

Abstract
Glycolipid glycosyltransferases catalyze the stepwise transfer of monosaccharides from sugar nucleotides to proper glycolipid acceptors. They are Golgi resident proteins that colocalize functionally in the organelle, but their intimate relationships are not known. Here, we show that the sequentially acting UDP-GalNAc:lactosylceramide/GM3/GD3 β-1,4-N-acetyl-galactosaminyltransferase and the UDP-Gal:GA2/GM2/GD2 β-1,3-galactosyltransferase associate physically in the distal Golgi. Immunoprecipitation of the respective epitope-tagged versions expressed in transfected CHO-K1 cells resulted in their mutual coimmunoprecipitation. The immunocomplexes efficiently catalyze the two transfer steps leading to the synthesis of GM1 from exogenous GM3 in the presence of UDP-GalNAc and UDP-Gal. The N-terminal domains (cytosolic tail, transmembrane domain, and few amino acids of the stem region) of both enzymes are involved in the interaction because (i) they reproduce the coimmunoprecipitation behavior of the full-length enzymes, (ii) they compete with the full-length counterpart in both coimmunoprecipitation and GM1 synthesis experiments, and (iii) fused to the cyan and yellow fluorescent proteins, they localize these proteins to the Golgi membranes in an association close enough as to allow fluorescence resonance energy transfer between them. We suggest that these associations may improve the efficiency of glycolipid synthesis by channeling the intermediates from the position of product to the position of acceptor along the transfer steps.
The synthesis of higher glycolipid oligosaccharides is carried out in the lumen of the Golgi cisternae by a complex system of membrane-bound glycolipid acceptors, glycosyltransferases, and sugar nucleotide transporters (1). Glycolipid glycosyltransferases are Golgi resident type II membrane proteins (2) that transfer monosaccharide units from the cognate sugar nucleotide donor to glycolipid acceptors produced by the transferases acting in the preceding steps in the pathway of synthesis. Early studies of the topology of the synthesis indicated that glycolipid glycosyltransferases concentrate along the Golgi stack following gradients reminiscent of the order in which they act in the pathway of synthesis (3). However, later work found transferases acting beyond the synthesis of glucosylceramide functionally coupled in the trans-Golgi network of retina cells (4). More recently, subfractionation studies positioned all of these enzymes to the late Golgi compartment from rat liver cells (5). This topological organization raises questions about the way in which glycolipid glycosyltransferases relate between them to support coupling of successive transfer steps. The present study addresses this question for the distal Golgi located UDP-GalNAc:lactosylceramide/GM3/GD3 β-1,4-N-acetylgalactosaminyltransferase (GalNAc-T) and UDP-Gal:GA2/GM2/GD2 β-1,3-galactosyltransferase (Gal-T2), which act in succession in the synthesis of complex glycolipids and gangliosides (6). Coimmunoprecipitation experiments of epitope-tagged versions of these enzymes expressed in CHO-K1 cells disclosed a physical interaction between them in which the N-terminal domains were involved. Biochemical experiments showed that immunocomplexes were functional in vitro, and in vivo determination of fluorescence resonance energy transfer (FRET) between cyan and yellow fluorescent proteins fused to the N-terminal domains of the transferases showed that these domains associated closely (1–10 nm) in the Golgi membranes, so that energy transfer between the fluorophores was possible.
Materials and Methods
Cell Culture and Transfection.
CHO-K1 cell culture, liposome transfection (Lipofectamine, Life Technologies, Grand Island, NY), and selection of stable transfectants was as indicated (7). Clone 3 constitutively expresses moderate levels of the c-myc tagged version of GalNAc-T (7). Clone IGT2 is a stable mouse Gal-T2-HA transfectant that expresses the enzyme under the control of an ecdysone-inducible promoter. When used, levels of expression were regulated by inducing with 1 μM ponasterone during 24 h (8). To obtain a membrane fraction from these cells containing Golgi membranes, cell homogenates in 10 mM Tris·HCl, pH 7.2/0.25 M sucrose were freed from debris by centrifugation at 2,000 × g for 3 min, and the membrane fraction was collected by pelleting the supernatant at 13,500 × g for 10 min.
Expression Plasmids.
The human GalNAc-T cDNA (9) was subcloned into the eukaryotic expression vector pCI-neo (Promega), and the sequence coding for the c-myc epitope was introduced at the C terminus before the stop codon (7). The mouse full-length Gal-T2 bearing a 9-aa hemagglutinin (HA) epitope at the C terminus was obtained by digesting pCEFL-Gal-T2-HA (8) with EcoRI/NotI and subcloning into the pCI-neo vector (pCI-Gal-T2-HA). The cDNA coding for amino acids 1–52 of Gal-T2 tagged with the HA epitope was amplified by PCR under standard conditions using as template the plasmid pCEFL-Gal-T21–52-HA-GFP (8). A XhoI site was introduced before the ATG codon at the 5′ end primer (5′-ttactcgagATGcccctcagcctcttcc-3′) and an SalI site was introduced in the 3′ end primer after the stop codon (5′-accgtcgactgttatgcgtaatccggtacgtc-3′). The PCR product was digested with XhoI and SalI and subcloned into the expression vector pCI-neo (pCI-Gal-T21–52-HA).
Constructs containing the cDNA coding for the N-terminal domains (cytosolic tail, transmembrane domain, and few amino acids of the stem region) of GalNAc-T, Gal-T2, and α-mannosidase II (ManII) fused to the N terminus of the enhanced cyan fluorescent protein (ECFP) or of the enhanced yellow fluorescent protein (EYFP) were obtained by subcloning the corresponding cDNA fragments into the plasmids pECFP-N1 and pEYFP-N1 (CLONTECH), which contain coding sequences for the cyan and the yellow fluorescent proteins, respectively. To generate GalNAc-T1–27-ECFP and GalNAc-T1–27-EYFP, a XhoI/SmaI fragment obtained by digestion of pCI-GalNAc-T-c-myc construct (7) was subcloned in the same restriction sites of the plasmids containing the fluorescent proteins.
Gal-T21–52-HA-ECFP and Gal-T21–52-HA-EYFP were prepared by PCR amplification of the sequence coding for the N-terminal domain (amino acids 1–52) of the Gal-T2. The 5′ end primer and template were the same as used to prepare pCI-Gal-T21–52HA except that the stop codon at 3′ end primer was removed (5′-accgtcgactgtgcgtaatccggtacgtc-3′). The PCR product digested with XhoI/SalI was subcloned into the cyan and yellow fluorescent protein expression vectors. Finally, to engineer ManII1–76-ECFP and ManII1–76-EYFP constructs, a SacII fragment coding for the first 76 amino acids was obtained by digesting the plasmid pBsK-ManII, which carries the full-length mouse mannosidase II cDNA (kindly provided by K. Moremen, University of Georgia). The SacII DNA fragment was subcloned in the same restriction site of plasmids coding for the fluorescent proteins. pEYFP-Golgi, encoding amino acids 1–81 of human glycoprotein GlcNAc β 1→4 Gal-T fused to the NH2 terminus of EYFP, was from CLONTECH.
Immunoprecipitation.
Cells (200 μg of protein) were lysed during 60 min on ice with 500 μl of lysis buffer (50 mM Tris·HCl, pH 7.2/1.0% Triton X-100/300 mM NaCl/3 μg/ml leupeptin/1 mM PMSF/3 μg/ml aprotinin/2 μM pestatin/1 mM EDTA/0.05% sodium azide). Lysates were preabsorbed with protein G-Sepharose beads (Amersham Pharmacia, 75% suspension washed with lysis buffer before use) for 60 min on ice. Aliquots of supernatants were incubated overnight on a rotating wheel at 4°C with specific monoclonal antibodies as will be indicated in each case. Dilutions were as follows: anti-HA (12CA5, Babco, Richmond CA), 1:150; anti-c-myc (CRL1729 immunopurified supernatant), 1:50; anti-green fluorescent protein (GFP, mixture of clones 7.1 and 13.1, Boehringer Mannheim), 1:200; polyclonal antibody anti-ManII (from K. Moremen), 1:50 and with 100 μl of protein G-Sepharose beads (50% slurry). Immunocomplexes were pelleted by centrifugation at 2,500 × g for 10 s and then washed five times at 4°C with lysis buffer and three times with 150 mM phosphate buffer, pH 7.2, 10 mM EDTA for immunoblotting or with 100 mM HCl cacodylate, pH 7.2, for glycosyltransferase activity assays.
Western Blotting.
The membrane fraction (100 μg of protein) or immunocomplexes obtained as indicated above were taken in Laemmli sample buffer (10) and subjected to SDS/PAGE as described (7). For immunoblotting, the following antibodies were used: polyclonal antibodies anti-HA (1:800), anti-c-myc (1:400, Babco), anti-ManII (1:400), and monoclonal antibody anti-GFP (1:1,000) followed by horseradish peroxidase-protein A (1:85,000, Sigma). For monoclonal antibodies, a previous step of rabbit anti-mouse IgG was run. All incubations were carried out in 0.25% polyvinylpirrolidone/0.25% BSA/0.05% Tween 20 in TBS (TTBS) for 1 h at room temperature, followed by three 15-min washes with TTBS. Blots were processed by using the Renaissance Chemiluminescence Reagent Plus (NEN) and exposed to Kodak BioMax MS x-ray film. Resulting bands were quantified by densitometry (SCION IMAGE 1 software, National Institutes of Health).
Microscopic Measurement of FRET in Living Cells.
Cells stably expressing N-terminal domains of Golgi proteins fused to ECFP or EYFP were grown in DMEM on coverslips adhered with Sylgard 182 silicone elastomer (Dow-Corning) to the bottom of perforated culture dishes. For FRET determinations, cells washed three times with PBS were observed in a Zeiss Axiovert 135M inverted microscope equipped with a 63×, 1.4 NA oil immersion objective while kept at 37°C and in an atmosphere of 5% CO2 in air. Cells were photographed with a Princeton Instrument Micromax CCD camera controlled with Metamorph 3.0 Imaging System (Universal Imaging, Dowsington, PA). The following filter sets (Omega Optical, Brattleboro, VT) were used: ECFP, filter set XF 114 [excitation (ex) 440DF20 nm, emission (em) 480DF30 nm, and dichroic mirror (dm) 455DRLPnm]; EYFP, filter set XF104 (ex 500RDF25 nm, em 545RDF35 nm, and a dm of 525DRLPnm); FRET, filter set XF88 (ex 440DF20 nm, em 535DF35 nm, and a dm of 455DRLPnm). Images were processed with Adobe PHOTOSHOP 5.0.
For quantitative FRET (FRETN), the three-filter set procedure was followed to correct for possible spillover due to overlapping of fluorochrome spectra and for dependence of FRET on the concentration of both donor and acceptor fluorophores (11, 12). The quantification procedure was applied in cells expressing the ECFP constructs alone, the EYFP constructs alone, and the ECFP plus EYFP constructs together. Single cells were selected, and a constant fluorescence intensity threshold was established for each filter set. The integrated morphometric analysis function of the METAMORPH 3.0 software was used to calculate the total gray value within the area above the threshold established for each filter set. Data were used for FRETN calculations as described (11).
Results
GM3 to GM1 Transfer Steps Are Functionally Coupled in CHO-K1 Cell Golgi Membranes.
CHO-K1 cells synthesize mainly the simple ganglioside GM3 due to the lack of GalNAc-T activity. However, they express GM2, GM1, and GD1a upon transfection with GalNAc-T (double arrow in Fig. 1A), which provides the necessary GM2 substrate to the endogenous, active Gal-T2 and Sial-T4 present in these cells (13). Using a clone of CHO-K1 cells that stably express moderate levels of c-myc tagged GalNAc-T (clone 3) (7), we first examined whether the transfer steps leading to the synthesis of GM1 from GM3 colocalize in the Golgi membranes of these cells. The membrane fraction of clone 3 was incubated with CMP-[3H]NeuAc, and the pattern of radioactive endogenous gangliosides was analyzed. Fig. 1B (left lane) shows that [3H]NeuAc was mostly incorporated into endogenous lactosylceramide to form [3H]GM3. A minor incorporation also occurred into endogenous GM1 to form [3H]GD1a. A second incubation of the membranes carrying [3H]GM3 with the nonradioactive sugar nucleotides necessary for conversion of GM3 into GM1 resulted in near 70% conversion of [3H]GM3 into [3H]GM2 and of near 60% conversion of [3H]GM2 into [3H]GM1 (Fig. 1B, right lane). Because standard conditions for vesicular coupling between compartments were not provided in these experiments, the simplest interpretation of the result is that GalNAc-T and Gal-T2 colocalize functionally in the Golgi membranes, allowing the product of GalNAc-T (GM2) to couple with Gal-T2 as substrate for conversion into GM1.
Figure 1.

Functional coupling of GM3, GM2, and GM1 synthesis in CHO-K1 cells. (A) LacCer is mainly used for synthesis of GM3, the precursor of “a” series gangliosides, but these are not synthesized due to the lack of GalNAc-T activity in these cells. Transfection of GalNAc-T (double arrow) provides the substrate GM2 to the endogenous Gal-T2 and Sial-T4. (B) A membrane fraction containing Golgi membranes, obtained from clone 3 as indicated in Materials and Methods, was incubated in a first step with 10 μM CMP-[3H]NeuAc (7,500 cpm/pmol) in 20 μl of 15 mM MnCl2/25 mM Hepes-KOH buffer, pH 7.0/25 mM KCl/2.5 mM Mg acetate. After 1 h at 37°C, membranes were washed by pelleting in 10 mM Tris·HCl buffer, pH 7.2/0.25 M sucrose and suspended in 50 μl of the same buffer. Two 20-μl aliquots were incubated in a second step for 1 h at 37°C, one without any addition (left lane) and the other supplemented with 10 μM each of UDP-GalNAc and UDP-Gal (right lane). Lipids were extracted, subjected to HPTLC, and visualized by phosphorimaging. Positions of cochromatographed standard gangliosides are indicated at right.
GalNAc-T Specifically Coimmunoprecipitates Gal-T2 and Vice Versa.
The experiment of Fig. 1 raises the question of whether a physical association between the participating transferases may facilitate the coupling of the product of one transfer step with the transferase catalyzing the next one. To examine whether GalNAc-T interacts with Gal-T2, clone 3 was transiently transfected with pCI-Gal-T2-HA. Lysates of the double transfectants were immunoprecipitated with either anti-c-myc (Fig. 2A, lanes 1 and 5) or anti-HA (Fig. 2A, lanes 2 and 4) antibodies, and the immunocomplexes were subjected to SDS/PAGE. Immunoblotting with either anti-HA or anti-c-myc antibodies showed that a fraction of Gal-T2 coimmunoprecipitated with GalNAc-T (Fig. 2A, lane 1) and that a fraction of GalNAc-T coimmunoprecipitated with Gal-T2 (Fig. 2A, lane 4). For each enzyme, densitometry of immunoblots indicated that about 25% of the total in the immunoprecipitate coimmunoprecipitated with the other enzyme (compare lanes 1 and 2 and lanes 4 and 5 in Fig. 2A). Similar percentages of coimmunoprecipitation were obtained from determinations of the corresponding enzyme activities (not shown). Applying the whole procedure to mixtures of lysates prepared from clone 3 and the stably expressing mouse Gal-T2-HA clone IGT2, each one contributing the same enzyme activity as in double transfectants, resulted in no coimmunoprecipitation of GalNAc-T and Gal-T2 (Fig. 2A, lanes 3 and 6). These results indicate that the interaction between both enzymes originated when they were coexpressed in vivo.
Figure 2.

GalNAc-T and Gal-T2 interact physically. (A) Immunoprecipitation (ip) of lysates of clone 3 transiently transfected with an HA-tagged Gal-T2 was performed by using either anti-c-myc (lanes 1 and 5) or anti-HA (lanes 2 and 4) antibodies. Immunocomplexes were analyzed by SDS/PAGE and Western blotting using anti-HA or anti-c-myc antibodies, as indicated. Lanes 3 and 6 correspond to a mixture of lysates from the single transfectant clones 3 and IGT2 (induced with 1 μM ponasterone) carrying enzyme activities comparable to those present in the double transfectants (specific activity, 1 and 2 nmol × mg prot−1 × h−1 of, respectively, GalNAc and Gal transferred to the corresponding exogenous acceptors) and subjected to the whole immunoprecipitation procedure. (B) GalNAc-T does not coimmunoprecipitate ManII. Lysates of clone 3 were immunoprecipitated with anti-ManII (lanes 1 and 3) or anti-c-myc (lanes 2 and 4) antibodies and Western blotted with anti-ManII or anti-c-myc antibodies, as indicated. Three independent experiments were run, which gave essentially the same results.
To discard a generalized association of Golgi resident proteins, the ability of the medial/trans Golgi concentrated ManII (14) of coimmunoprecipitate GalNAc-T was investigated. A lysate of clone 3 was immunoprecipitated with anti-ManII (Fig. 2B, lanes 1 and 3) or anti-c-myc (Fig. 2B, lanes 2 and 4) antibodies, and the immunoprecipitates were immunoblotted with either anti-c-myc or anti-ManII antibodies. In no case was coimmunoprecipitation of these two Golgi resident proteins observed, thus indicating that the GalNAc-T/Gal-T2 interaction shown in Fig. 2A is specific. The same results were obtained when the Gal-T2/ManII interaction was assessed (data not shown).
The N-Terminal Domain of Gal-T2 Participates in the Interaction with GalNAc-T.
It has been shown that particular regions of the N-terminal domain of glycoprotein oligosaccharide processing enzymes are relevant for their heterooligomerization and Golgi retention (15). To examine whether the amino terminus of Gal-T2 was involved in the interaction with GalNAc-T, a truncated Gal-T2 construct, containing amino acids 1–52 and the HA epitope (Gal-T21–52-HA, 6.2 kDa) was transfected to clone 3. It was verified by immunocytochemistry that this construct localizes to the Golgi complex (not shown). The putative association of the construct with GalNAc-T was assessed by immunoprecipitation and Western blotting. GalNAc-T coimmunoprecipitated about 30% of GalT21–52-HA (Fig. 3A, lanes 1 and 2). Conversely, Gal-T21–52-HA coimmunoprecipitated about 30% of the full-length (67 kDa) c-myc-tagged GalNAc-T (Fig. 3A, lanes 3 and 4). No coimmunoprecipitation between GalNAc-T and the N-terminal domain of a distal Golgi-located glycoprotein galactosyltransferase (16) fused to EYFP (β1,4 Gal-T1–81-EYFP) was detected in similar experiments (Fig. 3B). Immunoprecipitation with anti-c-myc antibodies of lysates from clone 3 cotransfected transiently with both the full-length and the truncated Gal-T2 forms resulted in coimmunoprecipitation of both Gal-T2 forms (Fig. 3C, lane 3). Gal-T21–52-HA competed with the full-length Gal-T2 for the GalNAc-T interaction, because about 40% less full-length Gal-T2 (43 kDa) coimmunoprecipitated with GalNAc-T in the triple transfectant (Fig. 3C, compare lanes 2 and 3). As levels of Gal-T2-HA expression were very similar in double (lane 1) and triple (lane 4) transfectants, the simplest interpretation of this result is that at least the N-terminal domain of Gal-T2 is relevant for its interaction with GalNAc-T.
Figure 3.

The N-terminal domain of Gal-T2 interacts with GalNAc-T. (A) Clone 3 was transfected with a truncated version of Gal-T2 (Gal-T21–52-HA, 6.2 kDa), immunoprecipitated with anti-c-myc (lanes 1 and 3) or anti-HA (lanes 2 and 4), and immunoblotted as indicated. (B) Lysates from GalNAc-T and β1,4Gal-T1–52-EYFP double transfectants were immunoprecipitated with anti-c-myc (lanes 1 and 3) or anti-GFP (lanes 2 and 4) and Western blotted with anti-GFP (lanes 1 and 2) or anti-c-myc (lanes 3 and 4) antibodies. (C) Clone 3 was transfected with the full-length Gal-T2-HA (43 kDa, lane 2) or cotransfected with both Gal-T21–52-HA and Gal-T2-HA cDNAs (lane 3). Expression levels of Gal-T2-HA in double (lane 1) and triple (lane 4) transfectants are shown. Lysates were immunoprecipitated with anti-c-myc (lanes 2 and 3) or anti-HA (lanes 1 and 4) antibodies and Western blotted with anti-HA antibodies.
EGFP Derivatives Fused at the C Terminus of N-Terminal Domains of GalNAc-T and Gal-T2 Undergo FRET.
Microscopic FRET increases the resolution of conventional fluorescence microscopy to the molecular level, allowing quantitative examination of protein–protein interactions or molecular clustering based on the nonradiative energy transfer between donor and acceptor molecules (17). The rate of energy transfer decays with the sixth power of the distance, so FRET assesses molecular proximity on a length scale of 1–10 nm (11, 18–20). To examine GalNAc-T/Gal-T2 interactions using FRET, a set of plasmids encoding chimeric proteins bearing the EGFP derivatives with spectral overlap (ECFP and EYFP) fused at the C terminus of the N-terminal domains of GalNAc-T, Gal-T2, β1,4 Gal-T, and ManII was constructed. The HA epitope was maintained at the C terminus of Gal-T2 to allow immunoprecipitation experiments.
The stable expression of the fusion proteins GalNAc-T1–27-EYFP (30.3 kDa) and Gal-T21–52-HA-ECFP (33.5 kDa) in single (Fig. 4A, lanes 1 and 3) and double (Fig. 4A, lane 2) transfectants was confirmed by Western blotting with anti-GFP monoclonal antibody. To control the presence of disulfide-bonded heterooligomers of ECFP and EYFP chimeras that would also undergo FRET, gels run in the absence of 2-mercaptoethanol were also examined (Fig. 4B). Although minor bands of disulfide-bonded homodimers were detected (arrows), disulfide-bonded heterodimers were essentially absent from the expected position (asterisk, lane 2). Fusion chimeras behaved as their full-length transferase counterparts in subcellular localization and immunoprecipitation assays. Both chimeras localize to the Golgi complex, displaying a strong yuxtanuclear/perinuclear staining in the region of the cell in which the endogenous Golgi marker ManII concentrate (Fig. 4E). Gal-T21–52-HA-ECFP coimmunoprecipitated about 15% of GalNAc-T1–27-EYFP (Fig. 4C, compare lanes 2 and 3). Contrarily, Gal-T21–52-HA-ECFP was unable to coimmunoprecipitate ManII1–76-EYFP (Fig. 4D, compare lanes 2 and 3).
Figure 4.

Expression of EGFP derivative fusion proteins. Membranes from CHO-K1 cells stably expressing Gal-T21–52-HA-ECFP (33.5 kDa, lane 1) or GalNAc-T1–27-EYFP (30.3 kDa, lane 3) or both fusion proteins (lane 2) were subjected to SDS/PAGE in the presence (A) or in the absence (B) of 2-mercaptoethanol and Western blotted with anti-GFP antibody. Arrows in B mark the positions of homodimers, and an asterisk marks the expected position of the band corresponding to heterodimers. (C) Lysates from single Gal-T21–52-HA-ECFP (lane 1) and from double Gal-T21–52-HA-ECFP and GalNAc-T1–27-EYFP (lanes 2 and 3) transfectants were immunoprecipitated (ip) with anti-HA (lanes 1 and 2) or with anti-GFP (lane 3) antibodies, subjected to SDS/PAGE, and Western blotted with anti-GFP antibody. (D) Lysates from single Gal-T21–52-HA-ECFP (lane 1) and from double Gal- T21–52-HA-ECFP and ManII1–76-EYFP (lanes 2 and 3) were immunoprecipitated with anti-HA (lanes 1 and 2) and with anti-GFP (lane 3) antibodies, subjected to SDS/PAGE, and Western blotted with anti-GFP antibody. (E) Visualization of Gal-T21–52-HA-ECFP, endogenous ManII, and GalNAc-T1–27-EYFP. Cells were fixed, immunostained for ManII, and observed under the fluorescence microscope using the filter for EYFP (GalNAc-T), rhodamine (ManII), and ECFP (Gal-T21–52). Bar, 20 μm.
Living cells stably expressing Gal-T21–52-HA-ECFP and GalNAc-T1–27-EYFP were examined for FRET (Fig. 5). As shown before, both constructs colocalize in the Golgi complex (Fig. 5 A and B), and it was clear that in particular zones they are close enough as to undergo FRET (Fig. 5C, arrowheads). In contrast, although the pair ManII1–76-ECFP (Fig. 5D) and GalNAc-T1–27-EYFP (Fig. 5E) also showed Golgi colocalization, it did not display regions of FRET (Fig. 5F). The quantitative analysis of FRET (FRETN, Table 1), which normalizes the FRET signal to the concentration of donor and acceptor fluorophores, gave a value for the Gal-T21–52-HA-ECFP/GalNAc-T1–27-EYFP pair about 1 order of magnitude higher than for the equivalent pairs ManII/GalNAc-T, ManII/Gal-T2, and GalNAc-T/(glycoprotein)Gal-T, for which no coimmunoprecipitation with the glycolipid glycosyltransferases was detected (Figs. 2–4). This indicated that the association between glycolipid glycosyltransferase N-terminal domains is relatively specific. The extent of FRET, estimated as the fraction of the total protein that undergoes FRET, was about 50% for the pair Gal-T21–52-HA-ECFP/GalNAc-T1–27-EYFP (Table 1). For pairs showing low FRETN, the extent of FRET was 14–17%, indicating a more randomized distribution of the participating proteins.
Figure 5.

Gal-T21–52-ECFP and GalNAc-T1–27-EYFP but not ManII1–76-ECFP and GalNAc-T1–27-EYFP undergo FRET in living cells. Cells expressing Gal-T21–52-ECFP (A) and GalNAc-T 1–27-EYFP (B) or ManII1–76-ECFP (D) and GalNAc-T1–27-EYFP (E) fusion proteins were observed with the filters for ECFP (A and D), EYFP (B and E), or FRET (C and F). Arrows indicate colocalization of the truncated forms of Gal-T2 and GalNAc-T (A and B) or of ManII and GalNAc-T (D and E) in the Golgi region. Arrowheads indicate regions of the Golgi in which truncated Gal-T2 and GalNAc-T forms were close enough as to undergo FRET as shown in C. No FRET between ManII and GalNAc-T was observed in F. (Bar, 20 μm.)
Table 1.
FRETN values for EGFP derivatives coexpressed in single cells
| Donor/acceptor pair* | Mean FRETN ± SD† | FRET extent, %‡ |
|---|---|---|
| Gal-T2/GalNAc-T§ | 1.1 × 10−5 ± 5 × 10−6 | 49 |
| Man II/Gal-T2 | 9.5 × 10−7 ± 6 × 10−7 | 17 |
| Man II/GalNAc-T | 8.0 × 10−7 ± 4 × 10−7 | 14 |
| GalNAc-T/β1,4Gal-T | 2.0 × 10−6 ± 5 × 10−7 | 17 |
In each pair, donor stands for the fusion to ECFP and acceptor for the fusion to EYFP.
FRETN, FRET normalized for the expression levels of both donor and acceptor fluorophores.
FRET extent was estimated by the formula
FRETN = FRET1/(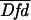 ×
×
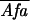 ), which approximates to [bound]/[total
donor] × [total acceptor] (11).
), which approximates to [bound]/[total
donor] × [total acceptor] (11).
Value significantly higher (P < 0.02, Student's t test) than the other analyzed pairs. Results are mean of four independent experiments, each one resulting from the observation of 50 cells.
GalNAc-T/Gal-T2 Immunocomplexes Efficiently Convert GM3 into GM1.
To examine whether immunocomplexes support the two transfer steps necessary for conversion of GM3 into GM1 (Fig. 1A), lysates of clone 3 transiently transfected with the full-length Gal-T2 were immunoprecipitated with anti-c-myc antibodies, and the immunocomplexes were incubated at 37°C in the presence of exogenous GM3, UDP-GalNAc, and UDP-[3H]Gal. Fig. 6 shows that [3H]Gal was incorporated into a lipid product and that the incorporation depended on the addition of the acceptor GM3 (double transfectant). The radioactive product was identified as [3H]GM1 by high-performance thin-layer chromatography (HPTLC, Inset, lane 1). A mixture of immunoprecipitates from lysates of single transfectants carrying equivalent enzyme activities produced negligible amounts of [3H]GM1 under the same incubation conditions (mix of single transfectants). Interestingly, an immunoprecipitate from lysates from clone 3 cotransfected with both the full-length and the truncated forms of Gal-T2 (triple transfectant) produced about half the amount of [3H]GM1 produced by the double transfectant (Inset, lane 2). Because GalNAc-T activity immunoprecipitated from double and triple transfectants was very similar while coimmunoprecipitating Gal-T2 activity was reduced (0.08 and 0.05 nmol of Gal transferred × h−1, respectively), this result suggests a competition between Gal-T21–52 and Gal-T2 for the interaction with GalNAc-T, as already evidenced in the experiment of Fig. 3B.
Figure 6.

Immunocomplexes from double transfectants efficiently convert GM3 to GM1. Truncated Gal-T2 affects the efficiency of conversion. To assess for the two-step conversion of GM3 to GM1, immunocomplexes from double (GalNAc-T/Gal-T2) or triple (GalNAc-T/Gal-T2/Gal-T21–52) transfectants or from mixtures of immunoprecipitates from single transfectants, as indicated, were incubated in a final volume of 20 μl with 100 μM UDP-GalNAc, 10 μM UDP-[3H]Gal (1 × 106 cpm), 20 mM MnCl2, 100 mM sodium cacodylate, pH 7.2, 3 mM CDP-Choline, 20 μg Triton CF-54/Tween 80 (2:1, wt/wt) for 2 h at 37°C in the presence or absence of 400 μM GM3 (see Fig. 1A). Reaction products were processed for lipid-bound radioactivity determination and HPTLC as indicated (4). Results are the mean ± SD of three independent experiments. (Inset). [3H]GM1 was identified as the main radioactive product of incubates with immunocomplexes of double (lane 1) or triple (lane 2) transfectants, after HPTLC of the lipid extracts and phosphorimaging. Lane 3 shows the positions of cochromatographed radioactive glycolipid standards obtained from clone 3 metabolically labeled from [3H]Gal.
Discussion
Here, we show that two successively acting glycolipid glycosyltransferases, GalNAc-T and Gal-T2, are physically associated in Golgi membranes of CHO-K1 cells and that the association is functional in vitro. Immunoprecipitation of GalNAc-T resulted in coimmunoprecipitation of about 25% Gal-T2, and conversely, Gal-T2 coimmunoprecipitated about 30% GalNAc-T. A truncated form of Gal-T2 containing the cytosolic, transmembrane domain and few amino acids from the stem region mimicked the coimmunoprecipitation behavior of the full-length Gal-T2 and competed with the full-length Gal-T2 for its interaction with GalNAc-T, indicating that the N-terminal domain was involved in the interaction. Tagged with ECFP and EYFP, these domains conveyed the tags to the Golgi complex membranes and positioned them in close spatial proximity (1–10 nm) so as to allow FRET between the tags in vivo.
GalNAc-T/Gal-T2 immunocomplexes formed in vivo were far more efficient than the sum of individually expressed enzymes for catalyzing the two-step conversion of exogenous GM3 into GM1 in vitro. The truncated form of Gal-T2 exerted a dominant negative effect on GM1 formation, consistent with its competence with the full-length Gal-T2 for the association with GalNAc-T observed in the Western blot experiments. This suggests that the amount of GalNAc-T sites available for interaction with Gal-T2 are limiting and can be saturated by an excess of the Gal-T2 N-terminal sequences.
Taken together, results indicate that the functional colocalization of GalNAc-T and Gal-T2 observed here (Fig. 1) and previously (4) may involve a physical association between them. Although we cannot discard the participation of the luminal domains (21), it was clear that the N-terminal domains are relevant to the association. That not all GalNAc-T and Gal-T2 or their respective N-terminal domains participate of the association may reflect, among other possibilities, the affinity constant of the complexes in the context of the mobility and recycling of these enzymes within the Golgi stacks (22).
Although other glycolipid glycosyltransferases remain to be investigated with regard to their ability to participate in this type of association, the present results give experimental support to the idea of “cooperative sequential specificity” in the synthesis of complex oligosaccharides by multiglycosyltransferase systems advanced many years ago (23). A mechanism of channeling the intermediates along multienzyme complexes, so that the product of each glycosyltransferase reaction becomes the substrate for the next enzyme in the sequence, would be more efficient than a synthesis based on random encounters of membrane-bound lipid acceptors diffusing laterally in the Golgi membranes with the membrane-bound, laterally diffusing transferases (24) that recognize them as substrate. Analogously, two multiprotein complexes having α-1,6 mannosyltransferase activity have been described in the yeast Saccharomyces cerevisiae, and the suggestion was made that these complexes may facilitate the rapid synthesis of polymannose structures (25, 26). In addition, the possibility that glycolipid glycosyltransferase complexes participate in the formation of kin oligomers conferring Golgi residence to the involved proteins, as has already been suggested for the N-acetylglucosaminyltransferase I and mannosidase II (15) involved in processing of N-linked glycoprotein oligosaccharides, is worth considering.
Acknowledgments
We thank Dr. José A. Martina for helpful discussions, Dr. K. Moremen for the expression clone encoding mouse mannosidase II, Dr. Andreas Girod for advice on FRET technology, and Dr. Gustavo Nores for standard glycolipid samples. We also thank Drs. G. A. Nores and J. Valdez for critical reading of the manuscript. The excellent technical assistance of G. Schachner and S. Deza with cell cultures is further acknowledged. This work was supported in part by Grants PMT-PICT 0181 from the Consejo Nacional de Investigaciones Científicas y Tecnicas, 4122 from the Consejo de Investigaciones Científicas y Tecnológicas, 89/96 from the Secretaría de Ciencia y Técnica de la Universidad Nacional de Córdoba of Argentina, and 75197-554001 from the Howard Hughes Medical Institute. H.J.F.M. and J.L.D. are Career Investigators, and C.G.G. is a fellow of Consejo Nacional de Investigaciones Científicas y Tecnicas.
Abbreviations
- FRET
fluorescence resonance energy transfer
- HA
hemagglutinin
- ECFP
enhanced cyan fluorescent protein
- EGFP
enhanced green fluorescent protein
- EYFP
enhanced yellow fluorescent protein
- GalNAc-T
UDP-GalNAc:lactosylceramide/GM3/GD3 β-1,4-N-acetyl-galactosaminyltransferase
- Gal-T2
UDP-Gal:GA2/GM2/GD2 β-1,3-galactosyltransferase
- Gal-T
UDP-Gal:GlcNAc β 1→4 galactosyltransferase
- GFP
green fluorescent protein
- HPTLC
high-performance thin-layer chromatography
- ManII
α-mannosidase II
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 1321.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.031458398.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.031458398
References
- 1.van Echten G, Sandhoff K. J Biol Chem. 1993;268:5341–5344. [PubMed] [Google Scholar]
- 2.Colley K J. Glycobiology. 1997;7:1–13. doi: 10.1093/glycob/7.1.1-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maccioni H J F, Daniotti J L, Martina J A. Biochim Biophys Acta. 1999;1437:101–118. doi: 10.1016/s1388-1981(99)00002-5. [DOI] [PubMed] [Google Scholar]
- 4.Maxzúd M K, Daniotti J L, Maccioni H J F. J Biol Chem. 1995;270:20207–20214. doi: 10.1074/jbc.270.34.20207. [DOI] [PubMed] [Google Scholar]
- 5.Lannert H, Gorgas K, Meissner I, Wieland F T, Jeckel D. J Biol Chem. 1998;273:2939–2946. doi: 10.1074/jbc.273.5.2939. [DOI] [PubMed] [Google Scholar]
- 6.van Echten-Deckert G, Sandhoff K. In: Organization and Topology of Sphingolipid Metabolism. Pinto B M, editor. Oxford: Pergamon; 1998. [Google Scholar]
- 7.Giraudo C G, Rosales Fritz V M, Maccioni H J. Biochem J. 1999;342:633–640. [PMC free article] [PubMed] [Google Scholar]
- 8.Martina J A, Daniotti J L, Maccioni H J. Neurochem Res. 2000;25:725–731. doi: 10.1023/a:1007527523734. [DOI] [PubMed] [Google Scholar]
- 9.Nagata Y, Yamashiro S, Yodoi J, Lloyd K, Shiku H, Furukawa K. J Biol Chem. 1992;267:12082–12089. [PubMed] [Google Scholar]
- 10.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 11.Gordon G W, Berry G, Liang X H, Levine B, Herman B. Biophys J. 1998;74:2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahajan N P, Linder K, Berry G, Gordon G W, Heim R, Herman B. Nat Biotechnol. 1998;16:547–552. doi: 10.1038/nbt0698-547. [DOI] [PubMed] [Google Scholar]
- 13.Rosales Fritz V M, Daniotti J L, Maccioni H J F. Biochim Biophys Acta. 1997;1354:153–158. doi: 10.1016/s0167-4781(97)00117-6. [DOI] [PubMed] [Google Scholar]
- 14.Rabouille C, Hui N, Hunte F, Kieckbusch R, Berger E G, Warren G, Nilsson T. J Cell Sci. 1995;108:1617–1627. doi: 10.1242/jcs.108.4.1617. [DOI] [PubMed] [Google Scholar]
- 15.Nilsson T, Rabouille C, Hui N, Watson R, Warren G. J Cell Sci. 1996;109:1975–1989. doi: 10.1242/jcs.109.7.1975. [DOI] [PubMed] [Google Scholar]
- 16.Llopis J, McCaffery J M, Miyawaki A, Farquhar M G, Tsien R Y. Proc Natl Acad Sci USA. 1998;95:6803–6808. doi: 10.1073/pnas.95.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jovin T M, Arndt-Jovin D J. In: Cell Structure and Function by Microspectrofluorimetry. Kohen E, Ploem J S, Hirschberg G J, editors. Orlando: Academic; 1989. pp. 99–117. [Google Scholar]
- 18.Jovin T M, Arndt-Jovin D J. Annu Rev Biophys Biophys Chem. 1989;18:271–308. doi: 10.1146/annurev.bb.18.060189.001415. [DOI] [PubMed] [Google Scholar]
- 19.Kenworthy A K, Edidin M. J Cell Biol. 1998;142:69–84. doi: 10.1083/jcb.142.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cacciatore T W, Brodfuehrer P D, Gonzalez J E, Jiang T, Adams S R, Tsien R Y, Kristan W B, Jr, Kleinfeld D. Neuron. 1999;23:449–459. doi: 10.1016/s0896-6273(00)80799-0. [DOI] [PubMed] [Google Scholar]
- 21.Opat A S, Houghton F, Gleeson P A. J Biol Chem. 2000;275:11836–11845. doi: 10.1074/jbc.275.16.11836. [DOI] [PubMed] [Google Scholar]
- 22.Zaal K J, Smith C L, Polishchuk R S, Altan N, Cole N B, Ellenberg J, Hirschberg K, Presley J F, Roberts T H, Siggia E, et al. Cell. 1999;99:589–601. doi: 10.1016/s0092-8674(00)81548-2. [DOI] [PubMed] [Google Scholar]
- 23.Roseman S. Chem Phys Lipids. 1970;5:270–297. doi: 10.1016/0009-3084(70)90024-1. [DOI] [PubMed] [Google Scholar]
- 24.Cole N B, Smith C L, Sciaky N, Terasaki M, Edidin M, Lippincott-Schwartz J. Science. 1996;273:797–801. doi: 10.1126/science.273.5276.797. [DOI] [PubMed] [Google Scholar]
- 25.Jungmann J, Munro S. EMBO J. 1998;17:423–434. doi: 10.1093/emboj/17.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jungmann J, Rayner J C, Munro S. J Biol Chem. 1999;274:6579–6585. doi: 10.1074/jbc.274.10.6579. [DOI] [PubMed] [Google Scholar]