

Abstract
A deficiency of functional dystrophin protein in muscle cells causes muscular dystrophy (MD). More than 50% of missense mutations that trigger the disease occur in the N-terminal actin binding domain (N-ABD or ABD1). We examined the effect of four disease-causing mutations—L54R, A168D, A171P, and Y231N—on the structural and biophysical properties of isolated N-ABD. Our results indicate that N-ABD is a monomeric, well-folded α-helical protein in solution, as is evident from its α-helical circular dichroism spectrum, blue shift of the native state tryptophan fluorescence, well-dispersed amide crosspeaks in 2D NMR 15N-1H HSQC fingerprint region, and rotational correlation time calculated from NMR longitudinal (T1) and transverse (T2) relaxation experiments. Compared to WT, three mutants—L54R, A168D, and A171P—show a decreased α-helicity and do not show a cooperative sigmoidal melt with temperature, indicating that these mutations exist in a wide range of conformations or in a “molten globule” state. In contrast, Y231N has an α-helical content similar to WT and shows a cooperative sigmoidal temperature melt but with a decreased stability. All four mutants experience serious misfolding and aggregation. FT-IR, circular dichroism, increase in thioflavin T fluorescence, and the congo red spectral shift and birefringence show that these aggregates contain intermolecular cross-β structure similar to that found in amyloid diseases. These results indicate that disease-causing mutants affect N-ABD structure by decreasing its thermodynamic stability and increasing its misfolding, thereby decreasing the net functional dystrophin concentration.
Keywords: actin binding domain, Becker muscular dystrophy, calponin homology domain, Duchenne muscular dystrophy, protein aggregation
Muscular dystrophy (MD) refers to a group of degenerative muscle diseases that cause progressive muscle weakness (1). Duchenne muscular dystrophy (DMD) is the most common and most severe type of MD and accounts for more than half of all MDs (2–4). DMD is an X-linked, recessive neuromuscular disorder found in 1 in 3,500 live male births. This disease is associated with continuous cycles of muscle cell regeneration and degeneration, ultimately resulting in a failure of muscle regeneration. Symptoms that include constant falling, waddling, and out-turned knees appear as early as age two. Patients’ life spans rarely exceed early to mid-twenties due to cardiac or respiratory failure. During the extreme phase of DMD, patients are unable to sit upright, move their arms or legs, or breathe on their own. A milder form of DMD is Becker muscular dystrophy (BMD) (4, 5). The pattern of symptom development in BMD resembles that of DMD, but with a slower rate of progression. At present, there is no cure available for MD.
DMD and BMD are triggered primarily by the decreased function of a vital muscle protein known as dystrophin (1, 6–9). DMD patients lack functional dystrophin, whereas BMD patients have a reduced efficiency of the protein. Dystrophin stabilizes the cell membrane of muscle cells against the mechanical forces associated with muscle contraction and stretch. It acts as a link between the sarcolemma transmembrane glycoprotein complex and actin fibers (Fig. 1A) (10–12). Upon binding, dystrophin protects actin filaments against depolymerization. Dystrophin-related defects lead to a loss of function at the sarcolemma, causing the muscle fibers to become brittle and susceptible to mechanical stress. This, in turn, leads to sarcolemmal fragility, muscle weakness, and may even cause muscle cell necrosis (8). In addition to its protective role, dystrophin has been proposed to act as a signaling molecule in cell signaling pathways such as muscle cell growth, cytoskeleton organization, muscle homeostasis, and atrophy/hypertrophy (7, 13–15).
Fig. 1.

(A) Illustration of dystrophin function (8, 12, 47). Dystrophin contains four distinct domains: N-terminal actin-binding domain (N-ABD or ABD1); central rod domain containing the second actin binding domain (ABD2); cysteine-rich domain (CR); and the C-terminal domain (CT). Dystrophin binds to the actin-based cytoskeleton through its two actin binding domains (N-ABD and ABD2) and to the transmembrane glycoprotein complex with its CR and CT domains. (B) Crystal structure of dystrophin N-ABD (18). N-ABD contains two calponin homology (CH) domains and three actin binding surfaces (ABS), shown in yellow. The four disease-causing mutations studied here are shown in red. The molecular structure was generated using the UCSF Chimera software (48) (http://www.cgl.ucsf.edu/chimera/).
Dystrophin is a long (∼0.15 μm), rod-shaped molecule composed of 3,684 amino acids. It contains four distinct domains: an N-terminal actin binding domain (N-ABD or ABD1), a central rod domain containing a second ABD (ABD2), a cysteine-rich domain (CR), and a C-terminal domain (CT) (Fig. 1A) (8). The central rod domain consists of 24 spectrin repeats and four hinge regions. Dystrophin binds to the actin-based cytoskeleton through its two actin binding domains (N-ABD and ABD2) and to the transmembrane glycoprotein complex with its CR and CT domains. Our goal is to understand how the structural and molecular principles of dystrophin govern its function and the effect thereon of disease-causing mutations. Because of its large size, full-length protein is not amenable to current solution structural techniques such as NMR. Hence we focus on studying the individual domains.
Among the mutations in dystrophin that trigger muscular dystrophy [listed in the Leiden MD mutation database (http://www.dmd.nl/) and Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/)], a major fraction of missense mutations (> 50%) occurs in N-ABD (16–18). We probed the effect of four mutations on the structure and stability of N-ABD: L54R, which triggers DMD; and A168D, A171P, and Y231N, which trigger BMD (16, 18–20). Fig. 1B places these mutations in the N-ABD structure (18). Our results presented here show that these mutations decrease thermodynamic stability and cause protein aggregation.
Results
Isolated N-ABD Is a Monomeric, Well-Structured α-Helical Protein in Solution.
When expressed in Escherichia coli, the isolated WT N-ABD domain was found predominantly in the solution phase (Fig. 2A) and could be prepared at high purity (Fig. 2B). Although earlier X-ray studies show that this domain exists as a dimer in the crystal state (18), we confirmed (native gel experiment, Fig. 2C) that N-ABD is a monomer in solution, as indicated in previous biochemical studies (10, 21–24). No higher molecular weight bands were seen when the native gel was overloaded with the protein, even after keeping the protein for one month at room temperature. Moreover, ABDs of distantly related proteins, such as fimbrin and α-actinin, exist as monomers (23, 25–27).
Fig. 2.

Characterization of WT N-ABD. (A) Denaturant gel (SDS/PAGE) of protein expression in E coli. Lane M corresponds to the protein molecular weight markers (bottom to top: 11, 17, 26, 34, 43, and 56 kDa, respectively). Lanes 1 and 2 show the protein content expressed in solution phase and in inclusion bodies respectively. (B) SDS/PAGE of protein purification profile. Lane M is the protein ladder, and lanes 1–5 are successive samples eluted from the Ni-sepharose column. (C) Native PAGE of freshly prepared WT N-ABD and after storing the protein for 1 month at room temperature. (D) CD spectrum, (E) tryptophan fluorescence spectra of native (N) and unfolded (U) states, and (F) 2D 15N-1H TROSY-HSQC NMR spectrum of WT N-ABD. (G) T1, T2, and τc values for individual amide crosspeaks (arbitrarily numbered, because the residue assignments are not known). In this calculation, only strong and nonoverlapping peaks were considered.
We studied the quality of N-ABD structure. The circular dichroism (CD) spectrum shows −ve bands at 208 and 222 nm characteristic of an α-helical protein (Fig. 2D). Tryptophan fluorescence emission in the native state showed a considerable blue shift with maximum at 338 nm compared to 356 nm for the unfolded state (Fig. 2E). For tryptophans exposed to solvent, the fluorescence maximum occurs at longer wavelengths, around 355 nm, whereas for those buried from the solvent, fluorescence emission occurs at shorter wavelengths around 325 nm. N-ABD contains eight tryptophans distributed all across the protein structure. The observed shift in the fluorescence emission maximum indicates that some of these are well buried in the native state, consistent with a well-folded structure. In addition, 2D NMR confirms that N-ABD has a well-folded protein core. The crosspeaks between amide nitrogens and hydrogens in the 2D 15N-1H HSQC spectrum are well-dispersed (Fig. 2F). We calculated the average rotational correlation time (τc) of the molecule as 18 ns from the longitudinal (T1) and transverse (T2) relaxation times of individual crosspeaks (Fig. 2G), which again indicates that N-ABD is a monomer in solution. In general, τc can be empirically calculated for a spherical protein from the number of amino acids (28), and the expected value for dystrophin N-ABD is 16 ns. The slightly higher τc observed suggests that the protein is not spherical but somewhat asymmetric.
Mutants Have a Strong Tendency to Aggregate.
In contrast to WT, which was predominantly (∼80%) expressed in solution (Fig. 2A), nearly 100% of all four disease-causing mutants L54R, A168D, A171P, and Y231N resulted in inclusion bodies under identical growth conditions (Fig. 3A). Varying expression and growth conditions did not improve the expression pattern. We purified mutants by centrifuging out the inclusion bodies and dissolving in high denaturant (Fig. 3B). Upon dilution of the denaturant, ∼99% of the mutants aggregate, whereas under identical refolding conditions WT refolds to about 30%.
Fig. 3.

(A) SDS/PAGE of N-ABD mutants expression in E coli. IB and S represent the protein content expressed in inclusion bodies and in the solution phase respectively. Lane M shows the protein molecular weight markers. (B) SDS/PAGE of purified mutants. Lane M is the protein molecular weight markers. (C) Fraction of refolded protein of WT N-ABD and four disease-causing mutants from their unfolded states. (D and E) Aggregation kinetics of WT and mutants as measured by the right-angle light scattering. Panels D and E show the aggregation kinetics without and with 0.5 M L-arginine, respectively.
The mutants also aggregate at a faster rate. When measured by right-angle light scattering, they seem to aggregate at the same rate as that of the WT (Fig. 3D). However, a clear distinction can be seen in the presence of arginine, which is known to stabilize protein native structures against protein aggregation (29). Within 150 sec, WT did not show much aggregate formation, whereas all the mutants aggregated within 150 sec (Fig. 3E). Also, considerable difference was seen in the aggregation kinetics of A168D mutant without and with arginine. With the addition of arginine, the A168D mutant aggregates at a slower rate (compare Fig. 3 D and E).
Mutants Have Decreased Stability.
Fig. 4 compares the secondary structure and stability of refolded WT and mutants. The CD spectrum of refolded WT exactly overlaps with that of the freshly purified WT (Fig. 4A Top). Also, refolded WT melted with a midpoint temperature (Tm) of 58 °C, like freshly purified WT (Fig. 4B Top). Evidently N-ABD reaches the same authentic native conformation after refolding from the unfolded state.
Fig. 4.

Characterization of soluble N-ABD mutants. (A) CD spectra of WT and mutants refolded from their unfolded states. (B) Thermal melts as measured by CD recorded as a function of temperature. In the top panels of A and B, solid and dashed curves show the data of freshly purified WT from E coli and refolded WT from its unfolded state.
Although 99% of the mutants aggregate under refolding conditions, we were able to recover 1% in soluble form. When compared to WT, the CD spectra of three mutants L54R, A168D, and A171P showed a decrease in the 222 nm band intensity indicating decreased α-helicity (Fig. 4A). A171P mutant is completely disordered as seen from the presence of a negative CD band at 200 nm, which is characteristic of a random coil. The three mutants do not show a cooperative sigmoidal melt with temperature, and they aggregate faster than the WT protein again indicating reduced stability. In contrast, the Y231N mutant has a similar CD spectrum and hence identical secondary structure as the WT but reduced stability. It shows a cooperative melting curve indicating a well-folded structure but with a Tm of 43 °C, which is 15 °C less than that of the WT.
N-ABD Aggregates Contain Amyloid-Like Cross-β Structure.
We probed the structure of WT and mutant N-ABD aggregates. We observe two types of structures: fibers with two or more fibrils twisted around one another in a helical fashion, and bundles of fibrils forming large aggregates (Fig. 5A). CD spectra of all the aggregates show a strong +ve peak at 197 nm and a −ve peak at 227 nm (Fig. 5B), which indicates that the aggregates contain significant β-structure. We examined the amide I region of the FT-IR spectra to determine the structure of aggregates (Fig. 5C). Although individual secondary structures absorb over multiple wavelengths, each type of secondary structure has characteristic IR bands (30). α-helix absorbs at 1,654 cm-1, which is obvious in the WT protein. Aggregates of both WT and mutants show 1,654 cm-1 band but with a decreased intensity, indicating significant α-helical structure but less than that of the WT. In addition, the aggregates show a band at 1,694 cm-1, which is a characteristic of intermolecular β-sheet (31, 32).
Fig. 5.

Characterization of WT and mutant N-ABD aggregates. (A) EM picture, (B) CD spectra of aggregates. (C) Second derivative FTIR spectra of aggregates (dashed curves) compared to the soluble WT monomer (solid curve). (D) Increased ThT fluorescence when bound to protein aggregates. The figure shows the fluorescence in buffer before and after adding aggregates. (E) Red shift in CR absorption spectra upon binding to protein aggregates. The figure shows the spectra before and after adding aggregates. In panels B–E, individual curves were not labeled because the minor differences in intensity can be accounted by experimental errors. (F) Congo red birefringence images of dystrophin N-ABD fibrils.
To further test for intermolecular cross-β structure of N-ABD aggregates, we performed Thioflavin T (ThT) and Congo red (CR) dye binding assays. ThT is fluorescent when bound to cross-β fibrils (33). Upon adding ThT to preformed WT and mutant N-ABD fibrils, ThT fluorescence increases by more than 10 times compared to that in buffer (Fig. 5D). Another characteristic of cross-β structure is a red shift in the CR absorption spectrum and apple-green birefringence (34) which arise because of the ordered oriented array of CR molecules along the cross-β structure (35). When CR was added to the N-ABD WT and mutant aggregates, its absorption maximum shifted from 482 nm in buffer to 525 nm (Fig. 5E). Also these aggregates show the characteristic birefringence (Fig. 5F) as observed by the light microscopy with polarization filters. Some of these birefringence images also show that smaller fibers bundle together in a helical fashion to form larger aggregates, which might correspond to very late stage disease-causing fibers.
Discussion
Effects of Disease-Causing Mutations on Dystrophin Stability.
Dystrophin was identified as a key protein in triggering MD 23 years ago (2), but little is known about the mechanisms of disease trigger at the molecular level. Here, we examined the effect of disease-causing mutations on protein structure and stability. N-ABD is the first dystrophin domain for which the molecular structure was determined (18). Superficial examination of the N-ABD structure shows no obvious correlation between the location of individual residues and disease trigger (Fig. 1B). The disease-causing mutations are scattered, not located in a single region or in a single CH domain or at the three actin binding surfaces (ABS). However, structural considerations suggest that the mutations should be destabilizing. They replace apolar residues in different hydrophobic pockets with charged or polar residues. The L54R mutation places a charged arginine residue in the hydrophobic core formed by A80, V83, and L84. The Y231N mutation places a polar residue in the hydrophobic cluster formed by F200, L213, and L234. The A168D mutation places a negatively charged residue in a hydrophobic cluster formed by the residues V154, V156, and Trp164. In addition, A168 is part of an α-helix. Ala has the highest helix propensity compared to any other amino acid (36), and hence mutating it will decrease the helix stability. Also, Asp at position 168 is three residues from another Asp at position 165 in the same helix, which will result in electrostatic repulsion leading to helix destabilization (37). The A171P mutation, in the same helix, replaces the strongest helix promoter with the strongest helix breaker (36). Accordingly, we tested the effect of these mutations on protein stability and misfolding.
Our results indicate that these mutations decrease protein stability and promote protein misfolding. Protein stability is a measure of the concentration of the functional protein. For mutants that do not show a clear cooperative melt (Fig. 4B), in particular L54R, A168D, and A171P, there exists a significant nonfunctional unfolded population that leads to decreased protein function. In addition, decrease in stability increases the protein’s susceptibility to its degradation by proteases in vivo. If a protein cannot fold to its correct native state after it has been synthesized by the ribosomal machinery, protein concentration and function will be decreased. In support of this argument, the first MD patient in whom the L54R mutation was detected had only 20% of normal levels of dystrophin (19). In terms of mutation effects on actin binding, the four disease-causing mutations studied here are not part of the three actin binding surfaces in N-ABD (Fig. 1B) and hence may not directly affect actin binding. However, if the actin binding involves a conformational change that brings the three ABSs together as proposed earlier (18) and the mutations are involved in such a transition, they may affect actin binding.
In previous work, Legardiner et al. found that the DMD-associated double mutant E2910V-N2912D in spectrin repeat 23 of the central rod domain (Fig. 1A) lowers the protein melting temperature and causes slowed folding (38). In another study, a large fragment deletion in the central rod domain, which is found in BMD patients, leaving a fused protein of the first three and the last five repeats of the rod, melted at a lower temperature (21). These studies together with the present work strongly suggest that the effect of mutations on dystrophin stability can trigger muscular dystrophy.
Stability and Protein Aggregation in Other Pathologies.
A similar situation is thought to occur in other proteins. In the cystic fibrosis transmembrane regulator (CFTR), the ΔF508 mutation that occurs in 70% of cystic fibrosis patients has a decreased stability and folds slowly compared to its WT, leading to a decrease in the functional protein concentration in vivo and net decreased function (39, 40). Carcinogenic mutations in the tumor suppressor protein p53 reduce the protein stability and perturb the protein structure affecting its DNA binding affinity. Folded p53 levels in vivo were correlated with changes in protein stability (41, 42). Sickle cell disease occurs when, after birth, the gamma chain in fetal hemoglobin is replaced by the beta chain carrying the Glu6Val mutation (43), leading to the concentration-dependent formation of sickle hemoglobin aggregates. A similar situation occurs in MD. There exists a homologous protein known as utrophin that has a 60% sequence similarity to that of dystrophin (8, 10). Utrophin is also a rod-shaped domain, contains similar four distinct domains, and functions similarly to dystrophin. It binds to actin and interacts with dystrophin-related proteins. It is confined specifically to the sarcolemma in fetal and regenerating muscle cells (8). As for fetal hemoglobin, utrophin levels decrease with human growth and dystrophin levels increase, leading to the onset of MD symptoms due to mutations in dystrophin. This can be possibly because defective dystrophin starts forming aggregates after it has reached a critical concentration in vivo.
We find that dystrophin tends to aggregate during folding. It is now understood that proteins unfold and refold even under native conditions (44, 45) in a stability-dependent manner. Thus there always exists a constant steady-state concentration of unfolded protein, given by [Unfolded] = [Native] exp(-ΔGeq/RT), where ΔGeq is the stabilization free energy, R is the gas constant, and T is absolute temperature. This relationship shows that protein destabilization increases the steady-state unfolded concentration exponentially, labilizing the protein to concentration-dependent aggregation during refolding. The presence of mutations that cause the refolding process to pause and populate partially folded species will exacerbate the tendency to aggregate. In this work we show that disease-causing mutations do in fact destabilize dystrophin N-ABD and lead to aggregation during the folding process. It appears that this pathogenic effect may well be much more general than for muscular dystrophy alone.
Materials and Methods
Expression and Purification
Dystrophin N-ABD (residues 1-246; C10S, C188S) plasmid was a kind gift from Steven Winder. We improved the protein yield and shortened the purification protocol by cloning the cDNA into a pET vector. The four mutations, L54R, A168D, A171P, and Y231N, were made using Stratagene’s Quick Mutagenesis protocol. WT and the four mutants were expressed in Escherichia coli (BL21-DE3) and purified using a Ni-Sepharose column. For mutants, inclusion bodies were solubilized in 8 M Urea prior to loading onto the Ni-Sepharose column. Homogeneity of native N-ABD sample was checked by loading 30 μg of the protein on a 6% native PAGE gel (Fig. 2C).
NMR
N-ABD was labeled with 15N by expressing in minimal medium supplemented with 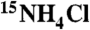 . 2D 15N/1H TROSY-HSQC NMR spectra were recorded in the Rocky Mountain Regional 900 MHz NMR Facility. For T1, T2 measurements, modified pulse sequences available with the Varian Biopack were used. Data was processed using NMRPipe software (46). τc was calculated using the equation,
. 2D 15N/1H TROSY-HSQC NMR spectra were recorded in the Rocky Mountain Regional 900 MHz NMR Facility. For T1, T2 measurements, modified pulse sequences available with the Varian Biopack were used. Data was processed using NMRPipe software (46). τc was calculated using the equation, 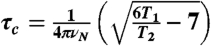 where νN is the 15N resonance frequency.
where νN is the 15N resonance frequency.
Refolding Yield
WT and mutant aggregates (50 μM) were solubilized in 100 mM phosphate, 150 mM NaCl, pH 7 (“PBS”) , 6 M GdmCl, and were refolded by diluting 10 times into PBS buffer and incubated overnight to reach equilibrium. Refolded solution was centrifuged at 30,000 g for 1 h at 4 °C and soluble protein content in the supernatant was quantified using absorbance at 280 nm (Fig. 3C). For measuring aggregation kinetics (Fig. 3 D and E), unfolded proteins were diluted by 100 times and changes in light scattering were measured at 450 nm where the protein doesn’t absorb.
Circular Dichroism
WT and mutant aggregates were solubilized in PBS buffer with 6 M GdmCl at a protein concentration of 100 μM. This solution was diluted 100 times into PBS buffer in a pulsatile manner (50 μl/minute) on ice with slow stirring. Refolded solution was incubated at 4 °C overnight, solution was centrifuged at 30,000 g for 1 h to remove any aggregates, clear supernatant was concentrated to 1 μM and used for measuring CD on a Chirascan Plus spectrometer. Thermal melts were performed by measuring CD spectra as a function of temperature. The CD melt was plotted at the wavelength where there was a maximum difference between the native and unfolded signals.
Dye Binding Assays
For ThT assay, approximately 2 μM of protein aggregates and 50 μM dye concentration was used. Changes in ThT fluorescence were measured using a SPEX Fluoromax-3 fluorometer using 440 nm as the excitation wavelength. For CR assay, 50 μM dye concentration was used with 2 μM of protein aggregates and the changes in the dye absorbance was measured using an Agilent 8453 UV-Visible spectrophotometer. For congo red birefringence, a dilute solution of preformed aggregates was dried over a glass slide and the images were taken on a Leica DME microscope with polarization filters.
FTIR
IR spectra of WT and mutant aggregates were recorded using a Biomem MB-series FTIR spectrometer (ABB Biomem Inc. Canada) and a Biocell CaF2 cell (Biotools). Spectra were processed using GRAMS/AI 7.00 (Thermo Galactic, Thermo Electron, CO). Area normalized second derivative of absorbance spectra were compared for analyzing secondary structure differences.
Acknowledgments.
We thank Steven Winder for his kind gift of the dystrophin N-ABD plasmid. We gratefully acknowledge the help of Geoffrey Armstrong (Rocky Mountain Regional 900 MHz NMR Facility), Shelley Reed (congo red birefringence), Dorothy Dill (EM core), and Biophysics Core in carrying out this work. We thank Steven Winder, Steven Ringel, John Carpenter, David Bain, Deborah Wuttke, David Jones, James Ervasti for helpful discussions, Roland Roberts for providing figure 3 of ref. 12, from which Fig. 1A was generated, and Walter Englander for a critical reading of the manuscript. This work was funded by the School of Pharmacy, University of Colorado, Denver, and a Jane and Charlie Butcher grant in Genomics and Biotechnology. Rocky Mountain Regional 900 MHz NMR Facility was funded by the National Institutes of Health instrumentation Grant P41GM068928.
Note Added in Proof.
While this manuscript was under review, a similar study was published by Henderson et al. (49) using the full-length dystrophin protein. Our results agree. Disease-causing mutations decrease thermodynamic stability, lead to noncooperative unfolding, and induce aggregation, although more so for the isolated N-ABD, suggesting that the aggregation propensity might originate from this domain. We also found that the aggregates contain significant amyloid-like cross-β structure and that small molecules such as arginine can rescue dystrophin from misfolding.
Footnotes
The authors declare no conflict of interest.
References
- 1.Emery AEH. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman EP, Brown J, Robert H, Kunkel LM. Dystrophin: The protein product of the Duchenne Muscular Dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 3.Emery A. Population frequencies of inherited neuromuscular disorders: A world survey. Neuromuscular Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 4.Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 5.Bakker E, van Ommen GB. In: Neuromuscular disorders: Clinical and molecular genetics. Emery AEH, editor. New York: John Wiley & Sons; 1998. pp. 59–85. [Google Scholar]
- 6.Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- 7.Batchelor CL, Winder SJ. Sparks, signals and shock absorbers: How dystrophin loss causes muscular dystrophy. Trends Cell Biol. 2006;16:198–205. doi: 10.1016/j.tcb.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Ervasti JM. Review: Dystrophin, its interactions with other proteins, and implications. Biochim Biophys Acta. 2007;1772:108–117. doi: 10.1016/j.bbadis.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 9.Vainzof M, Zatz M. In: Protein misfolding, aggregation, and conformational diseases. Part B: Molecular mechanisms of conformational diseases. Uversky VN, Fink AL, editors. New York: Springer; 2007. [Google Scholar]
- 10.Winder SJ. Dystrophin and utrophin: The missing Links! FEBS Lett. 1995;369:27–33. doi: 10.1016/0014-5793(95)00398-s. [DOI] [PubMed] [Google Scholar]
- 11.Rybakova IN, Patel JR, Ervasti JM. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol. 2000;150:1209–1214. doi: 10.1083/jcb.150.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts RG. Protein family review: Dystrophins and dystrobrevins. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-4-reviews3006. reviews3006.3001-3006.3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rando TA. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve. 2001;24:1575–1594. doi: 10.1002/mus.1192. [DOI] [PubMed] [Google Scholar]
- 14.Oak SA, Zhou YW, Jarret HW. Skeletal muscle signaling pathway through the dystrophin glycoprotein complex and Rac1. J Biol Chem. 2003;278:39287–39295. doi: 10.1074/jbc.M305551200. [DOI] [PubMed] [Google Scholar]
- 15.Glass DJ. A signaling role for dystrophin: Inhibiting skeletal muscle atrophy pathways. Cancer Cell. 2005;8:351–352. doi: 10.1016/j.ccr.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 16.Roberts RG, Borrow M, Bentley DR. Point mutations in the dystrophin gene. Proc Natl Acad Sci USA. 1992;89:2331–2335. doi: 10.1073/pnas.89.6.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sitnik R, et al. Novel point mutations in the dystrophin gene. Hum Mutat. 1997;10:217–222. doi: 10.1002/(SICI)1098-1004(1997)10:3<217::AID-HUMU7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 18.Norwood FL, Sutherland-Smith AJ, Keep NH, Kendrick-Jones J. The structure of the N-terminal actin-binding domain of human dystrophin and how mutations in this domain may cause Duchenne or Becker muscular dystrophy. Structure. 2000;8:481–491. doi: 10.1016/s0969-2126(00)00132-5. [DOI] [PubMed] [Google Scholar]
- 19.Prior TW, et al. A missense mutation in the dystrophin gene in a Duchenne muscular dystrophy patient. Nat Genet. 1993;4:357–360. doi: 10.1038/ng0893-357. [DOI] [PubMed] [Google Scholar]
- 20.Roberts RG, Gardner RJ, Bobrow M. Searching for the 1 in 2,400,000: A review of dystrophin gene point mutations. Hum Mutat. 1994;4:1–11. doi: 10.1002/humu.1380040102. [DOI] [PubMed] [Google Scholar]
- 21.Kahana E, Flood G, Gratzer WB. Physical properties of dystrophin rod domain. Cell Motil Cytoskel. 1997;36:246–252. doi: 10.1002/(SICI)1097-0169(1997)36:3<246::AID-CM5>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 22.Rybakova IN, Ervasti JM. Dystrophin-glycoprotein complex is monomeric and stabilizes actin filaments in vitro through a lateral association. J Biol Chem. 1997;272:28771–28778. doi: 10.1074/jbc.272.45.28771. [DOI] [PubMed] [Google Scholar]
- 23.Broderick MJF, Winder SJ. Towards a complete atomic structure of spectrin family proteins. J Struct Biol. 2002;137:184–193. doi: 10.1006/jsbi.2002.4465. [DOI] [PubMed] [Google Scholar]
- 24.Winder SJ, Gibson TJ, Kendrick-Jones J. Low probability of dystrophin and utrophin coiled coil regions forming dimers. Biochem Soc Trans. 1996;24:280S. doi: 10.1042/bst024280s. [DOI] [PubMed] [Google Scholar]
- 25.McGough A, Way M, DeRosier D. Determination of the α-actinin-binding site on actin filaments by cryoelectron microscopy and image analysis. J Cell Biol. 1994;126:433–443. doi: 10.1083/jcb.126.2.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldsmith SC, et al. The structure of an actin-crosslinking domain from human fimbrin. Nat Struct Biol. 1997;4:708–712. doi: 10.1038/nsb0997-708. [DOI] [PubMed] [Google Scholar]
- 27.Franzot G, Sjöblom B, Gautel M, Carugo KD. The crystal structure of the actin binding domain from α-actinin in its closed conformation: Structural insight into phospholipid regulation of α-actinin. J Mol Biol. 2005;348:151–165. doi: 10.1016/j.jmb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 28.Daragan VA, Mayo KH. Motional model analyses of protein and peptide dynamics using 13C and 15N NMR relaxation. Prog NMR Spectroscopy. 1997;31:63–105. [Google Scholar]
- 29.Baynes BM, Wang DIC, Trout BL. Role of arginine in the stabilization of proteins against aggregation. Biochemistry. 2005;44:4919–4925. doi: 10.1021/bi047528r. [DOI] [PubMed] [Google Scholar]
- 30.Dong A, Huang P, Caughey WS. Protein secondary structures in water from second-derivative amide I infrared spectra. Biochemistry. 1990;29:3303–3308. doi: 10.1021/bi00465a022. [DOI] [PubMed] [Google Scholar]
- 31.Dong A, Matsuura J, Manning MC, Carpenter JF. Intermolecular β-sheet results from trifluoroethanol-induced nonnative α-helical structure in β-sheet predominant proteins: Infrared and circular dichroism spectroscopic study. Arch Biochem Biophys. 1998;355:275–281. doi: 10.1006/abbi.1998.0718. [DOI] [PubMed] [Google Scholar]
- 32.Dong A, Randolph TW, Carpenter JF. Entrapping intermediates of thermal aggregation in α-helical proteins with low concentration of guanidine hydrochloride. J Biol Chem. 2000;275:27689–27693. doi: 10.1074/jbc.M005374200. [DOI] [PubMed] [Google Scholar]
- 33.Levine-III H. Thioflavine T interaction with synthetic Alzheimer's disease {beta}-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klunk WE, Pettegrew JW, Abraham DJ. Quantitative evaluation of congo red binding to amyloid-like proteins with a beta-pleated sheet conformation. J Histochem Cytochem. 1989;37:1273–1281. doi: 10.1177/37.8.2666510. [DOI] [PubMed] [Google Scholar]
- 35.Childers WS, Mehta AK, Lu K, Lynn DG. Templating molecular arrays in amyloid’s cross-β grooves. J Am Chem Soc. 2009;131:10165–10172. doi: 10.1021/ja902332s. [DOI] [PubMed] [Google Scholar]
- 36.Chakrabartty A, Kortemme T, Baldwin RL. Helix propensities of the amino acids measured in alanine-based peptides without helix-stabilizing side-chain interactions. Protein Sci. 1994;3:843–852. doi: 10.1002/pro.5560030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ebert G, Ebert C, Paudjojo L. Effect of water and electrostatic interactions on the conformation conversion of polypeptides and proteins. Prog Coll Pol Sci S. 1978;65:60–67. [Google Scholar]
- 38.Legardinier S, et al. A two-amino acid mutation encountered in Duchenne muscular dystrophy decreases stability of the rod domain 23 (R23) spectrin-like repeat of dystrophin. J Biol Chem. 2009;284:8822–8832. doi: 10.1074/jbc.M805846200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thibodeau PH, Brautigam CA, Machius M, Thomas PJ. Side chain and backbone contributions of Phe508 to CFTR folding. Nat Struct Mol Biol. 2005;12:10–16. doi: 10.1038/nsmb881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du K, Sharma M, Lukacs GL. The ΔF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol. 2005;12:17–25. doi: 10.1038/nsmb882. [DOI] [PubMed] [Google Scholar]
- 41.Mayer S, Rüdiger S, Ang HC, Joerger AC, Fersht AR. Correlation of levels of folded recombinant p53 in Escherichia coli with thermodynamic stability in vitro. J Mol Biol. 2007;372:268–276. doi: 10.1016/j.jmb.2007.06.044. [DOI] [PubMed] [Google Scholar]
- 42.Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 43.Mehanna AS. Sickle cell anemia and antisickling agents then and now. Curr Med Chem. 2001;8:79–88. doi: 10.2174/0929867013373778. [DOI] [PubMed] [Google Scholar]
- 44.Bai Y, Sosnick TR, Mayne L, Englander SW. Protein folding intermediates: Native-state hydrogen exchange. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Englander SW, Mayne L, Krishna MMG. Protein folding and misfolding: Mechanism and principles. Q Rev Biophys. 2007;40:287–326. doi: 10.1017/S0033583508004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 47.Blankinship MJ, Gregorevic P, Chamberlain JS. Gene therapy strategies for Duchenne muscular dystrophy utilizing recombinant adeno-associated virus vectors. Mol Ther. 2006;13:241–249. doi: 10.1016/j.ymthe.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 48.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 49.Henderson DM, Lee A, Ervasti JM. Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc Natl Acad Sci USA. 2010;107:9632–9637. doi: 10.1073/pnas.1001517107. [DOI] [PMC free article] [PubMed] [Google Scholar]