

Abstract
Activation of small conductance calcium-activated potassium (KCa2) channels can regulate neuronal firing and synaptic plasticity. They are characterized by their high sensitivity to the bee venom toxin apamin, but the mechanism of block is not understood. For example, apamin binds to both KCa2.2 and KCa2.3 with the same high affinity (KD ∼ 5 pm for both subtypes) but requires significantly higher concentrations to block functional current (IC50 values of ∼100 pm and ∼5 nm, respectively). This suggests that steps beyond binding are needed for channel block to occur. We have combined patch clamp and binding experiments on cell lines with molecular modeling and mutagenesis to gain more insight into the mechanism of action of the toxin. An outer pore histidine residue common to both subtypes was found to be critical for both binding and block by the toxin but not for block by tetraethylammonium (TEA) ions. These data indicated that apamin blocks KCa2 channels by binding to a site distinct from that used by TEA, supported by a finding that the onset of block by apamin was not affected by the presence of TEA. Structural modeling of ligand-channel interaction indicated that TEA binds deep within the channel pore, which contrasted with apamin being modeled to interact with the channel outer pore by utilizing the outer pore histidine residue. This multidisciplinary approach suggested that apamin does not behave as a classical pore blocker but blocks using an allosteric mechanism that is consistent with observed differences between binding affinity and potency of block.
Keywords: Calcium, Computer Modeling, Drug Action, Ion Channels, Neuron, Potassium Channels
Introduction
KCa2 channels (formerly known as SK channels) are characterized by their sensitivity to the highly specific toxin apamin (1). This 18-amino acid peptide, which has been isolated from the honeybee (Apis mellifera) venom (2), contains two disulfide bridges that provide a fairly rigid tertiary conformation (3), with two arginine residues (Arg-13 and Arg-14) being critical for its biological activity (4). The cloning of KCa2 channel subunits has revealed the existence of three subtypes (KCa2.1–KCa2.3, formerly SK1–SK3) (5) that bind apamin with very high affinity (KD ∼ 5–10 pm) (see Ref. 6 for a review). However, apamin is less potent at blocking KCa2 current and displays differential block of channel subtypes. For example, KCa2.2 (all species) displays the highest sensitivity, with IC50 values from 27 to 140 pm. Rat, human, and mouse KCa2.3-mediated currents show an intermediate sensitivity, with IC50 values ranging from 0.63 to 19 nm. Finally, human KCa2.1 is the least sensitive, with reported IC50 values ranging between 0.7 and 100 nm (6). These differences between binding and electrophysiological results suggest that the mechanism of block by apamin is complex and that binding and block by the toxin are not identical phenomena.
KCa2 channel subtypes are expressed throughout the CNS and periphery, displaying partially overlapping but distinct locations. This has led to the proposal that block of KCa2 channels may be a novel target for cognitive enhancement, depression, and dopamine-related disorders (7). However, blockers would be required to display significant selectivity for particular KCa2 channel subtypes. Differential block of KCa2 subunits by apamin, and even more so by the peptidic blocker Lei-Dab7 (8), has raised considerable interest. It is clear that understanding the mechanism of this differential block might contribute toward the synthesis of non-peptidic compounds that could selectively target a particular KCa2 subunit (7). In this study, we have used a multidisciplinary approach, including binding, mutagenesis, structural modeling, and patch clamp experiments with KCa2.2 and KCa2.3 channels, to gain a comprehensive understanding of how apamin works. Taken together, our results demonstrate that apamin operates with a mechanism that is not consistent with classical pore block.
EXPERIMENTAL PROCEDURES
Cell Culture and Cell and Membrane Preparation
Wild-type rat KCa2.2 (GenBankTM accession number NM_019314) and human KCa2.3 (GenBank accession number AF031815) channel DNAs were subcloned into the mammalian plasmid expression vectors pcDNA3 (Invitrogen, Paisley, UK) and pFLAGCMV2 (Sigma, Poole, UK), respectively. Point mutations in KCa2.2 (KCa2.2(H337N), KCa2.2(N337R), KCa2.2(N345G), and KCa2.2(N368H)) and KCa2.3 (KCa2.3(H522N) and KCa2.3(H491N)) were introduced using the QuikChange XL site-directed mutagenesis kit (Stratagene-Agilent, Stockport, UK) and subsequently confirmed by dye termination DNA sequencing.
Channels were transiently expressed in HEK293 cells. For each passage, cells were dissociated using an EDTA solution and maintained in modified essential medium (DMEM) (Invitrogen), supplemented with 10% fetal calf serum (Invitrogen) and 1% penicillin/streptomycin (Invitrogen) at 37 °C. They were plated onto 35-mm dishes (Falcon) 48 h before transfection. For electrophysiology, transient transfections of HEK293 cells were made using polyethyleneimine (Alfa Aesar, Inc.) by combining channel plasmid DNA with enhanced green fluorescent protein DNA in ratio of 1:1 to 1:10 (maximal plasmid content: 1 μg). Cells expressing enhanced green fluorescent protein were used for electrophysiology 12–24 h after transfection.
Membranes were prepared for binding experiments as follows. HEK293 cells were plated on a 100-mm dish for 2 days and then transfected with the corresponding plasmid using the polyethyleneimine method without enhanced green fluorescent protein. Cells were harvested after 48 h with cold PBS (4 °C) using 5 ml/dish and centrifuged twice for 10 min at 1000 × g (4 °C). The pellet was resuspended in a lysis buffer (50 mm Tris, BSA 1%, pH 7.4), mixed thoroughly, and centrifuged for 10 min at 200 × g (4 °C). The supernatant was centrifuged twice for 20 min at 16,000 × g (4 °C), and the pellet was resuspended in another buffer (5 mm Tris, 5.4 mm KCl, pH 8.5), using 1 ml/dish. Protein concentration was determined using a colorimetric protein assay with a bicinchoninic acid kit (Pierce). The absorbance was measured at 562 nm with a Multiskan Ascent (Thermo LabSystem, Waltham, MA) spectrophotometer. Glycerol (10%) was then added, and aliquots were stored at −80 °C.
Electrophysiology
Expressed KCa2 currents recorded in either the whole-cell or the excised outside-out patch configurations were evoked in symmetrical high (∼170 mm) K+ conditions using an internal solution that contained 1 μm free Ca2+. Pipettes were fabricated from KG-33 glass (Friedrich & Dimmock, Inc.) or from code 1403513 glass (Hilgenberg, Malsfeld, Germany) and filled with an internal solution composed of KCl (120 mm), HEPES (10 mm), EGTA (10 mm), Na2ATP (1.5 mm), CaCl2 (9.65 mm, calculated free [Ca2+]i 1 μm), MgCl2 (2.34 mm, calculated free [Mg2+]i 1 mm), pH 7.4, with ∼40 mm KOH. Cells were bathed in a control external solution that consisted of KCl (120 mm), HEPES (10 mm), EGTA (10 mm), CaCl2 (6.19 mm, calculated free [Ca2+]i 60 nm), MgCl2 (1.44 mm, calculated free [Mg2+]i 1 mm), pH 7.4, with ∼40 mm KOH. Some experiments were carried out using a physiological K+ external solution composed of NaCl (140 mm), KCl (5 mm), HEPES (10 mm), CaCl2 (2.5 mm), MgCl2 (1.2 mm), d-glucose (10 mm), pH 7.4 with NaOH. For kinetic experiments, solutions were rapidly exchanged using an RSC200 rapid switcher (Biologic, Claix, France) or a BPS-8 system from ALA Science (ALA Scientific Instruments, Farmingdale, NY).
Apamin, d-tubocurarine (d-TC),5 and UCL1684 were purchased from Tocris Biosciences (Bristol, UK). Apamin and d-TC solutions were prepared on the day of experiments from a frozen stock of 100 μm and 1 mm in water, respectively. UCL 1684 (Tocris Biosciences) stock was prepared by dissolving in dimethyl sulfoxide (DMSO) to 100 μm and stored in aliquots at −20 °C. Tetraethylammonium (TEA) (from Sigma) solutions were prepared from a 1 m stock solution in water. The reversible KCa blocker N-methyl-laudanosine (NML) (9) was synthesized in-house.
Binding Experiments
Saturation binding was carried out in a 10 mm Tris-HCl (pH 7.5) buffer solution containing 5.4 mm KCl and 0.1% BSA. 125I-apamin was obtained from PerkinElmer Life Sciences (Zaventem, Belgium), with a specific activity of 81.4 TBq/mmol. Glass fiber filters (Whatman GF/C, Maidstone, Kent, UK) used in these experiments were coated for 1 h in 0.5% polyethyleneimine (to prevent apamin from binding to the filter) and then washed with 2.5 ml of the ice-cold buffer just before use. Membrane preparations (final protein concentration: 10 μg/ml) were incubated with increasing concentrations of 125I-apamin for 1h at 0 °C. Binding experiments were terminated as follows: aliquots were filtered under reduced pressure through Whatman filters. Filters were rapidly washed twice with 2.5 ml of ice-cold buffer and placed into a vial containing Ecoscint A (7.5 ml) (National Diagnostics U.S.A, Atlanta, GA). The radioactivity remaining on the filters was measured using a Packard Tri-Carb 1600TR liquid scintillation analyzer with an efficacy of 69%. Nonspecific binding was determined in parallel experiments in the presence of an excess of unlabeled apamin (0.1 μm) and subtracted from the total binding to obtain the specific binding. Where 125I-apamin binding was not detected with some mutant channels, positive controls were carried out in tandem on wild-type channels. All binding data were obtained from a minimum of two batches of membranes.
Molecular Modeling
A homology model of KCa2.2 was created as described previously (10). Docking was performed using the software suite Sybyl (Tripos, St. Louis, MO). Docking simulations were produced by energy-minimizing the ligand using the MMF94s force field in Sybyl (Tripos) and then docking the ligand into the KCa2.2 tetramer model using the Surflex or FlexX docking modules in Sybyl. Interactions were accepted from the lowest energy binding mode. The NMR solved solution structure of apamin was donated by Dr. D. Wemmer (University of California, Berkeley).
Data Analysis
For saturation binding experiments, data were fit with a Hill equation of the form
with KD being the dissociation constant of the peptide and nh the Hill coefficient. Curve fitting was carried out using GraphPad Prism 5.02 (GraphPad Software, San Diego, CA). For all experiments, a 1/Y (where Y-Bound/total bound) weighting procedure was used, which gave more weight to the smaller values of radioactivity (i.e. those that are close to the KD).
For concentration-response relationships in electrophysiological experiments, data points representing current block were fit with a variable slope Hill equation of the form
where Icont is the amplitude of current in the absence of drug, I is the amplitude of current observed at a given concentration of blocker ([X], expressed in logarithmic units), IC50 is the concentration of blocker that blocks 50% of sensitive current, and nh is the Hill coefficient (expressed as negative values, but its absolute value is used in the text). Analysis of kinetic electrophysiological experiments is described under “Results.”
All numerical values are expressed as mean ± S.E. Statistical analysis was performed using Prism 5.02 (GraphPad Software). Data were analyzed with a parametric or non-parametric test where appropriate.
RESULTS
High Affinity Binding of Apamin to KCa2.2 and KCa2.3 Channels
The affinity of binding (KD) of apamin to KCa2.2 and KCa2.3 channels was assessed in saturation experiments on membranes prepared from transiently transfected HEK293 cells (see “Experimental Procedures”). Binding of 125I-apamin was saturable, with KD values of 7.5 ± 2.3 and 8.4 ± 1.7 pm for KCa2.2 and KCa2.3, respectively (n = 7) (Fig. 1, A and B). These values were not significantly different from each other (p = 0.77) and were consistent with previously published values (11). Obtained Hill coefficients were close to unity (0.87 ± 0.03 and 0.8 ± 0.1, respectively), indicating that binding was not cooperative.
FIGURE 1.

Binding and block of KCa2.2 and KCa2.3 channel current by apamin. A and B, representative examples of saturation relationships for 125I-apamin binding to expressed KCa2.2 (A) or KCa2.3 (B) subunits. C and D, whole-cell macroscopic currents derived from ramps from −80 to 80 mV imposed on voltage-clamped HEK293 cells expressing KCa2.2 (C) and (D) KCa2.3 subunits. The application of increasing concentrations of apamin inhibited macroscopic current. E, concentration-inhibition relationships for apamin inhibition of expressed KCa2.2 and KCa2.3 current.
Differential Block of KCa2.2 and KCa2.3 Currents by Apamin
Expression of either KCa2.2 or KCa2.3 subunits produced inwardly rectifying whole-cell currents in the presence of 1 μm intracellular Ca2+ (10, 12–14) (Fig. 1, C and D). Both KCa2.2-mediated and KCa2.3-mediated currents were blocked by the addition of apamin, but block was incomplete despite the addition of a supramaximal concentration of the bee venom toxin (15) (Fig. 1, C–E). Expressed KCa2.2 current was more sensitive to block by apamin than KCa2.3 current. Fig. 1E shows the fractional block of KCa2.2 and KCa2.3 currents measured at −80 mV in response to increasing concentrations of apamin. The data were fit with the Hill equation, giving the following for KCa2.2: IC50 = 107 ± 31 pm, nh = 1.8 ± 0.4 (n = 10), and giving the following for KCa2.3: IC50 = 6.1 ± 1.6 nm, nh = 1.4 ± 0.1 (n = 12) (6). The fitted Hill slope was significantly greater than unity for KCa2.3 (p < 0.02), but not for KCa2.2 (p < 0.08), although a trend was observed. These data suggested that some cooperativity might be utilized to achieve block. Finally, block of expressed KCa2.2 and KCa2.3 current was assessed under physiological K+ conditions to determine whether the concentration of external K+ affected the potency of block. KCa2.2 current was blocked by apamin with an IC50 = 70 ± 30 pm, nh = 0.91 ± 0.2 (n = 5), whereas KCa2.3 was blocked with an IC50 = 2.6 ± 0.4 nm, nh = 1.2 ± 0.1 (n = 9) under physiological conditions. These values were not significantly different from those obtained in symmetrical K+ conditions (KCa2.2, p = 0.16; KCa2.3, p = 0.25), indicating that the potency of block was not significantly affected by the concentration of external K+.
A Histidine Residue in the Channel Outer Pore Turret Was Essential for Block by Apamin
Particular amino acid residues within the outer pore have been reported to influence the sensitivity of KCa2 channel currents to block by apamin (16). We reported previously that the protonation of an outer pore histidine residue common to both KCa2.2 and KCa2.3 channels inhibited macroscopic current using an allosteric mechanism (10). It was possible that this His residue may interact with apamin to cause block. The sensitivity to block by apamin of the mutant channels KCa2.2(H337N) and KCa2.3(H491N) was investigated to examine this possibility, in which the common basic His residue had been replaced with an uncharged asparagine. Fig. 2, A and B, show representative KCa2.2(H337N) and KCa2.3(H491N) currents in symmetrical high K+ solutions before and after the application of 100 nm apamin. Currents were not significantly blocked by 100 nm apamin (Fig. 2D; KCa2.2(H337N) p = 0.22, KCa2.3(H491N) p = 0.11, n = 5). These data suggested that this common His residue located in the S5-PHelix loop was crucial to the inhibitory interaction of apamin with the channel. Radioligand binding experiments showed no specific binding of 125I-apamin (up to 300 pm) to either KCa2.2(H337N) or KCa2.3(H491N) channels (n = 3 for both mutants, data not shown). These data indicated that the His residue located in the S5-PHelix loop was critically required for the binding and subsequent block of KCa2.2 and KCa2.3 channels by apamin.
FIGURE 2.

His-337/His-491 residues are critical for block of KCa2.2 and KCa2.3 currents by apamin and organic blockers. A–C, outside-out patch (A and C) and whole-cell (B) macroscopic currents evoked by ramps from −80 to 80 mV in the absence (black trace) and the presence of a supramaximal concentration of apamin (100 nm) (gray trace). Mutation of the outer pore His residue in KCa2.2(H337N) (A) and KCa2.3(H491N) (B) produced currents that were insensitive to the bee venom toxin. Mutation of His-337 in KCa2.2 to the positively charged arginine (H337R) also produced currents that were apamin-insensitive (C). D, graph showing the mean ± S.E. inhibition by 100 nm apamin of macroscopic current from each mutant. E, graph showing the lack of block of KCa2.2(H337N) by the organic blockers UCL1684 (20 nm) and d-TC (100 μm) when compared with block of WT KCa2.2- and KCa2.3-mediated current.
It is possible that the proton acceptor property of the outer pore His residue, rather than the possibility of the residue possessing a net positive charge, was required for binding of apamin and subsequent channel block. Mutation of the His-337 residue to the positive arginine to yield KCa2.2(H337R) produced a channel current that was also resistant to inhibition by 100 nm apamin (Fig. 2, C and D; p = 0.43). These data indicated that binding and block by apamin required the electrostatic features of the un-ionized basic His residue in the channel outer pore.
The His Residue in the Outer Pore Turret Also Contributed to Block of KCa2.2 by d-Tubocurarine and UCL1684
The structural features of apamin proposed to confer high affinity binding have been used to design organic molecule blockers of KCa2 channels, providing a pharmacophore for KCa2 channel blockers (17, 18). A number of small organic molecule blockers of KCa2 channels, including dequalinium, d-TC, and several cyclic bis-quinolinium cyclophanes derived from dequalinium (e.g. UCL1684), possess two positively charged quinolinium groups that are spatially separated to be analogous to the separation of the positive guanidinium groups of the two arginine residues in apamin. These organic molecules displace 125I-apamin binding and are proposed to interact with the apamin binding site (19, 20). This would suggest that like block by apamin, block by these organic compounds would also require the outer pore His residue. This was tested by determining the sensitivity of KCa2.2(H337N)-mediated current to block by supramaximal concentrations of UCL1684 (21) (20 nm) and d-TC (100 μm) (Fig. 2E). The mutant channel was insensitive to both blockers (Fig. 2E; UCL1684 p = 0.47 n = 3, d-TC p = 0.12 n = 4). These data were consistent with binding displacement studies and indicated that the outer pore His residue in the S5-PHelix loop that is common to all KCa2 channel subtypes was essential to the binding and block by apamin and organic blockers.
The KCa2.2(H337N) Mutant Retained Sensitivity to TEA
It is possible that the lack of block by apamin, UCL1684, and d-TC of KCa2.2(H337N) and KCa2.3(H491N) channel currents resulted from the mutation affecting pore structure. We used TEA ions to probe for any inner pore structural changes that might have been caused by these mutations. TEA acts as a classic pore blocker, interacting at the extracellular mouth of the selectivity filter in K+ channels to obstruct K+ flux (22–25). TEA can therefore be used as a “molecular caliper” to probe the dimensions of the inner pore under different conditions. Different sensitivities are proposed to indicate differences in pore structure and dynamics (26, 27). Fig. 3, A and B, show current traces from outside-out patches recorded in low external K+ (to allow the iso-osmotic addition of TEA to the bath solution), showing that both WT and mutant KCa2.2 channel currents were blocked by TEA. A plot of the fractional current (I/Icont) measured at −40 mV against the concentration of TEA was fit with the Hill equation and revealed a sensitivity to block for KCa2.2 of IC50 = 1.9 ± 0.24 mm with a Hill slope nh = 1.1 ± 0.1 and for KCa2.2(H337N), IC50 = 7.7 ± 0.6 mm, nh = 0.9 ± 0.1 (Fig. 3C). These data showed that KCa2.2(H337N) was significantly less sensitive to TEA than WT KCa2.2 (p < 0.001, n = 3). The Hill slopes for both channels were not significantly different from unity (p > 0.05, n = 3), consistent with a one-to-one binding interaction that would be expected for a pore-blocking mechanism (28). The block of KCa2.2(H337N)-mediated current by TEA indicated that the general architecture of the inner pore was maintained by this mutation. However, it is likely that the ∼4-fold reduction in sensitivity to TEA suggests that the mutation of the outer pore His residue caused a slight change in the structure of the inner pore mouth where TEA is predicted to bind (see below).
FIGURE 3.

Block of KCa2.2 and KCa2.2(H337N) channels by extracellular TEA. A and B, representative traces from outside-out patches of expressed KCa2.2 (A) and KCa2.2(H337N) (B) channel currents evoked by voltage ramps from −100 to 100 mV in the absence and presence of increasing concentrations of extracellular TEA. Experiments were performed in low (5 mm) extracellular [K+], with increasing concentrations of TEA being substituted for NaCl. C, concentration-inhibition relationships for block of wild-type KCa2.2 and mutant KCa2.2(H337N) current by TEA (see “Results” for details).
Molecular Modeling of Apamin and TEA Interactions with the KCa2.2 Pore Region Suggested Distinct Binding Sites
Despite significant differences in functional properties, KCa2 channels are likely to share pore architecture with Kv channels (29). The mutation of an outer pore valine (Val-342) residue in KCa2.2 to the glycine that confers sensitivity of Kv channels to charybdotoxin created a charybdotoxin-sensitive KCa2.2 channel (29). The similarities in the effect of these mutations on KCa2.2 and Kv channels indicated that both the inner pore architecture and the outer pore architecture are similar in these two channel classes. We used our previously published homology model (10), based on the crystal structure of Kv1.2 (Protein Data Bank code 2a79B), to model the interactions of apamin and TEA with the KCa2.2 channel (see “Experimental Procedures” for details).
Fig. 4, A and B, show the results of a docking simulation of apamin targeted to the S5-PHelix loop region of the KCa2.2 pore region homology model that contains the important His-337 residue. Fig. 4A shows a top down view of the channel displayed in ribbon form and the apamin molecule in stick form. The loop regions of the outer pore are indicated by arrows. Apamin was found to interact with a number of the residues in the S5-PHelix loop, specifically forming H-bonds between the toxin residue Asn-2 and channel residue His-337, Arg-14 of apamin, and channel residues Gln-339 and Gln-340, the C1 thiol group of apamin and Asp-338 in the channel outer pore, Gln-17 residue of the toxin and Asn-345 of the channel and His-18 of the toxin, and Ser-344 of KCa2.2 (highlighted blue in the pore region sequence displayed). Other residues with electrostatic interactions are highlighted in red. Fig. 4B shows a transverse view of the interaction. The most striking feature of the docking simulation was the large distance between apamin and the conduction pathway for K+ ions.
FIGURE 4.

Structural modeling of the interaction between the KCa2.2 channel and apamin or TEA. A and B, top down (A) and side (B)views of apamin docked to the outer pore region of KCa2.2. Channel residues discussed under “Results” are highlighted as follows: His-337 (yellow), Gln-339 (orange), Asn-345 (green), and Asn-368 (cyan). Residues within the channel outer pore and apamin that make contact by hydrogen bonds are colored blue, whereas those channel residues making electrostatic interactions are in red. C and D, top down (C) and side (D) views of the interaction between TEA and KCa2.2. The quaternary ammonium ion is modeled to interact within the inner pore of the channel by electrostatic interactions with the C=O group of Tyr-362 (distance between C=O and N+ ∼4 Å), and the ethyl groups of TEA interact via van der Waals contacts with Gly-363 (orange), Asp-364 (yellow), and Val-366 (cyan).
Targeting residues that have been shown to affect TEA sensitivity through mutational studies in KCa2 (23) and Kv channels (30) allowed the interaction between the quaternary ammonium ion and KCa2.2 to be modeled (Fig. 4, C and D). Fig. 4C shows a top down view of the channel displayed in ribbon form and the TEA molecule in space-filled mode for display, with TEA modeled to interact directly above the selectivity filter (Fig. 4, C and D). TEA was modeled to interact within the channel pore by electrostatic interactions with the C=O group of Tyr-362, whereas the ethyl groups of TEA interact via van der Waals contacts with Gly-363 and Asp-364 within the selectivity sequence and Val-366 (highlighted red in the sequence displayed below the docking). These data suggested that apamin does not traverse the pore to cause block by occluding the passage of K+ ions through the selectivity filter, whereas TEA is likely to act as a pore blocker.
KCa2.2(N345G) Displayed a Reduced Sensitivity to Apamin but Retained High Binding Affinity
A mutational approach was used to determine whether the modeled interaction of KCa2.2 and apamin was accurate. The Gln-17 residue of apamin was modeled to interact with Asn-345 by formation of a hydrogen bond. This channel pore residue was mutated to the small uncharged glycine residue to neutralize the polar nature of the wild-type asparagine residue. Fig. 5A displays the concentration-response relationship for block of the KCa2.2(N345G) mutant by apamin. The data were fit with the Hill equation and gave values of IC50 = 4.5 ± 0.8 nm, nh = 1.30 ± 0.06 (n = 10), showing that mutation of Asn-345 significantly reduced sensitivity to apamin in comparison with WT KCa2.2 (mean IC50: 107 pm) (p < 0.001, Student's t test for unpaired values). The value of the Hill coefficient was significantly larger than 1 (p < 0.001, Student's t test). In contrast, TEA sensitivity was not significantly altered by this mutation (KCa2.2 IC50 = 2.2 ± 0.3 mm, nh = 0.97 ± 0.05 (n = 3), KCa2.2(N345G) IC50 = 3.8 ± 1 mm, nh = 0.99 ± 0.09 (n = 4, p = 0.84 for IC50 and p = 0.24 for Hill slope) (Fig. 5B), indicating that the inner pore region was unaltered.
FIGURE 5.

Reduced sensitivity to block by apamin displayed by KCa2.2(N345G). A, concentration-inhibition relationship for block of expressed KCa2.2(N345G) channel current by apamin. The dashed gray curve shows the sensitivity of block of wild-type KCa2.2 current for comparison. B, concentration-inhibition relationship for block of KCa2.2(N345G) current by extracellular TEA. The relationship of block by TEA of wild-type KCa2.2 current is shown for comparison (dashed gray line), showing that the channel mutation had little effect upon block by the quaternary ion.
Binding affinity of apamin for the KCa2.2(N345G) mutant (KD 8.9 ± 4.1 pm, n = 4) was not significantly different from binding to WT KCa2.2 channels (p = 0.75, Student's t test for unpaired values, data not shown). The Hill coefficient was 1.01 ± 0.13. These data supported the modeled interaction of apamin and KCa2.2 channel and suggested that the interaction of apamin with Asn-345 was involved in the translation between binding and channel block.
Apamin and TEA Blocked KCa2 Channels Using Non-interacting Binding Sites
The evidence presented so far suggested that apamin may not act as a classic pore blocker as is proposed for TEA. As both interact in the pore region, it was pertinent to further address the question of whether the binding sites for TEA and apamin overlap. Current remaining in the presence of a partially inhibiting concentration of TEA represented a measure of the probability that the channels were not occupied by a TEA molecule (p(Cun), with possible values between 0 and 1) (31).
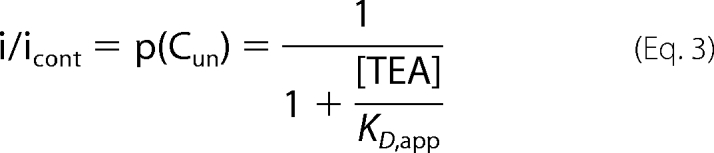 |
If TEA and apamin were to compete with each other for occupancy of the same binding site such that one could not bind if the other were already bound, the binding kinetics would be described by the following scheme.
 |
In this reaction, Cun represents the unblocked channel, C·TEA represents the channel bound with TEA, and C·APA represents the channel bound with apamin. It is clear that the on-rate for apamin block in the presence of TEA would be dependent on the probability (p(Cun)) of the channel being unoccupied by TEA. The equation for the relaxation time course to equilibrium for a bimolecular reaction following a rapid increase in the concentration of apamin would therefore be modified from
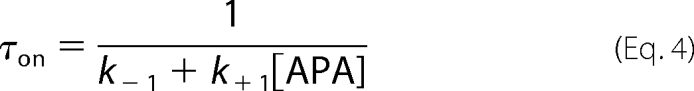 |
in the absence of TEA to
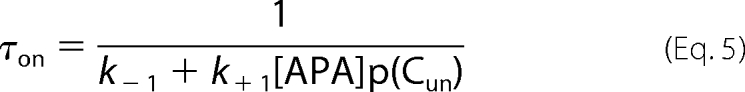 |
in the presence of a constant concentration of TEA. Therefore, the on-rate for apamin should be slower in the presence of TEA if the blocking sites of apamin and TEA overlapped.
The time course of block by apamin was well fit with a single exponential function. The rate of block by apamin was unchanged when applied after current was partly blocked by the application of TEA (Fig. 6A, B, and E). The current decays from separate experiments were fit with single exponentials that yielded time constants (τon) for block by 3 nm apamin of 0.70 ± 0.12 (n = 6) and 0.79 ± 0.08 s (n = 8) for apamin in the presence of a concentration of TEA that was close to the observed IC50 (1.8 mm). In contrast, the rate of block by apamin of current partly blocked by NML was slowed to 1.36 ± 0.08 s in the presence of 5 μm NML and 1.84 ± 0.09 s in 7.5 μm NML (n = 10 and 8, respectively) (Fig. 6, C, D, and E). The effect of NML was consistent with the KCa2 channel blocker (9) competing with apamin binding (32), whereas the lack of effect of TEA suggested that TEA and apamin block KCa2 channels by acting at non-interacting and likely separate sites.
FIGURE 6.

Apamin and TEA block KCa2.3 current by using non-interacting binding sites. A, example trace of whole-cell holding current recorded at −80 mV from a cell expressing KCa2.3 channels and bathed in high extracellular K+ solution. Fast application of apamin (3 nm) produced a block with a τon of 0.70 ± 0.12 s. Fits are shown as gray lines in all panels. B, membrane current recorded at −50 mV from a cell expressing KCa2.3 channel subunits. Fast application of TEA (1.8 mm) blocked ∼30% of current but had no effect on the time course of subsequent block by fast applied apamin (3 nm)(τon of 0.79 ± 0.08 s). C, fast application of NML (5 μm) blocked ∼40% of expressed KCa2.3 current, with the presence of this channel blocker slowing the rate of block by the subsequent fast application of apamin (τon slowed to 1.36 ± 0.08 s). D, a greater slowing of the rate of block by apamin was produced by a higher concentration of NML, with τon of block by apamin (3 nm) being slowed to 1.84 ± 0.09 s in 7.5 μm NML. E, graph showing individual values from all experiments of the τon (Tau) of block by apamin (3 nm) applied in the absence or presence of either TEA (1.8 mm) or NML (5 and 7.5 μm). A one-way analysis of variance showed that τon values were significantly different from each other (F = 37, p < 0.001). Tukey's post-hoc tests showed that the τon of apamin was unaffected by TEA (p > 0.05), but was significantly affected by both concentrations of NML (p < 0.05). n. s., not significant. ***, significant with p < 0.001.
Effect of the KCa2.2 Asn-368 Position on the Sensitivity of the Channels to Apamin
It was originally reported that Asn-368 within the outer pore region of KCa2.2 was an important contributor to providing a higher sensitivity to block by apamin than observed with hKCa2.1 (16). However, the role of this residue is less clear with the subsequent finding that hKCa2.1 current was not resistant to block by apamin (15). This residue is the only difference between the sequences in the SF-S6 loop region for KCa2.2 (Asn-368) and KCa2.3 (His-522) (Fig. 7A), and it is unknown whether this distinction underlies the different sensitivities (IC50) to apamin block of KCa2.2 (∼100 pm) and KCa2.3 (∼5 nm). Mutation of Asn-368 in KCa2.2 to mimic the pore sequence of KCa2.3 (KCa2.2(N368H)) produced a channel that bound 125I-apamin with a KD of 7.1 ± 0.59 pm (n = 8), which was not significantly different from binding to WT KCa2.2 (p = 0.85) or KCa2.3 (p = 0.47). In contrast, KCa2.2(N368H) current was blocked by apamin with a sensitivity that was not significantly different from block of WT KCa2.3 (IC50 = 2.96 ± 0.6 nm, nh = 1.47 ± 0.21, n = 7, p = 0.09). This value was significantly greater than that observed for WT KCa2.2 (Fig. 7, B and D; p < 0.001). These data suggested that Asn-368 was critical for the extra functional sensitivity to block by apamin of KCa2.2 when compared with KCa2.3. These findings suggested that KCa2.3 mutated to mimic the pore of KCa2.2 (KCa2.3(H522N)) would display a sensitivity to block by apamin like WT KCa2.2. Perhaps surprisingly, the sensitivity of KCa2.3(H522N) remained unchanged when compared with the WT channel ((p = 0.35, Fig. 7, C and D). The data were fit with the Hill equation and gave IC50 values of 4.2 ± 0.5 nm, nh = 1.67 ± 0.12 (n = 8, nh significantly different from 1, p = 0.0008, Student's t test) (see “Discussion”).
FIGURE 7.

Pore-mimicking mutants reveal differences between KCa2.2 and KCa2.3 structure. A, sequence alignment of the pore regions of KCa2.2 and KCa2.3 channels, with residues that differ highlighted in gray. SF, channel selectivity filter. B, concentration-inhibition relationships of KCa2.2(N368H), a mutation that produced a KCa2.2 channel whose pore mimicked KCa2.3. This mutation produced a channel current that was blocked by apamin with a sensitivity that was similar to that seen with wild-type KCa2.3 current. The relationships of block by apamin of wild-type KCa2.2 and KCa2.3 currents are shown for comparison in dashed gray. C, mutation of the outer pore of KCa2.3 to mimic KCa2.2 (KCa2.3(H522N)) produced a current that was blocked by apamin with a sensitivity that was not significantly different from wild-type KCa2.3 current. The relationships of block by apamin of wild-type KCa2.2 and KCa2.3 currents are shown for comparison in dashed gray. D, bar chart displaying the IC50 values showing the reduction in sensitivity in KCa2.2(N368H) when compared with WT KCa2.2(p < 0.0001, n = 7–10). No significant shift was observed for KCa2.3(H522N) when compared with WT KCa2.3 (p > 0.05, n = 8–12).
KCa2.2 Channels Can Be Blocked by Only One Apamin Molecule
Co-expression of KCa2.2 and KCa2.1 (16) or KCa2.2 and KCa2.3 subunits (23) produced functional heteromeric channels that displayed sensitivities to blockers that were intermediate to those observed for homomeric channels. We used the same approach using heteromeric channels containing the apamin-sensitive WT KCa2.2 subunits and apamin-insensitive KCa2.2(H337N) subunits to determine how subunit stoichiometry might affect apamin sensitivity. Expressed currents were sensitive to apamin (Fig. 8A). The mean data were best fit by the sum of two Hill equations (Fig. 8B). The high sensitivity component displayed an IC50 (IC50,a) of 271 pm with a Hill slope (nh,a) of 1.3. The lower sensitivity component had an IC50 (IC50,b) of 33 nm, with an nh,b of 0.85 (n = 6). The high sensitivity component made up 62% of the total block. These data indicated that channels were forming heteromers, displaying two distinct sensitivities to block by apamin. The kinetics of recovery from block of the heteromeric channels confirmed that two separate classes of apamin interaction sites existed, with the time course of recovery being best described by the sum of two exponential functions displaying a fast component, τoff,1 = 2.15 ± 0.17 s, and a slower component, τoff,2 = 56.1 ± 13.5 s (n = 5; Fig. 8C).
FIGURE 8.

Co-expression of apamin-sensitive wild-type KCa2.2 and apamin-insensitive KCa2.2(H337N) produced heteromeric channels that displayed distinct sensitivities to apamin. A, representative outside-out patch current traces evoked by voltage ramps from −100 to 100 mV, in the absence (con, control) and presence of increasing concentrations of apamin. B, concentration-inhibition relationship for block of current produced by co-expression of wild-type KCa2.2 and mutant KCa2.2(H337N) channels by apamin. Mean data were fit with a two component Hill equation, with IC50 values of ∼270 pm and 33 nm, demonstrating that heteromeric channels were expressed. frac, fraction. C, example trace of outside-out patch holding current (Vh −80 mV) from a cell co-expressing wild-type and mutant KCa2.2 channels during the rapid application and removal of apamin (3 nm). Current was blocked by apamin with an exponential time course (not shown), with the recovery from block being best described by the sum of two exponential components of taus: τoff,1, 1.8 s, and τoff,2, 36.4 s. These data suggest that apamin is interacting with two populations of channels that possess different sensitivities to block by the toxin. D, the probabilities of occurrence (Pocc) of different predicted stoichiometries of channel subunit assembly were calculated assuming that the probability of the inclusion of KCa2.2 and KCa2.2(H337N) subunits into the channel tetramer was equivalent.
The probability of the occurrence (Pocc) of different stoichiometries of the tetramer subunit composition can be predicted, assuming that the probability of incorporating either a WT KCa2.2 or a KCa2.2(H337N) channel subunit into the functional channel tetramer during channel assembly was equivalent. Fig. 8D illustrates the possible heteromeric stoichiometries and the Pocc of such combinations of subunits. Current derived from homomeric KCa2.2(H337N) channels would not contribute to the inhibition of the macroscopic current as the current was apamin-insensitive (Fig. 2). Therefore, the Pocc was adjusted to include current derived only from putatively apamin-sensitive heteromers that included WT subunits. The proportion of channels containing adjacent WT subunits channels gave a Pocc value of ∼0.6, leaving channels not containing adjacent WT subunits giving a Pocc value of ∼0.4 (Fig. 8D). Approximately 62% of expressed current was blocked by apamin with a high sensitivity (fraca), leaving ∼38% of current being blocked by the toxin with a lower sensitivity (fracb)(Fig. 8B). This suggested that apamin must bind to a channel containing at least two adjacent WT subunits to block with high sensitivity. The steep Hill slope (1.3) of the high sensitivity component of block suggested that the positive cooperative binding of more than one molecule of apamin to channels containing adjacent WT subunits provided block of high sensitivity. Therefore, it is likely that current blocked with a low sensitivity represented apamin interacting with channels that contained non-adjacent subunits or only a single WT subunit. This was supported by the lower sensitivity component displaying a Hill slope of 0.85, which suggested that no cooperativity of binding exists for apamin blocking these channels. These data suggested that the efficacy of apamin block is influenced positively by interactions between subunits but that it is also possible for apamin to interact with one sensitive subunit to produce inhibition, albeit at higher concentrations.
DISCUSSION
Point mutations within the outer pore region of KCa2 channels have been shown to affect the sensitivity of block by apamin (16, 29), which has led to the assumption that the bee venom toxin acts as a pore blocker. However, it has been recently reported that a point mutation in the S3-S4 extracellular loop had a major impact on the sensitivity of hKCa2.1 current to block by apamin (33). This information places doubt on whether apamin can act as a pore blocker, as it is unlikely that apamin is large enough to bind to an extracellular loop and traverse deep into the pore to cause block.
This study has identified two pore residues that influence apamin sensitivity (His-337/His-491 of KCa2.2 and KCa2.3, respectively, and Asn-345 of KCa2.2) to add to the formerly identified residues (16). Macroscopic currents from the mutants KCa2.2(H337N) and the equivalent KCa2.3(H491N) were both insensitive to 100 nm apamin, revealing the importance of the His residue located in the S5-PHelix loop of the outer pore in the apamin interaction. This lack of block arose from a loss of apamin binding. The KCa2.2(H337R) mutant was also insensitive, indicating that it was the proton acceptor property of the His residue that was crucial to both binding and block. We had previously found that external protons allosterically inhibited KCa2 current by interacting with the outer pore His residue (10), and it is possible that positively charged residues on apamin might mimic protons and interact with the His residue. Structural modeling of the interaction of apamin with KCa2.2 channel produced a lowest energy interaction that positioned apamin away from the selectivity filter, interacting with multiple residues in the S5-PHelix loop, including both the outer pore His and some already proposed to mediate the apamin-channel interaction (16). This modeled interaction was supported by the lower sensitivity to block by apamin of the KCa2.2(N345G) mutant. A previous study showing that a serine residue in the S3-S4 loop region contributed to high affinity block of KCa2.2 by apamin suggested that the outer pore region residues alone do not compose the complete binding site but do so in combination with the S3-S4 loop (33). Based on the orientation of the S3-S4 transmembrane segments within the Kv1.2 structure, it is possible that the S3-S4 loop may come into close contact with the S5-PHelix loop region. Our modeled interaction of apamin with KCa2.2 placed Arg-13 of the toxin projecting away from the pore region of the channel, making it possible that this residue could interact with residues outside the channel pore. Therefore, our modeled interaction is consistent with mutational studies and suggests that apamin is unlikely to physically occlude the pore.
The KCa2.2(H337N) channel was also found to be insensitive to supramaximal concentrations of KCa2.2 and KCa2.3 channel organic molecule blockers such as d-TC, UCL1684, and NML (data not shown for the latter). These molecules all displace 125I-apamin binding and must compete for either part of or the entire binding site used by the toxin (17–19, 32). Therefore, the lack of block by these compounds was not surprising based on the overlap of the binding sites. However, the implications of these data were surprising. It is likely that apamin does not block KCa2-mediated current by occluding the pore. Therefore, it is unlikely that d-TC, UCL1684, and NML are pore blockers. However, it is possible that the KCa2.2(H337N) mutation might perturb pore shape that would prevent binding of these organic blockers. We used TEA to investigate whether this might have been the case.
TEA has been used as a molecular caliper to probe the inner pore of Kv channels (27). TEA could be used in the same way for KCa2 channels, but first, it had to be determined whether apamin and TEA bound to non-overlapping sites. TEA was modeled to interact close to the selectivity filter of a KCa2 channel, in a region that has been previously proposed based on mutation studies (23). These data suggested that apamin and TEA bound to distinct sites within the pore region of these channels. This was supported by the finding that the presence of TEA did not affect the kinetics of apamin block. In contrast, the kinetics of block was slowed in a concentration-dependent manner by NML, a blocker known to compete for the same binding site as the toxin. These data provided strong evidence that apamin and TEA do not bind to overlapping binding sites. For comparison, an opposite conclusion was drawn concerning TEA and charybdotoxin block of single BK channels (31). It is clear that TEA can also be used with KCa2 channels to probe the channel inner pore. The apamin-insensitive mutant KCa2.2(H337N) was less sensitive to block by TEA (IC50 ∼8 mm) than the WT channel (IC50 ∼2 mm). The reduced sensitivity of KCa2.2(H337N) suggested that the inner pore region was somewhat altered by this mutation, but much less so than the extent to which the apamin site was perturbed. The absolute effect of this mutation on apamin binding indicated that this residue was a significant contributor to the binding site of the toxin. The lack of block of KCa2.2(H337N) by d-TC, UCL1684, and NML would suggest that these blockers also bind to the outer pore, a suggestion supported by the fact that these blockers displace apamin binding. Therefore, these compounds are not pore blockers and, like apamin, must inhibit macroscopic KCa2 current by an allosteric mechanism. This suggestion would help to explain the disparity between binding KD values (∼8 pm for both KCa2.2 and KCa2.3) and the functional inhibition of current by apamin, particularly in KCa2.3 (IC50 ∼5 nm). This proposed separation of binding and block was supported by the finding that no cooperativity of apamin binding was observed, whereas positive cooperativity was observed for functional inhibition.
Two residues within the pore sequence of KCa2 channels have been identified that might be involved in translating binding to block. The first was Asn-345 within KCa2.2, where mutation to glycine produced a channel that displayed a lowered sensitivity to block by apamin but retained high affinity binding for the toxin. The second was Asn-368 within KCa2.2, which corresponds to His-522 in KCa2.3. The sensitivity to block by apamin was reduced to that of KCa2.3 by mutation of Asn-368 to His, but high affinity binding for apamin was retained. These data were in accord with previous work, where mutation of the corresponding His in hKCa2.1 to Asn to mimic the pore sequence of KCa2.2 increased sensitivity to block by apamin (16). The Asn-368 within KCa2.2 is modeled to directly interact with Gln-340 and not apamin, with Gln-340 being modeled to interact with the toxin. Therefore, mutation of Asn-368 would be expected to affect block but not binding of apamin. In contrast, Asn-345 within KCa2.2 is modeled to directly interact with apamin by hydrogen bonding. Mutation of Asn-345 to glycine affected block but not binding of the toxin. It is clear that at least His-337 in the outer pore turret is essential for binding of apamin, whereas Ser-243 in the extracellular S3-S4 loop of KCa2.2 is suggested to contribute to high affinity binding (33). Therefore, it is likely that the lack of effect of mutation of Asn-345 on binding of apamin reflects that interaction of the toxin with this residue is crucial in translating binding to block, rather than it significantly contributing to high affinity binding. No residues modeled to interact with apamin will bind K+ ions because changes in external K+ concentration had no effect on the block of either KCa2.2 or KCa2.3 by apamin. Finally, care must be taken when considering apamin binding and block of KCa2.3 current. For example, mutation of KCa2.3 to mimic the KCa2.2 pore (KCa2.3(H522N)) did not have any effect on apamin sensitivity. These data support the suggestion either that the pore shape of KCa2.2 and KCa2.3 differs (10) or that differences might exist in the mechanism of transduction of binding to block between KCa2.2 and KCa2.3.
The presented modeling and mutagenesis data suggested that apamin binds to the channel outer pore rather than deep within the inner pore. Therefore, it is possible that multiple apamin molecules bind to the channel simultaneously to cause block. Concentration-response relationships for apamin block of KCa2.3 currents produced Hill slopes that were significantly greater than 1 (with a trend toward this also observed for KCa2.2). This suggested that more than one molecule of apamin binds to cause block and that positive cooperativity exists to produce block. The proposal that two or more molecules of apamin bind to produce high sensitivity block was supported by the finding that channel heteromers consisting of the apamin-sensitive WT KCa2.2 and apamin-insensitive KCa2.2(H337N) subunits displayed two distinct populations of sensitive current. The higher sensitivity site (IC50,a ∼ 270 pm) displayed a Hill slope of >1 and is proposed to correlate with those channels that contained adjacent WT KCa2.2 subunits. In contrast, the lower sensitivity component (IC50,b ∼ 33 nm) displayed a Hill slope of 0.85 and is proposed to comprise current from channels that contained two non-adjacent WT KCa2.2 subunits or only one WT KCa2.2 subunit. In contrast, Hill slopes of unity were produced for apamin binding, indicating no positive cooperativity (11, 34, 35). Clearly, this difference might indicate that although no cooperativity exists between the binding of two or more apamin molecules, adjacent subunits bound with apamin do interact to cause block. In summary, we suggest that apamin does not block KCa2 channels with a simple pore-blocking mechanism. It is proposed that apamin binding to the outer pore causes a disruption of the structural coupling between the outer pore region and the selectivity filter, causing collapse of the selectivity filter to impair conduction of K+ ions. This allosteric hypothesis provides a novel mechanistic basis for block of KCa2 current by apamin, thus aiding the search for subtype-specific non-peptidic inhibitors of the KCa2 channel subfamily.
Acknowledgment
We thank Christelle Gillissen for technical assistance.
This work was supported by a grant from the Belgian Science Policy Interuniversity Attraction Poles program and by a “FIRST-Doctorant Entreprise International” grant (Grant 516131) from the Walloon region of Belgium (to C. L.).
- d-TC
- d-tubocurarine
- TEA
- tetraethylammonium
- NML
- N-methyl-laudanosine.
REFERENCES
- 1.Blatz A. L., Magleby K. L. (1986) Nature 323, 718–720 [DOI] [PubMed] [Google Scholar]
- 2.Vincent J. P., Schweitz H., Lazdunski M. (1975) Biochemistry 14, 2521–2525 [DOI] [PubMed] [Google Scholar]
- 3.Pease J. H., Wemmer D. E. (1988) Biochemistry 27, 8491–8498 [DOI] [PubMed] [Google Scholar]
- 4.Labbé-Jullié C., Granier C., Albericio F., Defendini M. L., Ceard B., Rochat H., Van Rietschoten J. (1991) Eur. J. Biochem. 196, 639–645 [DOI] [PubMed] [Google Scholar]
- 5.Köhler M., Hirschberg B., Bond C. T., Kinzie J. M., Marrion N. V., Maylie J., Adelman J. P. (1996) Science 273, 1709–1714 [DOI] [PubMed] [Google Scholar]
- 6.Pedarzani P., Stocker M. (2008) Cell. Mol. Life Sci. 65, 3196–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liégeois J. F., Mercier F., Graulich A., Graulich-Lorge F., Scuvée- Moreau J., Seutin V. (2003) Curr. Med. Chem. 10, 625–647 [DOI] [PubMed] [Google Scholar]
- 8.Shakkottai V. G., Regaya I., Wulff H., Fajloun Z., Tomita H., Fathallah M., Cahalan M. D., Gargus J. J., Sabatier J. M., Chandy K. G. (2001) J. Biol. Chem. 276, 43145–43151 [DOI] [PubMed] [Google Scholar]
- 9.Scuvee-Moreau J., Liegeois J. F., Massotte L., Seutin V. (2002) J. Pharmacol. Exp. Ther. 302, 1176–1183 [DOI] [PubMed] [Google Scholar]
- 10.Goodchild S. J., Lamy C., Seutin V., Marrion N. V. (2009) J. Gen. Physiol. 134, 295–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finlayson K., McLuckie J., Hern J., Aramori I., Olverman H. J., Kelly J. S. (2001) Neuropharmacology 41, 341–350 [DOI] [PubMed] [Google Scholar]
- 12.Soh H., Park C. S. (2001) Biophys. J. 80, 2207–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stocker M. (2004) Nat. Rev. Neurosci. 5, 758–770 [DOI] [PubMed] [Google Scholar]
- 14.Li W., Aldrich R. W. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 1075–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dale T. J., Cryan J. E., Chen M. X., Trezise D. J. (2002) Naunyn Schmiedebergs Arch. Pharmacol 366, 470–477 [DOI] [PubMed] [Google Scholar]
- 16.Ishii T. M., Maylie J., Adelman J. P. (1997) J. Biol. Chem. 272, 23195–23200 [DOI] [PubMed] [Google Scholar]
- 17.Dilly S., Graulich A., Farce A., Seutin V., Liegeois J. F., Chavatte P. (2005) J. Enzyme Inhib. Med. Chem. 20, 517–523 [DOI] [PubMed] [Google Scholar]
- 18.Sørensen U. S., Strøbaek D., Christophersen P., Hougaard C., Jensen M. L., Nielsen E. Ø., Peters D., Teuber L. (2008) J. Med. Chem. 51, 7625–7634 [DOI] [PubMed] [Google Scholar]
- 19.Cook N. S., Haylett D. G. (1985) J. Physiol. 358, 373–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castle N. A., Haylett D. G., Morgan J. M., Jenkinson D. H. (1993) Eur. J. Pharmacol. 236, 201–207 [DOI] [PubMed] [Google Scholar]
- 21.Rosa J. C., Galanakis D., Ganellin C. R., Dunn P. M., Jenkinson D. H. (1998) J. Med. Chem. 41, 2–5 [DOI] [PubMed] [Google Scholar]
- 22.Heginbotham L., MacKinnon R. (1992) Neuron 8, 483–491 [DOI] [PubMed] [Google Scholar]
- 23.Monaghan A. S., Benton D. C., Bahia P. K., Hosseini R., Shah Y. A., Haylett D. G., Moss G. W. (2004) J. Biol. Chem. 279, 1003–1009 [DOI] [PubMed] [Google Scholar]
- 24.Lenaeus M. J., Vamvouka M., Focia P. J., Gross A. (2005) Nat. Struct. Mol. Biol. 12, 454–459 [DOI] [PubMed] [Google Scholar]
- 25.Ahern C. A., Eastwood A. L., Lester H. A., Dougherty D. A., Horn R. (2006) J. Gen. Physiol. 128, 649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Molina A., Castellano A. G., López-Barneo J. (1997) J. Physiol. 499, 361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bretschneider F., Wrisch A., Lehmann-Horn F., Grissmer S. (1999) Biophys. J. 76, 2351–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hille B. (2001) Ion Channels of Excitable Membranes, pp. 539–574, Sinauer Associates, Sunderland, MA [Google Scholar]
- 29.Jäger H., Grissmer S. (2004) Toxicon 43, 951–960 [DOI] [PubMed] [Google Scholar]
- 30.Heginbotham L., Abramson T., MacKinnon R. (1992) Science 258, 1152–1155 [DOI] [PubMed] [Google Scholar]
- 31.Miller C. (1988) Neuron 1, 1003–1006 [DOI] [PubMed] [Google Scholar]
- 32.Graulich A., Mercier F., Scuvée-Moreau J., Seutin V., Liégeois J. F. (2005) Bioorg. Med. Chem. 13, 1201–1209 [DOI] [PubMed] [Google Scholar]
- 33.Nolting A., Ferraro T., D'hoedt D., Stocker M. (2007) J. Biol. Chem. 282, 3478–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hugues M., Duval D., Kitabgi P., Lazdunski M., Vincent J. P. (1982) J. Biol. Chem. 257, 2762–2769 [PubMed] [Google Scholar]
- 35.Grunnet M., Jensen B. S., Olesen S. P., Klaerke D. A. (2001) Pflugers Arch. 441, 544–550 [DOI] [PubMed] [Google Scholar]