER2/neu-binding affibody ligands. Prot Eng Des Sel. 2004;17:455–462. doi: 10.1093/protein/gzh053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.