

Abstract
Activating transcription factor 3 (ATF3) is a common stress sensor, and its rapid induction by cellular stresses (e.g. DNA damage) is crucial for cells to mount appropriate responses (e.g. activating the tumor suppressor p53) and maintain homeostasis. Although emerging evidence suggests that dysregulation of ATF3 contributes to occurrences of human diseases including cancer, the mechanism(s) by which ATF3 expression is regulated is largely unknown. Here, we demonstrate that mouse double minute 2 (MDM2) is a bona fide E3 ubiquitin ligase for ATF3 and regulates ATF3 expression by promoting its degradation. MDM2 via its C-terminal RING finger can bind to the Basic region of ATF3 and mediate the addition of ubiquitin moieties to the ATF3 leucine zipper domain. As a consequence, ATF3, but not a mutant deficient in MDM2 binding (Δ80–100), is degraded by MDM2-mediated proteolysis. Consistent with these results, ablation of MDM2 in cells not only increases basal ATF3 levels, but results in stabilization of ATF3 in late stages of DNA damage responses. Because ATF3 was recently identified as a p53 activator, these results suggest that MDM2 could inactivate p53 through an additional feedback mechanism involving ATF3. Therefore, we provide the first evidence demonstrating that ATF3 is regulated by a posttranslational mechanism.
Keywords: DNA Damage, Protein Stability, Protein-Protein Interactions, Transcription Factors, Ubiquitin Ligase, Ubiquitination, ATF3, DNA Damage, MDM2, Stress Response
Introduction
ATF32 is a member of the ATF/CREB family of transcription factors, and its expression is rapidly induced by a large variety of cellular stresses including DNA damage, wounds, and cellular injury (1). ATF3 can bind to DNA (via the ATF/CREB consensus sequence, 5′-TGACGTCA-3′) (1) and other proteins (e.g. Smad3, p53, and E6) (2–4), resulting in alterations in gene expression and cellular functions. Although consequences of stress-induced ATF3 expression are not well understood, recent evidence links ATF3 to several important pathways, including TGFβ signaling (2), the Toll-like receptor 4 pathway (5), the eIF2 kinase-mediated endoplasmic reticulum stress response (6), as well as the p53 activation pathway (3), suggesting that dysregulation of ATF3 could contribute to occurrences of many human diseases including cancer. Indeed, although we previously showed that ATF3 deficiency promotes oncogenic transformation (3, 7), recent unbiased cDNA array studies have revealed that ATF3 expression is down-regulated in common human cancers (for a review of these data, see Ref. (8).
Although induction of ATF3 expression is a common characteristic of stress responses (1), the mechanisms by which ATF3 expression is regulated during these processes remain largely unknown. It has been shown that ATF3 expression can be regulated by transcription factors such as ATF2, Smad3, and NF-κB (2, 9, 10) and controlled by signaling mediated by p38 or JNK/SAPK (9, 11). Moreover, an atypical p53-binding site was identified in the ATF3 promoter (12). However, whether p53 regulates ATF3 expression still remains to be firmly validated, even though a marginal effect of p53 on ATF3 expression was reported in specific cells and in response to specific stress (13–15). Interestingly, ATF3 expression induced by stress is often transient, and both ATF3 mRNA and protein levels return to basal levels in late stages of stress responses (1), suggesting that temporal expression of ATF3 could be important for cells to mount appropriate stress responses. Because the ATF3 promoter contains an ATF/CREB cis-element that can be bound by ATF3 itself, stress-induced ATF3 transcription can be turned down through autorepression (16). However, the mechanism(s) governing rapid degradation of ATF3, a protein lacking a PEST degradation signal peptide, in late stages of stress responses is currently unknown.
In an effort to elucidate the mechanisms regulating ATF3 expression in response to DNA damage, we identified mouse double minute 2 (MDM2) as the first E3 ubiquitin ligase for ATF3. The C terminus of MDM2 contains a RING finger that can direct p53 ubiquitination (17) and subsequent proteosomal degradation (18, 19), and thus MDM2 serves as a major p53 repressor (20). Given that ATF3 can stabilize p53 (3), the identification of ATF3 as a novel substrate for MDM2 unveils an additional mechanism for a tight control of p53 function by MDM2.
EXPERIMENTAL PROCEDURES
Plasmids and Cells
The ATF3 constructs, MDM2 C464A, and ubiquitin constructs were obtained from Drs. Tsonwin Hai, Karen Vousden, and Toshiaki Suzuki, respectively. We used PCR to fuse a FLAG sequence to the ATF3 N terminus to construct the FLAG-ATF3 plasmid. HCT116 p53−/− cells and p53−/−/mdm2+/+ and p53−/−/mdm2−/− mouse embryonic fibroblasts (MEFs) were kindly provided by Drs. Bert Vogelstein and Guillermina Lozano.
Protein Purification and in Vitro Translation
Histidine-tagged ATF3 and Δ102–139 and GST-MDM2 proteins were prepared as described previously (3). The TNT Quick Coupled Transcription/Translation System and the S30 T7 High Yield Protein Expression System (Promega) were used for in vitro translation of ATF3 and MDM2 proteins, respectively, following the manufacturers' protocols.
Glutathione S-Transferase (GST) Pulldown Assays
The plasmids encoding GST protein fused with truncated MDM2 and ATF3 were described previously (3, 21). These plasmids were transformed into BL21 strain, and expression of GST or GST fusion proteins was induced by isopropyl-β-d-thiogalactopyranoside. GST pulldown assays were carried out as described previously (3, 4). Immobilized GST fusion proteins were incubated with in vitro translated ATF3 or MDM2 at 4 °C overnight, and bound proteins were detected by immunoblotting as described (22).
In Vitro Ubiquitination Assays
These were performed using a reconstituted ubiquitination reaction system as described previously (3). Briefly, 10 ng of purified ATF3 or Δ102–139 was incubated with 400 ng of MDM2, 25 ng of E1, 100 ng of E2, and 5 μg of ubiquitin in a 30-μl reaction containing 40 mm Tris-HCl, pH 7.5, 5 mm MgCl2, 2 mm DTT, and 2 mm ATP at 37 °C for 90 min. E1, E2, and ubiquitin were purchased from Boston Biochem. Ubiquitination reactions were terminated by boiling the samples in SDS-loading buffer, and the samples were subjected to immunoblotting for detection of ubiquitinated ATF3 proteins using an ATF3 antibody (C-19; Santa Cruz Biotechnology).
In Vivo Ubiquitination Assays
We used two slightly different conditions to carry out these experiments. Cells were transfected with plasmids encoding ATF3, FLAG-Ub, and MDM2, or with plasmids encoding FLAG-ATF3, HA-Ub, and MDM2, and then treated with 25 μm MG-132 for 4 h before being lysed in a buffer containing 2 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5 mm EDTA, 1% Nonidet P-40, 1% sodium deoxycholate, 0.025% SDS, and proteinase inhibitors. For cells transfected with FLAG-Ub, cell lysates were subjected to immunoprecipitation (IP) using an ATF3 antibody, and detection of ubiquitinated proteins was carried out using an anti-FLAG antibody. For cells transfected with HA-Ub, IP was performed using the anti-FLAG antibody, and ubiquitination was detected with an anti-HA antibody.
shRNA Knockdown and Lentiviral Infections
MDM2 knockdown was performed using a lentivector-based shRNA system (pSIH-H1 shRNA Cloning and Lentivector Expression system; System Biosciences) according to the manufacturer's recommendation. The MDM2-targeted sequences were 5′-AGG AAT TTA GAC AAC CTG A-3′ and 5′-AGC CAT TGC TTT TGA AGT T-3′, based on previous publications (21, 23). For negative controls, a luciferase-targeted sequence (5′-CTT ACG CTG AGT ACT TCG A-3′) was cloned into the lentivector.
Immunoblotting
This was performed as described previously (24). Antibodies for p53 (DO-1), ATF3 (C-19), and MDM2 (SMP-14 and N-20) were purchased from Santa Cruz Biotechnology. Antibodies for GFP and β-actin were obtained from Invitrogen and Sigma, respectively.
Real-time RT-PCR
Total RNA was prepared, reverse transcribed, and subjected to real-time PCR assays as described previously (25). The sequences of primers used for amplifying human and mouse ATF3, human GADPH, and mouse β-actin cDNA are available upon request.
RESULTS
ATF3 Levels Correlate Inversely with MDM2 Levels in the DNA Damage Response
DNA-damaging agents including doxorubicin (DOX), camptothecin (CPT), and actinomycin D (AD) can induce ATF3 expression (3). Although the three DNA-damaging agents similarly induced a transient increase of ATF3 mRNA level as expected (16), we noticed a difference in the kinetics of changes in ATF3 protein levels, i.e. the ATF3 protein levels were rapidly decreased 4–8 h after treatments with DOX and AD, but remained higher even 24 h after the CPT treatments (Fig. 1, A and B). ATF3 was even reduced to a level lower than the basal level 16 h after the DOX treatment. These results suggest that there is a mechanism(s) that regulates ATF3 protein degradation in the DNA damage response and such a mechanism could not be functional in cells treated with CPT. Although p53 can bind to ATF3 (22) and might regulate ATF3 stability, it is unlikely that p53 is involved in such a mechanism because p53 was similarly induced by the three DNA-damaging agents (Fig. 1A). However, MDM2, a p53 transcriptional target, was induced by DOX and AD, but its induction by CPT was negligible (Fig. 1A), consistent with an early report (26). Because MDM2 is a well characterized E3 ubiquitin ligase and can mediate ubiquitin-dependent degradation of many proteins other than p53 (27), there was a possibility that MDM2 induced by AD or DOX promotes ATF3 degradation whereas lack of MDM2 induction by CPT results in ATF3 stabilization. Interestingly, the time when ATF3 protein levels started to decrease coincided with the time when the MDM2 induction approached maximum (Fig. 1A). The inverse correlation between the ATF3 levels and the MDM2 levels thus suggests that MDM2 could regulate ATF3 levels in response to DNA damage.
FIGURE 1.

MDM2 regulates ATF3 levels in the DNA damage response. A and B, A549 cells were treated with 0.4 μg/ml DOX, 2 nm AD, or 1.5 μm CPT and lysed at the indicated times for immunoblotting (A). Densitometry was used to quantitate ATF3 levels (B). C, A549 cells were treated as in A. Total RNA was prepared and subjected to real-time RT-PCR to quantify ATF3 mRNA levels. D–F, mdm2-wild-type (p53−/−/mdm2+/+) or -deficient (p53−/−/mdm2−/−) MEF cells were treated with 0.4 μg/ml DOX for the indicated times and then subjected to immunoblotting (D) or real-time RT-PCR (F). ATF3 levels were quantitated by densitometry, and the results are shown in E. G and H, indicated cells were treated with 0.4 μg/ml DOX for 8 h. After removal of DOX, 100 μg/ml cycloheximide (CHX) was added into culture medium. Cells were harvested at the indicated times for immunoblotting. ATF3 levels were quantitated by densitometry, and the results are shown in H. I, p53−/−/mdm2+/+ or p53−/−/mdm2−/− MEF cells were treated with 0.4 μg/ml DOX for 24 and 48 h and stained with propidium iodide for flow cytometry analysis as described previously (3). Percentages of subG0/G1 cells were used to calculate the folds of increase in apoptosis rates after DOX treatments.
Deficiency of MDM2 Leads to Stabilization of ATF3 in the DNA Damage Response
To test this possibility, we determined whether deficiency of MDM2 in cells could affect ATF3 levels after DNA damage. Toward this end, we employed p53 and mdm2 double knock-out MEFs (p53−/−/mdm2−/−) because mdm2-knock-out mice are embryonic lethal (28, 29) and there are no mdm2−/− MEF cells available. We thus treated p53−/−/mdm2+/+ MEF and p53−/−/mdm2−/− MEF cells with DOX and subjected the cells to immunoblotting and real-time PCR assays. Both MEFs are deficient in p53, and we confirmed that the p53−/−/mdm2−/− MEF cells did not express MDM2 (supplemental Fig. S1). As expected, DOX induced a transient increase of ATF3 mRNA level in both mdm2-wild-type and -knock-out MEF cells (Fig. 1F), and accordingly, ATF3 protein levels were increased within 2 h of treatments (Fig. 1, D and E). Consistent with Fig. 1A and the transient nature of induction of ATF3 transcription (Fig. 1F), the ATF3 protein levels were rapidly decreased in the mdm2-wild-type cells. In striking contrast, after an initial decrease occurred during 2–4 h, the ATF3 protein levels remained unchanged even after 48 h in the p53−/−/mdm2−/− MEF cells, suggesting that MDM2 is required for ATF3 degradation in late stages of the DNA damage response. Indeed, cycloheximide chase experiments showed that the half-life of ATF3 induced by DOX was significantly extended in the p53−/−/mdm2−/− MEF cells (Fig. 1, G and H). These results thus strongly suggest that MDM2 could participate in regulating ATF3 stability in the DNA damage response. Interestingly, compared with the mdm2-wild-type MEFs, the p53−/−/mdm2−/− cells were more sensitive to DOX-induced apoptosis (Fig. 1I). Given that ATF3 is a well characterized proapoptotic protein (10), these results are consistent with a notion that MDM2-mediated destabilization of ATF3 could be involved in a mechanism to ensure cell survival after DNA damage has been repaired.
Knockdown of MDM2 Expression Increases the Basal ATF3 Level
ATF3 is a short lived protein. In LNCaP cells, ATF3 was partly ubiquitinated (supplemental Fig. S2A), suggesting that the basal ATF3 level could be maintained through ubiquitin-dependent degradation. Interestingly, it appeared that MDM2 could also regulate basal ATF3 expression because the ATF3 protein level was higher in p53−/−/mdm2−/− MEFs than mdm2-wild-type MEF cells (Fig. 2A), although the two cell lines expressed a similar level of ATF3 mRNA (Fig. 2B). To provide evidence supporting that MDM2 regulates basal ATF3 expression, we knocked down MDM2 expression in LNCaP cells using two independent MDM2-specific shRNA (shM2). LNCaP cells were chosen because they express MDM2 and ATF3 at detectable levels. As expected, the two MDM2 shRNA increased the p53 level (Fig. 2C) as a consequence of prevention of p53 degradation (18, 19). Importantly, in line with the notion that MDM2 could regulate ATF3 stability, the two MDM2 shRNA dramatically increased ATF3 levels (Fig. 2C, lanes 3–4 versus lanes 1–2). Although the ATF3 promoter contains an atypical p53-binding site (12), it is important to note that the increase of ATF3 levels by shM2 was unlikely caused by p53-mediated transactivation of the promoter because shM2–1 did not alter the ATF3 mRNA level (Fig. 2D) but still largely increased the ATF3 protein level (Fig. 2C, lane 3). Moreover, the same MDM2 shRNA increased the ATF3 protein level in p53-deficient HCT116 cells (Fig. 2E, lane 2). Also, as shown in Fig. 2A, the ATF3 level was elevated in p53−/−/mdm2−/− MEF cells. Therefore, these results indicate that MDM2 may regulate ATF3 protein levels by modulating its proteolysis.
FIGURE 2.

MDM2 regulates the basal level of ATF3. A and B, p53−/−/mdm2+/+and p53−/−/mdm2−/− MEF cells were harvested and subjected to immunoblotting (A) or real-time RT-PCR (B). C, LNCaP cells were infected with lentiviruses expressing MDM2-specific shRNA (shM2-1 or shM2-2) for 3 days and then lysed for immunoblotting. D, LNCaP cells expressing shM2-1 or shLuc were lysed for real-time RT-PCR assays. E, p53-deficient (p53−/−) HCT116 cells were infected with lentiviruses expressing shM2 or shLuc and subjected to immunoblotting as in C.
MDM2 Promotes Degradation of ATF3
To provide direct evidence supporting that MDM2 contributes to ATF3 proteolysis, we co-expressed ATF3 with increasing amounts of MDM2 in p53-deficient H1299 cells and determined protein levels using immunoblotting. Indeed, expression of MDM2 led to decrease of ATF3 levels in a dose-dependent manner (Fig. 3A). Such an effect was unlikely caused by possible effects of MDM2 on the activity of the CMV promoter that directed ATF3 transcription because expression of GFP, a protein expressed under control of the same promoter, remained at the same level (Fig. 3A). Interestingly, expression of C464A, a MDM2 mutant lacking the E3 ubiquitin ligase activity (17), failed to induce decrease in the ATF3 level (Fig. 3B, lane 3). Therefore, the ubiquitin ligase activity of MDM2 appeared to be required for MDM2-mediated regulation of ATF3 degradation. We also measured the ATF3 half-life in the absence or the presence of MDM2 using cycloheximide chase assays. The results showed that ATF3 was destabilized by MDM2 but not the C464A mutant (Fig. 3, C and D). Taken together, these results demonstrate that MDM2 mediates degradation of ATF3.
FIGURE 3.

MDM2 promotes ATF3 degradation. A, H1299 cells were transfected with ATF3, GFP, and increasing amounts of MDM2 as indicated. Cells were lysed, and GFP expression levels were quantitated using a fluorescence spectrophotometer to normalize transfection efficiencies. Normalized cell lysates were then subjected to immunoblotting. B, H1299 cells were transfected as indicated and subjected to immunoblotting as in A. C and D, H1299 cells were transfected and treated with 100 μg/ml cycloheximide for different time. ATF3 levels were quantitated by densitometry, and the results are shown in D.
ATF3 Binds Directly to MDM2
MDM2 is an E3 ubiquitin ligase and might promote ATF3 proteolysis by binding to this protein and mediate its ubiquitination. To test this possibility, we first determined whether ATF3 could interact with MDM2. We thus co-expressed MDM2 and ATF3 in H1299 cells and performed co-IP assays. The results show that the ATF3 antibody, but not IgG, could precipitate MDM2 only in a condition when exogenous ATF3 protein was present (Fig. 4A, lane 5 versus lane 3), suggesting that ATF3 bound to MDM2 in the cells. Although both ATF3 and MDM2 can bind to p53 (3, 30), the precipitation of MDM2 by the ATF3 antibody was unlikely mediated by p53 because H1299 cells used in the experiments are p53-deficient. Interestingly, the endogenous MDM2 protein could be co-precipitated with the endogenous ATF3 protein by the ATF3 antibody in LNCaP cells (Fig. 4B, lane 3), indicating that the ATF3-MDM2 interaction occurred indeed in vivo. Of note, the endogenous p53 protein was also precipitated by the ATF3 antibody, consistent with our previous finding that ATF3 is a p53-binding partner (3). To demonstrate that MDM2 could directly bind to ATF3, we incubated purified recombinant ATF3 protein with purified GST-MDM2 protein (3) and subjected the incubated proteins to GST pulldown assays. The glutathione-agarose pulled down purified ATF3 protein only after it was incubated with GST-MDM2 (Fig. 4C, lane 4 versus lane 3). These results thus demonstrate that ATF3 can directly bind to MDM2.
FIGURE 4.
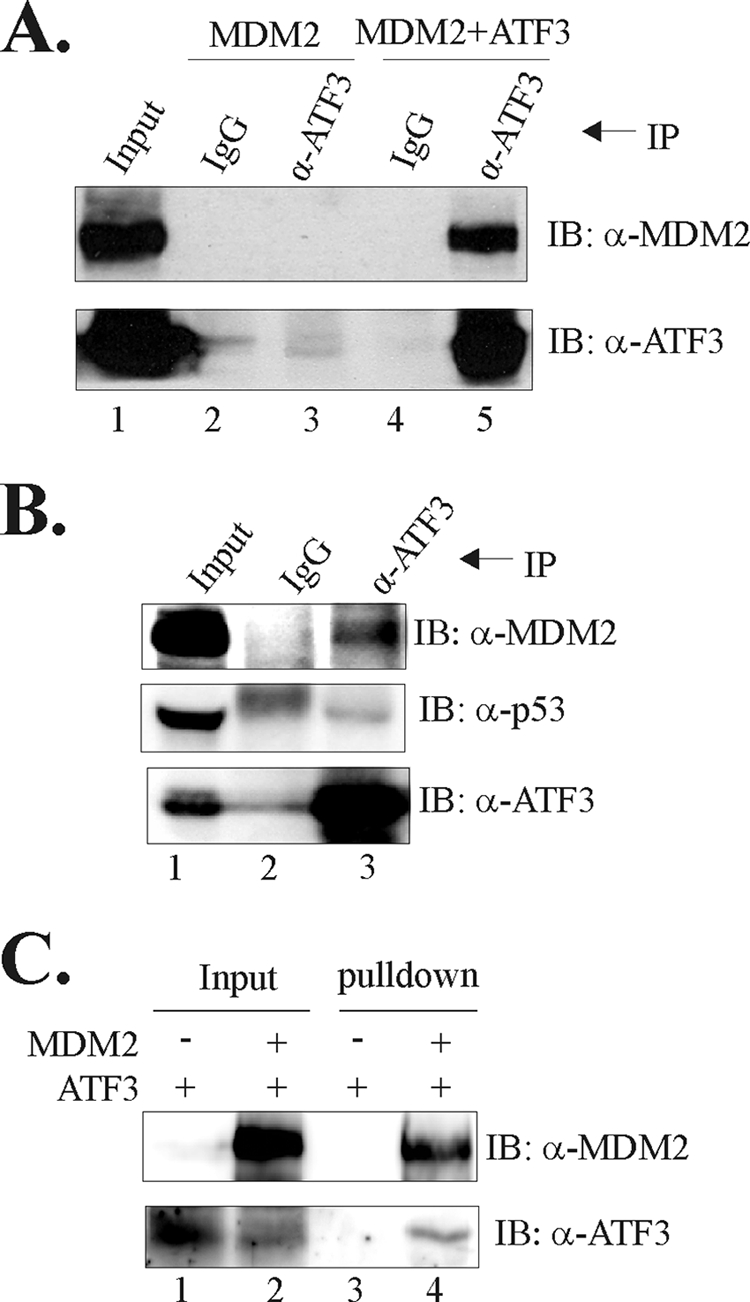
MDM2 binds directly to ATF3. A, H1299 cells were transfected as indicated and lysed for IP assays using the ATF3 antibody (α-ATF3) or IgG. Precipitated proteins were subjected to immunoblotting (IB) to detect MDM2 and ATF3. B, LNCaP cell lysates were incubated with the ATF3 antibody or IgG at 4 °C overnight. Immunoprecipitated proteins were then subjected to immunoblotting. C, 100 ng of purified ATF3 protein was incubated with 200 ng of purified GST-MDM2 protein or BSA at 4 °C overnight. Protein complexes were pulled down by glutathione-agarose and subjected to immunoblotting.
ATF3 Is a Bona Fide Substrate for MDM2
Because MDM2 is a well characterized E3 ubiquitin ligase, we asked whether ATF3 could be ubiquitinated by MDM2. We thus incubated purified ATF3 protein with MDM2, E1, E2, ubiquitin, and other required ubiquitination reaction components (3) to determine whether ATF3 could be ubiquitinated by MDM2 in vitro. In the presence of all of the required ubiquitination reaction components, MDM2 catalyzed the addition of ubiquitin moieties to ATF3, resulting in the appearance of an array of slower migratory bands (Fig. 5A, lane 6), whereas omission of E1, E2, MDM2, or ubiquitin from the reactions abolished ATF3 ubiquitination (Fig. 5A, lane 2-5). These results thus provided direct evidence demonstrating that MDM2 is indeed an E3 ubiquitin ligase for ATF3. ATF3 contains 16 lysine residues, and 9 of them are clustered in the leucine zipper (Zip) domain that is required for binding to DNA and proteins (e.g. p53, E6) (3, 4). Interestingly, MDM2 failed to catalyze ubiquitin modification of an ATF3 mutant (Δ102–139) lacking this domain (Fig. 5B, lane 3 versus lane 4) although deletion of the Zip domain did not abolish the ATF3-MDM2 interaction (Fig. 5C, lane 6). Therefore, it is very likely that MDM2 catalyzed the addition of ubiquitin moieties to the 9 lysine residues in the Zip domain. We also determined whether MDM2 could promote ATF3 ubiquitination in vivo. We thus co-expressed ATF3, MDM2, and ubiquitin in PC3 and H1299 cells. Under both experimental conditions, co-expression of MDM2 promoted modification of ATF3 by ubiquitin (Fig. 5D and 5E, lane 2). The low level of ATF3 ubiquitination in the absence of transfected MDM2 (Fig. 5D, lane 4; Fig. 5E, lane 5) could due to the endogenous MDM2, or an unidentified ubiquitin ligase(s). As expected, the C464A mutant failed to induce ATF3 ubiquitination (supplemental Fig. S2B). We concluded that MDM2 could serve as a bona fide E3 ubiquitin ligase for ATF3 thereby promoting proteolysis of the latter protein.
FIGURE 5.

MDM2 is a bona fide E3 ubiquitin ligase for ATF3. A, purified ATF3 protein was incubated with E1, E2, MDM2, and/or ubiquitin as indicated and then subjected to immunoblotting using the ATF3 antibody. B, purified ATF3 or Δ102–139 protein was subjected to in vitro ubiquitination assay as in A. C, FLAG-tagged ATF3 or Δ102–139 protein was expressed with MDM2 in H1299 cells as indicated. Cell lysates were immunoprecipitated with the anti-FLAG antibody. D, PC3 cells were transfected with ATF3, MDM2, and/or FLAG-Ub as indicated. Cell lysates were immunoprecipitated with the ATF3 antibody and subjected to immunoblotting (IB) for ubiquitinated proteins using the FLAG antibody. E, H1299 cells were transfected with FLAG-ATF3, MDM2, and/or HA-Ub as indicated, followed by IP assays using the FLAG antibody. Ubiquitinated proteins were detected with an anti-HA antibody.
ATF3 via Its Basic Region Binds to the RING Finger of MDM2
MDM2 contains several conserved domains that can mediate protein-protein interactions (27). To determine the region(s) responsible for MDM2 binding to ATF3, we immobilized truncated MDM2 proteins onto glutathione-agarose, incubated them with in vitro translated ATF3 protein, and performed GST pulldown assays. Interestingly, the MDM2 C-terminal fragments including the RING finger region (amino acids 425–491), but not the N-terminal fragments (amino acids 1–150 and 1–301), pulled down the ATF3 protein (Fig. 6A, lanes 4–6 versus lanes 2–3), indicating that ATF3 bound to the RING finger domain of MDM2. On the other hand, because the ATF3 Zip domain was dispensable for MDM2 binding (Fig. 5C), we performed GST pulldown assays to characterize ATF3 region(s) required for the ATF3-MDM2 interaction. In line with our previous results that ATF3 interacted with MDM2 (Fig. 4), the full-length ATF3 protein pulled down in vitro-translated MDM2 protein (Fig. 6B, lane 3). This interaction required the ATF3 C terminus (81–181) but not the N terminus (1–80) (Fig. 6B, lane 5 versus lane 4). However, a C-terminal fragment (102–181) containing the Zip domain failed to pull down MDM2 (Fig. 6A, lane 9), consistent with Fig. 5B showing that the Zip domain was dispensable for ATF3 binding to MDM2. These results suggest that MDM2 could bind to the basic region (amino acids 80–101), which was previously shown to mediate the ATF3-Smad3 interaction (2). Indeed, deletion of the Basic region (Δ80–100) diminished the interaction between ATF3 and MDM2 (Fig. 6B, lane 10). Taken together, our results indicate that ATF3 binds to the RING finger of MDM2 via its Basic region.
FIGURE 6.

ATF3 via its basic region binds to the RING finger of MDM2. A, GST-MDM2 fusion proteins were immobilized on glutathione-agarose and incubated with in vitro translated ATF3 protein. After extensive washes, bound proteins were eluted, subjected to SDS-PAGE, and detected by immunoblotting. The lower panel shows Coomassie Blue staining of the fusion proteins. B, GST-ATF3 fusion proteins were immobilized and incubated with in vitro translated MDM2 as in A. The bound MDM2 protein was detected by immunoblotting. The lower panel shows Ponceau S staining of the blot.
ATF3-MDM2 Interaction Is Indispensable for MDM2-mediated Degradation of ATF3
Finally, we determined whether ATF3 degradation mediated by MDM2 was indeed caused by ATF3 binding to the E3 ubiquitin ligase. We first performed co-IP experiments to confirm that the ATF3 Basic region is required for the ATF3-MDM2 interaction in vivo. Because the Basic region contains a putative nuclear localization signal (NLS), deletion of this region resulted in cytoplasmic localization of ATF3 (supplemental Fig. S3). We therefore fused a SV40 NLS (3) with the Δ80–100 mutant to drive the latter protein to nuclei (supplemental Fig. S3). Although fusion with this exogenous NLS did not affect the interaction of MDM2 with wild-type ATF3 (Fig. 7A, lane 2), deletion of the Basic region indeed abolished the ATF3-MDM2 interaction (Fig. 7A, lane 3 versus lane 2). We thus co-expressed the NLS-Δ80–100 protein with MDM2 in H1299 cells to determine whether this mutant protein could be resistant to MDM2-mediated degradation. Indeed, MDM2 decreased the level of wild-type ATF3 but not the mutant deficient in MDM2 binding (Fig. 7B, lane 2 versus lane 4). Therefore, we demonstrate that MDM2 mediated ATF3 degradation through binding to the latter. Of note, we found that the MDM2 C464A mutant bound to ATF3 (supplemental Fig. S4), and thus the inability of this mutant protein to degrade ATF3 (Fig. 3B) was indeed due to its lack of E3 ubiquitin ligase activity.
FIGURE 7.

MDM2-mediated ATF3 degradation requires the binding of MDM2 to ATF3. A, H1299 cells were transfected with the indicated plasmids and subjected to IP using the ATF3 antibody. B, ATF3 or Δ80–100 was expressed with or without MDM2 and subjected to immunoblotting as in Fig. 3A. C, schematic representation of negative-feedback loops for tight controls of p53 activity in cellular stress responses is shown.
DISCUSSION
Although rapid induction of ATF3 expression is a marked characteristic of cellular responses to a wide range of stresses, neither the consequences of this induction nor the mechanisms regulating this induction are well understood. ATF3 is an immediate early gene, and it is generally believed that ATF3 induction is achieved mainly through transcriptional activation (1). In this study, we presented the first evidence demonstrating that ATF3 expression can also be regulated at posttranslational levels. In this regard, MDM2 serves as a bona fide E3 ubiquitin ligase for ATF3 and promotes ATF3 degradation. As a consequence, ATF3 protein levels were rapidly decreased in late stages of DNA damage response when MDM2 was induced. Because p53 is commonly activated by cellular stresses and can consequently induce MDM2 expression (31), the significance of our findings extends beyond the identification of a novel mechanism for regulation of the DNA damage response. Moreover, because MDM2 itself can be posttranslationally modified (e.g. phosphorylation) and degraded under certain circumstances such as severe DNA damage (32), our results suggest that down-regulation of MDM2 expression under such conditions could lead to a rapid increase of ATF3 expression. Indeed, although severe DNA damage induced by a high concentration (4 μg/ml) of DOX repressed ATF3 transcription in early hours (1–8 h) after the DNA-damaging treatment (supplemental Fig. S5C), a likely consequence of general transcription inhibition caused by widespread DNA damage (33), the ATF3 protein level was rapidly increased (supplemental Fig. S5, A, lanes 2–5, versus lane 1, and B). Interestingly, such a transcription-independent induction of ATF3 expression was accompanied by a decrease in the MDM2 level as expected (supplemental Fig. S5A, lanes 2–5 versus lane 1). Therefore, our finding that ATF3 is a substrate for MDM2 suggests a novel role of MDM2 that may play in cellular stress response. This role is separate from its regulation of p53 activity and thus adds to a growing list of novel MDM2 functions that are independent of p53 (27).
MDM2 is a major p53 repressor. Upon DNA damage, activated p53 induces expression of MDM2, which in turn represses p53 and forms a negative-feedback loop for a tight control of p53 activity (34). We have previously demonstrated that ATF3 interacts with p53 and can prevent p53 from MDM2-mediated ubiquitination and degradation in response to DNA damage (3). Considering that ATF3 could also be a transcriptional target of p53 (12), our current findings that MDM2 promotes degradation of ATF3 thus reveal an additional negative-feedback mechanism for fine-tuning p53 activity thereby ensuring cells to mount appropriate responses to DNA damage (Fig. 7C). Interestingly, whereas ATF3 binds to MDM2 at the RING finger domain that is distinct from the p53-binding region, MDM2 and p53 bind to ATF3 at different but adjacent regions. Therefore, it is likely that ATF3 forms a complex with p53 and MDM2. Because MDM2 can repress p53 transcriptional activity (30), it would be interesting to investigate whether the formation of the ATF3·MDM2·p53 complex could alter expression of ATF3 target genes or confer ATF3 with an ability to regulate expression of genes lacking the ATF/CREB motif. On the other hand, because ATF3 is a bona fide substrate for MDM2, ATF3 could directly inhibit the MDM2 E3 ubiquitin ligase activity through substrate competition and consequently prevent MDM2 from promoting ubiquitination and degradation of its substrates including p53 (3).
Interestingly, MDM2 binds to ATF3 at the Basic region but catalyzes the addition of ubiquitin moieties to the adjacent Zip domain. It is not uncommon that MDM2 binds to its substrate in one region but ubiquitinate lysine residues in another region. Indeed, p53 is mainly ubiquitinated at the C terminus but bound by MDM2 at the N terminus (35, 36). It is hypothesized that the binding of MDM2 to its substrates alters their conformations, thereby allowing the RING finger to access lysine residues far removed from binding regions for ubiquitination (37). Interestingly, the 9 lysine residues residing in the Zip domain are highly conserved across species, suggesting that MDM2-mediated ATF3 ubiquitination could be an evolutionally conserved mechanism for regulating stress responses. In summary, we have identified ATF3 as a novel substrate for MDM2. MDM2 interacts with ATF3 and promotes its ubiquitination and degradation and thereby contributes to regulation of ATF3 in cellular stress responses.
Supplementary Material
Acknowledgments
We thank Drs. Tsonwin Hai, Guillermina Lozano, Karen Vousden, Bert Vogelstein, and Toshiaki Suzuki for providing reagents and materials.
This work was supported in part by Department of Defense Grant PC061106 (W81XWH-07-1-0095) (to C. Y.) and National Institutes of Health/NCI Grants CA127724, CA095441, and CA129828 (to H. L.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
- ATF3
- activating transcription factor 3
- AD
- actinomycin D
- CPT
- camptothecin
- CREB
- cAMP-responsive element-binding protein
- DOX
- doxorubicin
- IP
- immunoprecipitation
- MDM2
- mouse double minute 2
- MEF
- mouse embryonic fibroblast
- NLS
- nuclear localization signal
- RT
- reverse transcription
- shM2
- MDM2-specific shRNA
- Ub
- ubiquitin
- Zip domain
- leucine zipper domain.
REFERENCES
- 1.Hai T., Wolfgang C. D., Marsee D. K., Allen A. E., Sivaprasad U. (1999) Gene Expression 7, 321–335 [PMC free article] [PubMed] [Google Scholar]
- 2.Kang Y., Chen C. R., Massagué J. (2003) Mol. Cell 11, 915–926 [DOI] [PubMed] [Google Scholar]
- 3.Yan C., Lu D., Hai T., Boyd D. D. (2005) EMBO J. 24, 2425–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H., Mo P., Ren S., Yan C. (2010) J. Biol. Chem. 285, 13201–13210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilchrist M., Thorsson V., Li B., Rust A. G., Korb M., Roach J. C., Kennedy K., Hai T., Bolouri H., Aderem A. (2006) Nature 441, 173–178 [DOI] [PubMed] [Google Scholar]
- 6.Jiang H.-Y., Wek S. A., McGrath B. C., Lu D., Hai T., Harding H. P., Wang X., Ron D., Cavener D. R., Wek R. C. (2004) Mol. Cell. Biol. 24, 1365–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu D., Wolfgang C. D., Hai T. (2006) J. Biol. Chem. 281, 10473–10481 [DOI] [PubMed] [Google Scholar]
- 8.Yan C., Boyd D. D. (2006) Cell Cycle 5, 926–929 [DOI] [PubMed] [Google Scholar]
- 9.Kool J., Hamdi M., Cornelissen-Steijger P., van der Eb A. J., Terleth C., van Dam H. (2003) Oncogene 22, 4235–4242 [DOI] [PubMed] [Google Scholar]
- 10.Hartman M. G., Lu D., Kim M. L., Kociba G. J., Shukri T., Buteau J., Wang X., Frankel W. L., Guttridge D., Prentki M., Grey S. T., Ron D., Hai T. (2004) Mol. Cell. Biol. 24, 5721–5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu D., Chen J., Hai T. (2007) Biochem. J. 401, 559–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang C., Gao C., Kawauchi J., Hashimoto Y., Tsuchida N., Kitajima S. (2002) Biochem. Biophys. Res. Commun. 297, 1302–1310 [DOI] [PubMed] [Google Scholar]
- 13.Fan F., Jin S., Amundson S. A., Tong T., Fan W., Zhao H., Zhu X., Mazzacurati L., Li X., Petrik K. L., Fornace A. J., Jr., Rajasekaran B., Zhan Q. (2002) Oncogene 21, 7488–7496 [DOI] [PubMed] [Google Scholar]
- 14.Amundson S. A., Bittner M., Chen Y., Trent J., Meltzer P., Fornace A. J., Jr. (1999) Oncogene 18, 3666–3672 [DOI] [PubMed] [Google Scholar]
- 15.Robles A. I., Bemmels N. A., Foraker A. B., Harris C. C. (2001) Cancer Res. 61, 6660–6664 [PubMed] [Google Scholar]
- 16.Wolfgang C. D., Liang G., Okamoto Y., Allen A. E., Hai T. (2000) J. Biol. Chem. 275, 16865–16870 [DOI] [PubMed] [Google Scholar]
- 17.Honda R., Tanaka H., Yasuda H. (1997) FEBS Lett. 420, 25–27 [DOI] [PubMed] [Google Scholar]
- 18.Haupt Y., Maya R., Kazaz A., Oren M. (1997) Nature 387, 296–299 [DOI] [PubMed] [Google Scholar]
- 19.Kubbutat M. H., Jones S. N., Vousden K. H. (1997) Nature 387, 299–303 [DOI] [PubMed] [Google Scholar]
- 20.Freedman D. A., Levine A. J. (1999) Cancer Res. 59, 1–7 [PubMed] [Google Scholar]
- 21.Jin Y., Lee H., Zeng S. X., Dai M. S., Lu H. (2003) EMBO J. 22, 6365–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan C., Wang H., Boyd D. D. (2002) J. Biol. Chem. 277, 10804–10812 [DOI] [PubMed] [Google Scholar]
- 23.Dornan D., Wertz I., Shimizu H., Arnott D., Frantz G. D., Dowd P., O'Rourke K., Koeppen H., Dixit V. M. (2004) Nature 429, 86–92 [DOI] [PubMed] [Google Scholar]
- 24.Yan C., Wang H., Toh Y., Boyd D. D. (2003) J. Biol. Chem. 278, 2309–2316 [DOI] [PubMed] [Google Scholar]
- 25.Yan C., Boyd D. D. (2006) Mol. Cell. Biol. 26, 6357–6371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashcroft M., Taya Y., Vousden K. H. (2000) Mol. Cell. Biol. 20, 3224–3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Z., Zhang R. (2005) Curr. Cancer Drug Targets 5, 9–20 [DOI] [PubMed] [Google Scholar]
- 28.Jones S. N., Roe A. E., Donehower L. A., Bradley A. (1995) Nature 378, 206–208 [DOI] [PubMed] [Google Scholar]
- 29.Montes de Oca Luna R., Wagner D. S., Lozano G. (1995) Nature 378, 203–206 [DOI] [PubMed] [Google Scholar]
- 30.Momand J., Zambetti G. P., Olson D. C., George D., Levine A. J. (1992) Cell 69, 1237–1245 [DOI] [PubMed] [Google Scholar]
- 31.Vogelstein B., Lane D., Levine A. J. (2000) Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 32.Kranz D., Dohmesen C., Dobbelstein M. (2008) J. Cell Biol. 182, 197–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ljungman M., Lane D. P. (2004) Nat. Rev. Cancer 4, 727–737 [DOI] [PubMed] [Google Scholar]
- 34.Michael D., Oren M. (2003) Semin. Cancer Biol. 13, 49–58 [DOI] [PubMed] [Google Scholar]
- 35.Nakamura S., Roth J. A., Mukhopadhyay T. (2000) Mol. Cell. Biol. 20, 9391–9398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez M. S., Desterro J. M., Lain S., Lane D. P., Hay R. T. (2000) Mol. Cell. Biol. 20, 8458–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wallace M., Worrall E., Pettersson S., Hupp T. R., Ball K. L.(2006) Mol. Cell 23, 251–263 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.