

Abstract
The nitric-oxide synthases (NOS, EC 1.14.13.39) are modular enzymes containing attached flavoprotein and heme (NOSoxy) domains. To generate nitric oxide (NO), the NOS FMN subdomain must interact with the NOSoxy domain to deliver electrons to the heme for O2 activation during catalysis. The molecular basis and how the interaction is regulated is unclear. We explored the role of eight positively charged residues that create an electropositive patch on NOSoxy in enabling the electron transfer by incorporating mutations that neutralized or reversed their individual charges. Stopped-flow and steady-state experiments revealed that individual charges at Lys423, Lys620, and Lys660 were the most important in enabling heme reduction in nNOS. Charge reversal was more disruptive than neutralization in all cases, and the effects on heme reduction were not due to a weakening in the thermodynamic driving force for heme reduction. Mutant NO synthesis activities displayed a complex pattern that could be simulated by a global model for NOS catalysis. This analysis revealed that the mutations impact the NO synthesis activity only through their effects on heme reduction rates. We conclude that heme reduction and NO synthesis in nNOS is enabled by electrostatic interactions involving Lys423, Lys620, and Lys660, which form a triad of positive charges on the NOSoxy surface. A simulated docking study reveals how electrostatic interactions of this triad can enable an FMN-NOSoxy interaction that is productive for electron transfer.
Keywords: Enzyme Catalysis, Enzyme Mechanisms, Nitric Oxide, Nitric Oxide Synthase, Reductase
Introduction
Nitric oxide (NO) is a small molecule essential to many biological processes (1). NO is produced in animals from l-Arg by NOS (EC 1.14.13.39) (2). Due to the highly diffusible and short-lived nature of NO, its bioactivity is mainly regulated by the NO synthesis activity of NOS enzymes (3). Three main isozymes of NOS are present in mammals: inducible NOS (iNOS),2 endothelial NOS, and neuronal NOS (nNOS) (4–6). These NOS enzymes are active as homodimers, with dimer formation involving a large interface of the oxygenase domain (7, 8). Each NOS monomer consists of two domains: an N-terminal oxygenase domain (nNOSoxy), which binds the cofactors protoporphyrin IX (heme), (6R)-5,6,7,8-tetrahydro-l-biopterin (H4B) and the substrate l-Arg, and a C-terminal reductase domain with binding sites for FAD, FMN, and NADPH. The C-terminal domain shows high homology with cytochrome P450 reductase and related proteins (6, 9). A calmodulin (CaM) binding site is located in the connecting sequence between both domains (4–6).
The electron transfer chain in NOS enzymes involves both monomers (see Fig. 1A). Electrons are transferred from NADPH to FAD to FMN in one monomer and then from the FMN to the heme domain of the partner subunit (10). This FMN to heme electron transfer is usually the rate-limiting step of NOS catalysis (2). CaM binding increases the rate of FAD to FMN electron transfer (11), and, more importantly, it also triggers the FMN to heme electron transfer (12). The effect of CaM can be explained in terms of a model in which the FMN domain is in a conformational equilibrium between FMN-shielded and -deshielded conformations (see Fig. 1A) (13–16). CaM favors a FMN-deshielded conformation in which FMN can transfer electrons to artificial acceptors like cytochrome c. But, to make possible electron transfer to the NOS heme, a driving force also is needed to promote the interaction between heme and FMN domains. The existing evidence suggests that an electrostatic component of the interaction is important (17–19). A cluster of negatively charged residues on the surface of the FMN domain is apparent in the crystal structure of the nNOS reductase domain (19). Mutation of these negatively charged residues inhibited electron transfer between FMN and heme in some cases (20). These results, together with structural data, supported a model for domain interaction where the cluster of negative charges on the FMN domain may interact with a positively charged region on the surface of the nNOSoxy domain (18, 19) (see Fig. 1 and supplemental Fig. S1). However, information about the potential roles of the surface residues of NOSoxy is scarce, and studies have been carried out for only three residues: Arg410 (21), Trp421 (22), and Lys423 (17). To further assess the contribution of the positively charged residues on the FMN to heme electron transfer, we have characterized mutants of eight positive residues on the nNOSoxy surface that neutralize or reverse those positive charges (see Fig. 1B and supplemental Fig. S1).
FIGURE 1.

Conformational equilibrium and electrostatic interactions of the nNOS domains. A, to allow NO synthesis, the FMN domain has to shift from a position adequate to accept electrons from FAD (left, FMN-shielded conformation) to a conformation in which is able to reach the heme domain (right, FMN-deshielded conformation). Available evidence indicates that the FMN domain uses the same face to interact with FAD and heme domains (20). The signs indicate the charges on each surface. B, surface of the oxygenase domain of nNOS. The two subunits of the oxygenase dimer are shown in white and light gray. The positively charged residues forming a electropositive patch are shown in dark gray. The labels indicating residues fully conserved in vertebrate NOS proteins are shown in boldface type; residues partially conserved are shown in italics.
EXPERIMENTAL PROCEDURES
Reagents
H4B was purchased from Schircks Laboratories (Jona, Switzerland). CO gas was obtained from Praxair, Inc. (Danbury, CT). EPPS was purchased from Fisher Scientific. DTT was purchased from RPI Corp. (Mount Prospect, IL). All other reagents were purchased from Sigma.
Site-directed Mutagenesis
Mouse nNOS mutants were prepared by site-directed mutagenesis on a pCWori vector containing the cDNA of wild-type rat nNOS full-length enzyme construct and also, in some cases, the nNOS oxygenase domain construct (nNOSoxy) (23). Oligonucleotides for site-directed mutagenesis were obtained from Integrated DNA Technologies (Coralville, IA) and are listed in supplemental Table S1. Site directed mutagenesis was performed using the QuikChange XL mutagenesis kit (Stratagene, La Jolla, CA). Mutations were confirmed by DNA sequencing at the Cleveland Clinic Genomics Core Facility. Mutated plasmids were transformed into BL21(DE3) Escherichia coli cells using the TransformAid bacterial transformation kit (Fermentas, Hanover, MD).
Protein Expression and Purification
Wild-type and mutant nNOS proteins containing a His6 tag attached to their N termini (for full-length nNOS) or C termini (nNOSoxy) were overexpressed in E. coli strain BL21(DE3). Full-length proteins were purified in the presence of H4B and l-Arg as described previously (23) but after the Ni2+ nitrilotriacetic acid-Sepharose CL-4B column, 2 mm CaCl2 was added to the sample and the 2′,5′-ADP-Sepharose column step was replaced by a CaM-Sepharose column as reported for the purification of the reductase domain (14). NOSoxy proteins were expressed and purified as described (24). Protein concentration was determined from the absorbance at 444 nm of the ferrous heme-CO complex, using an extinction coefficient of 76 mm−1 cm−1 (Δϵ444 nm-500 nm) (25).
Dimer Formation
The native oligomerization state of the wild-type nNOS and mutant proteins was estimated by size exclusion chromatography relative to gel filtration standards as reported previously (26). Protein samples (50 μl and 25 μm) were loaded in a Superdex 200 HR column (Pharmacia) equilibrated with 40 mm EPPS buffer, pH 7.6, 10% glycerol, 0.5 mm DTT, and 200 mm NaCl.
Arginine Binding
l-Arg binding affinity was studied at 25 °C by perturbation difference spectroscopy according to methods described previously (27, 28). nNOS samples (∼5 μm) in 40 mm EPPS buffer, pH 7.6, with 10% glycerol, 1 mm DTT, 10 μm H4B, and 1 mm imidazole were titrated with l-Arg (final concentration, 1 mm). The samples were allowed to equilibrate for 30 min, and the titration was performed by stepwise addition of l-Arg (final concentration, 1 mm). Binding constants were determined by the difference spectra between 390 and 430 nm. Baseline correction was done by subtracting the absorbance at 700 nm. The data were fitted to a simple hyperbola, and an apparent Kd value was obtained. The dissociation constant Kd for l-Arg was calculated by the following equation,
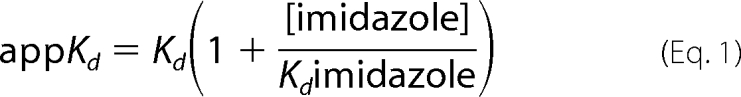 |
where a value of Kd imidazole = 160 μm was used (29).
Steady-state Assays
Cytochrome c reductase activity was determined as reported previously (20) by following the absorbance change for the reduction of cytochrome c by nNOS at 550 nm using an extinction coefficient of 21 mm−1 cm−1. Reaction mixtures (total volume, 400 μl) contained ≤0.01 μm nNOS, 25 μm FAD, 25 μm FMN, 0.1 mg/ml bovine serum albumin, 1 mm CaCl2, 0.2 mm EDTA, 1.0 μm CaM, 100 units/ml catalase, 40 units/ml superoxide dismutase, 65 μm cytochrome c, and 150 mm NaCl in 40 mm EPPS buffer, pH 7.6. The reaction was initiated by adding NADPH to a final concentration of 250 μm. In the assays without CaM, neither CaM nor CaCl2 were added, and EDTA concentration was 0.45 mm. NO synthesis and NADPH oxidation rates were determined as described previously (20). The NO synthesis activity was determined by the conversion of oxyhemoglobin to methemoglobin using an extinction coefficient of 38 mm−1 cm−1 at 401 nm. The NADPH oxidation rates were determined following the absorbance at 340 nm, using an extinction coefficient of 6.2 mm−1 cm−1. Reaction mixtures (total volume, 400 μl) contained ≤0.2 μm nNOS, 0.3 mm dithiotreitol, 4 μm FAD, 4 μm FMN, 4 μm H4B, 10 mm l-Arg, 0.1 mg/ml bovine serum albumin, 0.8 mm CaCl2, 0.2 mm EDTA, 1 μm calmodulin, 100 units/ml catalase, 60 units/ml superoxide dismutase, 5 μm oxyhemoglobin, and 150 mm NaCl in 40 mm EPPS buffer, pH 7.6. The reaction was initiated by adding NADPH to a final concentration of 250 μm. In the assays without calmodulin, neither calmodulin nor CaCl2 were added, and EDTA concentration was increased to 0.45 mm. NADPH oxidation in the presence of the l-Arg analogue l-agmatine was studied in the same conditions of the NO synthesis assay but replacing l-Arg by 2 mm l-agmatine. All steady-state assays were carried out at 25 °C. Reported values are means ± S.E. of three or more determinations.
Heme Reduction
The electron transfer from the reductase domain to the heme was studied at 10 °C as described previously (20, 30) using a Hi-Tech SF-61 stopped-flow instrument with a diode array detector (Hi-Tech Scientific, Salisbury, UK) equipped for anaerobic work. A protein solution containing 10 μm nNOS, 100 mm EPPS, pH 7.6, 100 mm NaCl, 10 μm H4B, 2 mm l-Arg, 0.5 mm DTT, 50 μm CaM, 5 mm CaCl2, and 1 mm EDTA, at least half-CO-saturated, was mixed with an anaerobic, CO-saturated solution containing 100 mm EPPS, pH 7.6, 100 mm NaCl, and 100 μm NADPH. Heme reduction was followed by the formation of the ferrous-CO complex with a maximum ∼444 nm. The time course of the absorbance changes was fitted to a biexponential equation, where the first phase (absorbance decrease) was assigned to the flavin reduction, and the subsequent absorbance increase was assigned to heme reduction. The signal-to-noise ratio was improved by averaging data from six or more individual mixing experiments.
Midpoint Potential Measurements
Spectroelectrochemical titrations were carried out in a glove-box (Belle Technology, Dorset, UK) under an N2 atmosphere, as described previously (16, 31). Briefly, nNOSoxy proteins were made anaerobic by gel filtration in a Sephadex G-25 column (PD-10, GE Healthcare) equilibrated with anaerobic buffer (100 mm phosphate buffer, pH 7.0, and 125 mm NaCl). Protein samples were diluted to a 3.5-ml final volume (final concentration ≈ 10 μm), and l-Arg (2.5 mm) and H4B (25 μm) were added. The following electron mediator dyes (5 μm) were used: phenosafranine (Em = −252 mV), benzyl viologen (Em = −358 mV) and anthraquinone-2-sulfonate (Em = −225 mV). The titration was carried out at 15 °C by bolus additions of sodium dithionite solution. Absorption spectra were recorded with a Cary 50 spectrophotometer equipped with a dip-probe detector, and the potentials were measured with an Accumet AB15 pH meter (Thermo Fisher Scientific) using a silver/silver chloride microelectrode saturated with 4 m KCl. Reported values for the redox potential measurements are mean values ± S.D. of three or more determinations. Actual errors may be larger as other factors such as electrode accuracy or equilibration errors are not factored in the calculations.
Homology Modeling of the nNOS FMN Domain-CaM Complex
A structure of the FMN domain of iNOS in complex with CaM as been reported (Protein Data Bank code 3HR4) (32). As iNOS and nNOS share a high sequence homology, we believe a homology model of the nNOS sequence based on the iNOS structure should yield reasonable results. The model was built using the Swiss-Model server (33) from the sequences of the wild-type rat nNOS (GenBankTM accession no. CAA42574.1) and human calmodulin (GenBankTM accession no. AAD45181.1) and the available iNOS-CaM structure (Protein Data Bank code 3HR4, chains A and B) as template. The coordinates of the FMN molecule (not modeled by the server) were copied from the 3HR4 structure (chain A) and added to the modeled Protein Data Bank file. The autoinhibitory helix is present in the nNOS sequence but not in iNOS and, therefore, its structure is highly tentative; it is included in the figure as it gives an idea of the location and size of this element. The CaM structure is unchanged from the original (Protein Data Bank code 3HR4, chain B) as the target and query sequences are identical.
Protein-Protein Docking of the Oxygenase and FMN Domains
To assess the possible structure of a functional complex between the oxygenase domain and the FMN domain a set of docking complexes were generated by the docking server ZDOCK (34). The structures used were 1ZVL for the oxygenase domain (35) and the homology model structure of the FMN domain described above. Based on our results and previous mutagenesis scanning of negatively charged residues in the FMN domain (20), we imposed the presence of the following residues in the protein-protein interface: Lys423, Lys620, Lys660 (oxy domain) and Glu762, Asp813, Glu816, and Glu819 (FMN domain). 12 structures were produced by the server, and its feasibility was evaluated according to two distance criteria: the FMN-heme distance should be reasonable for electron transfer (<15 Å), and the distance between the residues Trp716 (C terminus of the oxygenase structure) and Arg727 (N terminus of the FMN structure) should be within the range of the 10 missing residues that must connect both ends (<30 Å). Only one model satisfied both conditions (FMN-heme distance, 4 Å; Trp716–Arg727 distance, 19 Å). Solvent accessible surface calculations were carried out with Surface Racer software (version 5.0) (36) using a probe radius of 1.4 Å.
RESULTS
Selection of Mutated Residues
All positively charged residues indicated in Fig. 1B (Lys406, Arg410, Lys423, Lys447, Lys452, Arg456, Lys620, Lys645, Lys660, and Arg667) were replaced by Asn or Glu except for Lys406 and Arg410. These latter two residues are strictly conserved in all NOS enzymes (supplemental Fig. S2) and are close to the heme proximal ligand (Cys415). Mutagenesis of Arg410 was shown to compromise substrate binding (21) and thus reduces NOS activity in a way independent from FMN domain-heme domain interaction. Lys406 may have a similar effect, and its side chain is mostly buried, contributing only a small positively charged surface (Fig. 1B and supplemental Fig. S1).
General Properties of nNOS Oxygenase Domain Surface Mutants
The mutations did not alter significantly the protein expression levels or heme binding. Analysis of l-Arg binding affinity by UV-visible spectroscopy indicated that the binding in the mutants was similar to wild-type enzyme (supplemental Fig. S3). Dimer formation was studied by gel filtration chromatography (data not shown). The mutants were purified mostly in the dimeric form without major differences in dimer content as compared with wild-type enzyme, although in the case of the R456E and R456N mutants a significant amount of free, proteolyzed flavoprotein domain (≈25%) was found. Mutations of Lys620 did not alter dimer formation, even though this residue can be considered part of the dimer interface (8) and helps to form the positively charged patch on the partner subunit. We determined the cytochrome c reductase activity of the mutants (supplemental Table S2 and supplemental Fig. S4). This activity is dependent on FMN domain to cytochrome c electron transfer and therefore depends mostly on the flavoprotein domain. As expected, mutations in the oxygenase domain did not cause a large alteration in the cytochrome c reduction rates. Addition of calmodulin induces a roughly 10-fold increase in activity in both wild-type and mutant nNOS forms. The higher values for R456E and R456N mutants are most likely due to the presence of free flavoprotein domain.
Rates of Heme Reduction
To assess the effects on the FMN to heme electron transfer, we measured the rate of heme reduction in each nNOS mutant at 10 °C as described (20, 30). The reactions were initiated by mixing excess NADPH with each CaM-bound enzyme in the presence of CO under anaerobic conditions. The rate of heme reduction was determined from the resulting absorbance increase at 444 nm. The heme reduction rates are compared in Fig. 2 (values in supplemental Table S3). The majority of the mutants, including those at Lys447, Lys452, Arg456, Lys645, and Arg667 exhibited little or no change in heme reduction rate as compared with the wild-type enzyme. The K452N mutant was the only mutant whose rate was significantly increased relative to WT nNOS. Conversely, the K423E, K620E, and K660E mutations were detrimental for the heme reduction rates. This implicates at least three of the eight positive charged residues in aiding the FMN to heme electron transfer. For all of the residues we studied, the charge reversal mutation (negatively charged mutant) always had slower heme reduction rates than the neutral mutation counterpart (Fig. 2). This again suggests that the positive electrostatic potential on the nNOSoxy domain surface is important for the efficient docking of the FMN and heme domains and efficient electron transfer.
FIGURE 2.

Ferric heme reduction rates of wild-type nNOS and mutants. Rates were determined by stopped-flow spectroscopy at 10 °C under anaerobic conditions. Dotted lines indicate the rate for wild-type nNOS. Bars indicate means ± S.D.
Heme Midpoint Potential
There is a relatively modest thermodynamic driving force for the FMN to heme electron transfer in nNOS, as indicated by the midpoint potentials of the relevant flavin and heme couples (FMNH2/FMN semiquinone: −273 mV (16) or −274 mV (37) in isolated reductase systems, although values of −220 mV have been reported for the holoenzyme (38); FeIII heme/FeII heme: −248 mV (39) or −306 mV (40) for isolated oxygenase domains, −290 mV estimated for the holoenzyme (38); FeIII heme/FeII-CO heme: −229 mV (as determined at 0.5 mm CO, the NOS ferric heme does not bind CO, and, therefore, the value is a mixture of the the FeIII/FeII-CO redox couple and the FeII/FeII-CO binding equilibrium and is CO-dependent (40)). This makes the heme reduction rate in nNOS sensitive to decreases in the heme reduction potential (see Refs. 27 and 41 for an example). Because the charge reversal mutations we incorporated place electronegative charge near the heme, they could conceivably lower the heme midpoint potential and thus affect heme reduction. We therefore measured the heme midpoint potentials of the two charge reversal mutations that had the greatest impact on the heme reduction rate (K423E and K620E). The estimated midpoint potentials of the two mutants were −188 ± 5 mV for K423E and −201 ± 10 mV for K620E (see supplemental Fig. S5; values for the redox potential measurements are mean value ± S.D. of three or more determinations), which are more positive than the midpoint potential of −253 ± 3 mV here determined for WT nNOSoxy, in agreement with previous values (39). This indicates that they do not diminish the thermodynamic driving force for heme reduction in nNOS and so must slow the heme reduction rate by a different mechanism.
Steady-state Electron Flux through the Heme
Because l-agmatine is unable to support NO synthesis, there is no feedback inhibition by NO that occurs during steady-state catalysis of NADPH oxidation in the presence of l-agmatine. Under this circumstance, the steady-state NADPH oxidation rate is proportional to the electron flux through the heme to O2 (27, 42). We therefore measured the NADPH oxidation rates of the mutant NOS proteins in the presence of l-agmatine. Fig. 3 plots these steady-state rates (values in supplemental Table S2) against the corresponding heme reduction rates for each mutant determined by stopped-flow measurements (see Fig. 2). A good correlation is observed (r2 = 0.964), indicating that the surface mutations alter electron flux through the nNOS heme primarily by altering the heme reduction rate. The electron flux value we measured for nNOS with l-agmatine yields an estimate of 10.5 s−1 for the heme reduction rate at 25 °C, in good agreement with the 13 s−1 estimate for flavin to heme electron transfer recently derived from laser flash photolysis experiments done at room temperature (43).
FIGURE 3.

Heme reduction rates versus NADPH oxidation in the presence of agmatine. Relationship between the rates of heme reduction and the steady-state NADPH oxidation in the presence of the inhibitor l-agmatine. Open circles indicate charge reversal mutations, and closed circles indicate charge-neutralizing mutations.
NO Synthesis Activity and Corresponding NADPH Oxidation
The steady-state rates of NO synthesis and concurrent rates of NADPH oxidation for each CaM-bound enzyme are compared in Fig. 4. All mutants had a measurable rate of NO synthesis activity (Fig. 4, top; supplemental Table S2). The lowest activity was observed in the K423E mutant, confirming an earlier report (17). Apart from K423E, only K660N had an NO synthesis activity that was significantly lower than that of wild-type. On the other hand, four mutants showed super-normal NO synthesis activity: K660E (163% increase over wild-type), R456E (129% increase), K645E (68% increase), and R667E (61% increase). Mutations at three positively charged residues caused little change in NO synthesis activity: Lys447, Lys452, and Lys620. For several mutants, the NO synthesis activities did not correlate with their measured rates of heme reduction or with their electron flux rates in the presence of l-agmatine (for example, K660N and K620E).
FIGURE 4.

Steady-state NO synthesis and NADPH oxidation activities of nNOS mutants. Top, NO synthesis rates in the presence of CaM. Bottom, NADPH oxidation rates in the presence or absence of CaM. Assays were carried out at 25 °C. Dotted lines indicate the activity of wild-type nNOS. Bars indicate means ± S.D.
In general, the NADPH oxidation rates (Fig. 4, bottom and supplemental Table S2) correlate with the NO synthesis activities. A value of 2.4 molecules of NADPH oxidized per NO formed for WT nNOS is slightly higher but close to other reported values (20). The NADPH/NO ratios are shown in supplemental Table S4 and supplemental Fig. S6. The main alterations of the NADPH/NO ratio are found in K423E, K660N, R667N, K447E (all have greater uncoupled NADPH oxidation), K660E, and K645E (both have better coupled NADPH oxidation than WT). For K423E, the vast majority of NADPH oxidation is uncoupled (>200 per NO), whereas more moderate uncoupling is seen for K660N (4.0 NADPH per NO), R667N (3.3), and K447E (3.1). In contrast, the two mutants with better coupling than wild-type, K660E (1.6 NADPH per NO) and K645E (1.8), approach the theoretical minimal value of 1.5 NADPH per NO (2).
DISCUSSION
Electron transfer between the NOS FMN and heme is key for NO synthesis. Previous work identified some protein structural features that may influence this step in NOS enzymes (17, 19, 20, 30, 42, 44). The electron transfer may rely on complementary electrostatic interaction between charged surface residues (18, 19), as described for flavodoxins, dual-flavin enzymes, and other enzyme systems (32, 45–49). This work is the first to examine the individual roles of eight residues that create a positive charge cluster on the NOSoxy domain. Results from steady-state and stopped-flow measurements are in good agreement with one another and define a hierarchy of importance for the eight charged surface residues.
When judged under the most conservative criterion (mutation to neutralize each individual positive charge), the individual charge at only two residues (Lys423 and Lys620) is required for supporting heme reduction in nNOS. This group grows to include three additional residues (Lys645, Lys660, and Arg667) when judged by results obtained upon charge reversal rather than charge neutralization. Our findings regarding the K423E mutation match those in a previous study (17). Our results show the following: (i) individual charge neutralization or charge reversal at Lys423 and Lys620 were the most effective in inhibiting heme reduction in nNOS; and (ii) the mutational effects we observed on heme reduction were not associated with a change in the heme midpoint potential and thus were not due to an altered thermodynamic driving force for heme reduction. Together, this argues that Lys423 and Lys620 enable heme reduction primarily through electrostatic interactions.
Sequence Conservation
NOS protein sequence alignments (supplemental Fig. S2) show that the degree of sequence conservation among the positive patch residues does not correlate so well with their rank order in influencing nNOS heme reduction. For example, Arg456, Arg667, and Lys660 are the most conserved among the eight residues we studied, despite our finding that their individual charges have either no (Arg456, Arg667) or only moderate (Lys660) roles in supporting heme reduction in nNOS. Conversely, the two residues we judged as most important are not conserved highly among NOS enzymes, with Lys423 being sometimes replaced by Asn and Lys620 being often replaced by Thr. The lowest degree of conservation corresponds to Lys447 and Lys452, consistent in our case with their being of low relevance for nNOS heme reduction, but in sum, a global importance for positive charge at Lys423 and Lys620 is not apparent from the alignments. It is particularly notable that on iNOSoxy, several of the corresponding residues are of neutral charge. The iNOS FMN domain also has more neutral residues in place of the negatively charged residues on the complementary surface patch of the nNOS FMN domain (20). This implies nNOS may have a greater capacity for electrostatic interaction between its FMN and NOSoxy domains, a feature that is associated with the rate of heme reduction being four to seven times faster in nNOS than in iNOS (2).
Heme Reduction versus NO Synthesis Rates
There is an apparent mismatch between the NO synthesis activities and heme reduction rates in our study. For example, mutants that show increased NO synthesis activities relative to WT nNOS (R456E, K645E, K660E, and R667E) have slower heme reduction rates. Fig. 5 plots the measured NO synthesis activities against either the corresponding heme reduction rates (upper panel) or the steady state rates of electron flux through the heme as measured by NADPH oxidation with l-agmatine (lower panel). Both graphs show that the relationship approximates to a bell shape and reveals that WT nNOS has heme reduction and electron flux rates that are greater than the range associated with highest NO synthesis activity. A global model of NOS catalysis (Fig. 6), which we previously described in detail, explains why such a bell-shaped relationship exists between the heme reduction rate and the steady-state NO synthesis activity in NOS enzymes (2, 50). Basically, the relative rates of heme reduction (kr, kr′) and FeIII-NO dissociation (kd) determine the proportion of nNOS molecules that enter the productive (NO-releasing) cycle versus the futile (NO dioxygenase) cycle during steady-state catalysis (Fig. 6). The kr, kr′, and kd parameters thus strike a balance, because although increasing the kr value allows the nNOS to synthesize NO faster, it also causes a greater proportion of nNOS molecules to enter the futile cycle. According to this model, the kr, kr′ of WT nNOS are above the optimum regarding steady-state NO release (i.e. the steady-state NO synthesis activity). The WT nNOS has a lower activity because its rate of heme reduction is causing a more than optimal amount of the enzyme molecules to enter the futile cycle in unit time. In contrast, the NOSoxy surface mutations that slow heme reduction by a moderate amount (like K620N) bring the kr value down into the optimal range, whereas mutations that slow heme reduction by a greater extent (like K620E) fall below the optimal range for kr and in which case exhibit a lower NO synthesis activity because their actual NO biosynthesis is slow.
FIGURE 5.

Heme reduction rates and NADPH oxidation rates with agmatine versus NO synthesis activities. Top, relationship between the steady-state NO synthesis activity and the ferric heme reduction rate in the full-length nNOS proteins. Bottom, NADPH oxidation rates in the presence of agmatine versus NO synthesis rates. The line denotes a hypothetical fit of the data along with the trends predicted by the global model (2). Open circles indicate charge reversal mutations, and closed circles indicate charge neutralizing mutations.
FIGURE 6.

Global model of NOS catalysis. Global kinetic model for NOS catalysis. FMN to heme electron transfer (kr) is rate-limiting for the NO synthesis reactions. kcat1 and kcat2 are the conversion rates of the enzyme FeIIO2 species to products in the l-Arg and Nω-hydroxy-l-arginine (NOHA) reactions, respectively. Not all of the produced NO is released as the ferric heme-NO product complex (FeIIINO) can either release NO (kd) or become reduced (kr′) to a ferrous heme-NO complex (FeIINO), which reacts with O2 (kox) to regenerate ferric enzyme through a futile NO dioxygenase cycle without net NO release. The observed steady-state NO synthesis rates are determined by the relative rates of kr, kox, and kd.
The experimental data in Fig. 5 distribute approximately according to the theoretical relationship between the NO synthesis activity and heme reduction rate in nNOS as derived from a computer simulation of the global model (see Fig. 3 in Ref. 2). This relationship was demonstrated before, albeit to a less complete extent, in a study where the nNOS heme reduction rate was varied using a series of CaM protein structural analogues (30). In the current data, the apparent lack of outliers beyond what might be attributed to normal experimental variability suggests that the nNOSoxy surface mutations may largely or completely restrict their effects to the heme reduction rate (kr) in nNOS and may not significantly alter the other two main kinetic variables (kox and kd in Fig. 6), which otherwise would skew the relationship between the NO synthesis activity and heme reduction rate (for example, as occurs in E762N nNOS; see Ref. 51).
Docking of the Oxygenase and FMN Domains
The three Lys residues that were identified as electrostatic contributors for nNOS heme reduction (Lys423, Lys620, and Lys660) are positioned on the surface to triangulate the heme edge in NOSoxy (Fig. 1B and supplemental Fig. S1) and thereby could promote a specific docking of the negatively charged FMN domain for electron transfer. Indeed, placing together the two most important residues for nNOS heme reduction (Glu762 from the FMN domain and Lys423 of NOSoxy) was found in our previous modeling to position the bound FMN and heme groups within a reasonable distance for electron transfer (52). Additional alignment could presumably be achieved through concerted electrostatic interactions of Lys620 and Lys660. We took advantage of this acquired knowledge to assess the putative structure of an nNOSoxy-FMN complex. In silico docking of the putative nNOSoxy-FMN domain complex was carried out as described under “Experimental Procedures.” Briefly, we used a FMN-CaM complex modeled after the recent iNOS FMN-CaM complex structure (32) and a nNOSoxy structure and imposed restrictions to the docking server so as to include in the interface the residues Lys423, Lys620, and Lys660 from nNOSoxy and Glu762, Asp813, Glu816, and Glu819 from the FMN domain, whose importance was shown previously (20). From 12 docking solutions, only one was found that complied with the distance restrictions, namely, feasible distances for FMN-heme electron transfer and the distance of a linker within residues Trp716 (C terminus of the oxygenase structure) and Arg727 (N terminus of the FMN structure). The resulting model is shown in Fig. 7 and supplemental Fig. S7. The structure is quite similar to the putative model presented for iNOS (32). This fact is surprising as differences exist on the surface charges of iNOS and nNOS as noted earlier (20). (The iNOSoxy surface is less positively charged and its FMN subdomain surface is less negatively charged than in nNOS). Also, in our case, we did not impose the further restriction of superimposing the rest of the reductase domain (19) on the FMN domain structure. The interface area was close to 1200 Å2, slightly larger than previous models (725 Å2 (32) or 900 Å2 (53)). As another point in common with the model by Xia et al. (32), the nNOS Trp587, analogous residue to Trp372 in iNOS, is placed between the heme and FMN cofactors. The illustrated situation of the charged residues of nNOSoxy and the FMN domains is coherent with the results reported here and for the FMN domain (20), as a number of residues that have little effect on heme reduction are not in the interface, even though they were not excluded explicitly during the docking process (for instance, Lys447, Lys452, Glu789, and Glu790 (Fig. 7)). In summary, the model seems to be a reasonable working model for the FMN-heme interface and a guide for further experiments.
FIGURE 7.

Docking model of a nNOSoxy-FMN-CaM complex. Left, molecular surface of the complex. nNOSoxy monomers are shown in light blue and light green; the FMN domain is shown in light orange, and CaM is shown in red. The residues Trp716 and Arg727, which are linked by a 10-residue sequence are shown in green and orange, respectively; residues Lys423 and Glu762 are shown in blue and red, respectively. Right, location of selected residues in the model. α carbon traces are shown as thin lines. Residues mutated in this study and in Panda et al. (20) are shown as sticks. Oxy domain residues are labeled in blue/green (monomer A/B); FMN domain residues are labeled in orange. Heme (red) and FMN (orange) are shown as sticks.
Implications for the nNOS Mechanism
When CaM binding triggers the FMN to heme electron transfer in nNOS, its effect is at least 2-fold. First, its binding destabilizes the FAD-FMN domain interface in nNOS, and thereby stabilizes a “FMN-deshielded” or “open” conformation that makes the FMN domain available to interact with NOSoxy or with non-native electron acceptors like cytochrome c (15, 54–56). However, this shift in the nNOS conformational equilibrium is not enough to promote significant heme reduction and NO synthesis in nNOS (56). CaM is therefore thought to promote an additional step that brings the bound FMN and heme groups within a distance amenable for electron transfer. The exact nature of this step is uncertain and open to conjecture. For example, it might depend on the removal of a “hinge stop” that otherwise prevents conformational movement that brings the FMN and heme closer (15, 32) or could also involve CaM constraining the motions of the deshielded FMN domain to increase the likelihood of productive docking with NOSoxy (32, 56, 57). In any case, if the long range motion of the FMN domain (approximately a 60-Å distance) places it within the proximity of the NOSoxy surface, more specific short range motions (within 15–25 Å) are likely to occur that can enable a transition state to be achieved for FMN to heme electron transfer (45). The general distance constraints on electrostatic interactions dictate that they would only come into play during these short range motions, which likely involve rapid conformational sampling as described in other redox enzymes (58–60).
Conclusions
Our study provides new experimental support for structural models that invoke charge pairing in the domain-domain interactions associated with electron transfer in nNOS (15, 18–20, 32). During steady-state catalysis, the relatively slow long range motions of the FMN domain (16, 56), rather than the FMN-NOSoxy interaction per se, limits electron flux through the heme. Our current findings and working structural model suggest that the complementary electrostatic interactions involving the triad Lys423, Lys620, and Lys660 is most involved in directing short range conformational sampling motions that enable the FMN domain to achieve docked complexes with NOSoxy that are productive for electron transfer. Because the long range conformational motion of the FMN domain is relatively slow, it may inherently blunt the importance of the electrostatic interactions in heme reduction. In addition, ensemble effects of multiple positive surface charges were not tested here but might conceivably increase the importance of the individual residues beyond what we observed from their individual mutations. Finally, protein sequence comparisons suggest that heme reduction in various NOS isoforms may rely to different extents on electrostatic interactions at the domain interface.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants CA53914, GM51491, and HL76491 (to D. J. S.). This work was also supported by American Heart Association Postdoctoral Fellowship 0625632B (to J. T.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S4 and Figs. S1–S7.
- iNOS
- inducible nitric-oxide synthase
- CaM
- calmodulin
- EPPS
- 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid
- H4B
- (6R)-5,6,7,8-tetrahydro-l-biopterin
- nNOS
- neuronal nitric-oxide synthase
- WT
- wild-type.
REFERENCES
- 1.Pfeiffer S., Mayer B., Hemmens B. (1999) Angew. Chem. Int. Ed. 38, 1714–1731 [DOI] [PubMed] [Google Scholar]
- 2.Stuehr D. J., Santolini J., Wang Z. Q., Wei C. C., Adak S. (2004) J. Biol. Chem. 279, 36167–36170 [DOI] [PubMed] [Google Scholar]
- 3.Lancaster J. R., Jr. (1997) Nitric Oxide 1, 18–30 [DOI] [PubMed] [Google Scholar]
- 4.Alderton W. K., Cooper C. E., Knowles R. G. (2001) Biochem. J. 357, 593–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stuehr D. J. (1999) Biochim. Biophys. Acta 1411, 217–230 [DOI] [PubMed] [Google Scholar]
- 6.Gorren A. C., Mayer B. (2007) Biochim. Biophys. Acta 1770, 432–445 [DOI] [PubMed] [Google Scholar]
- 7.Fischmann T. O., Hruza A., Niu X. D., Fossetta J. D., Lunn C. A., Dolphin E., Prongay A. J., Reichert P., Lundell D. J., Narula S. K., Weber P. C. (1999) Nat. Struct. Biol. 6, 233–242 [DOI] [PubMed] [Google Scholar]
- 8.Crane B. R., Arvai A. S., Ghosh D. K., Wu C., Getzoff E. D., Stuehr D. J., Tainer J. A. (1998) Science 279, 2121–2126 [DOI] [PubMed] [Google Scholar]
- 9.Bredt D. S., Hwang P. M., Glatt C. E., Lowenstein C., Reed R. R., Snyder S. H. (1991) Nature 351, 714–718 [DOI] [PubMed] [Google Scholar]
- 10.Siddhanta U., Presta A., Fan B., Wolan D., Rousseau D. L., Stuehr D. J. (1998) J. Biol. Chem. 273, 18950–18958 [DOI] [PubMed] [Google Scholar]
- 11.Matsuda H., Iyanagi T. (1999) Biochim. Biophys. Acta 1473, 345–355 [DOI] [PubMed] [Google Scholar]
- 12.Abu-Soud H. M., Yoho L. L., Stuehr D. J. (1994) J. Biol. Chem. 269, 32047–32050 [PubMed] [Google Scholar]
- 13.Craig D. H., Chapman S. K., Daff S. (2002) J. Biol. Chem. 277, 33987–33994 [DOI] [PubMed] [Google Scholar]
- 14.Konas D. W., Zhu K., Sharma M., Aulak K. S., Brudvig G. W., Stuehr D. J. (2004) J. Biol. Chem. 279, 35412–35425 [DOI] [PubMed] [Google Scholar]
- 15.Welland A., Garnaud P. E., Kitamura M., Miles C. S., Daff S. (2008) Biochemistry 47, 9771–9780 [DOI] [PubMed] [Google Scholar]
- 16.Ilagan R. P., Tiso M., Konas D. W., Hemann C., Durra D., Hille R., Stuehr D. J. (2008) J. Biol. Chem. 283, 19603–19615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimanuki T., Sato H., Daff S., Sagami I., Shimizu T. (1999) J. Biol. Chem. 274, 26956–26961 [DOI] [PubMed] [Google Scholar]
- 18.Crane B. R., Rosenfeld R. J., Arvai A. S., Ghosh D. K., Ghosh S., Tainer J. A., Stuehr D. J., Getzoff E. D. (1999) EMBO J. 18, 6271–6281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcin E. D., Bruns C. M., Lloyd S. J., Hosfield D. J., Tiso M., Gachhui R., Stuehr D. J., Tainer J. A., Getzoff E. D. (2004) J. Biol. Chem. 279, 37918–37927 [DOI] [PubMed] [Google Scholar]
- 20.Panda K., Haque M. M., Garcin-Hosfield E. D., Durra D., Getzoff E. D., Stuehr D. J. (2006) J. Biol. Chem. 281, 36819–36827 [DOI] [PubMed] [Google Scholar]
- 21.Yadav J., Fujiwara S., Sagami I., Shimizu T. (2004) Chem. Lett. 33, 752–753 [Google Scholar]
- 22.Yumoto T., Sagami I., Daff S., Shimizu T. (2000) J. Inorg. Biochem. 82, 163–170 [DOI] [PubMed] [Google Scholar]
- 23.Adak S., Ghosh S., Abu-Soud H. M., Stuehr D. J. (1999) J. Biol. Chem. 274, 22313–22320 [DOI] [PubMed] [Google Scholar]
- 24.Panda K., Adak S., Aulak K. S., Santolini J., McDonald J. F., Stuehr D. J. (2003) J. Biol. Chem. 278, 37122–37131 [DOI] [PubMed] [Google Scholar]
- 25.Stuehr D. J., Ikeda-Saito M. (1992) J. Biol. Chem. 267, 20547–20550 [PubMed] [Google Scholar]
- 26.Ghosh D. K., Crane B. R., Ghosh S., Wolan D., Gachhui R., Crooks C., Presta A., Tainer J. A., Getzoff E. D., Stuehr D. J. (1999) EMBO J. 18, 6260–6270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adak S., Crooks C., Wang Q., Crane B. R., Tainer J. A., Getzoff E. D., Stuehr D. J. (1999) J. Biol. Chem. 274, 26907–26911 [DOI] [PubMed] [Google Scholar]
- 28.McMillan K., Masters B. S. (1993) Biochemistry 32, 9875–9880 [DOI] [PubMed] [Google Scholar]
- 29.Gorren A. C., Schmidt K., Mayer B. (2002) Biochemistry 41, 7819–7829 [DOI] [PubMed] [Google Scholar]
- 30.Adak S., Santolini J., Tikunova S., Wang Q., Johnson J. D., Stuehr D. J. (2001) J. Biol. Chem. 276, 1244–1252 [DOI] [PubMed] [Google Scholar]
- 31.Tejero J., Biswas A., Wang Z. Q., Page R. C., Haque M. M., Hemann C., Zweier J. L., Misra S., Stuehr D. J. (2008) J. Biol. Chem. 283, 33498–33507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xia C., Misra I., Iyanagi T., Kim J. J. (2009) J. Biol. Chem. 284, 30708–30717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwede T., Kopp J., Guex N., Peitsch M. C. (2003) Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen R., Li L., Weng Z. (2003) Proteins 52, 80–87 [DOI] [PubMed] [Google Scholar]
- 35.Matter H., Kumar H. S., Fedorov R., Frey A., Kotsonis P., Hartmann E., Fröhlich L. G., Reif A., Pfleiderer W., Scheurer P., Ghosh D. K., Schlichting I., Schmidt H. H. (2005) J. Med. Chem. 48, 4783–4792 [DOI] [PubMed] [Google Scholar]
- 36.Tsodikov O. V., Record M. T., Jr., Sergeev Y. V. (2002) J. Comput. Chem. 23, 600–609 [DOI] [PubMed] [Google Scholar]
- 37.Noble M. A., Munro A. W., Rivers S. L., Robledo L., Daff S. N., Yellowlees L. J., Shimizu T., Sagami I., Guillemette J. G., Chapman S. K. (1999) Biochemistry 38, 16413–16418 [DOI] [PubMed] [Google Scholar]
- 38.Gao Y. T., Smith S. M., Weinberg J. B., Montgomery H. J., Newman E., Guillemette J. G., Ghosh D. K., Roman L. J., Martasek P., Salerno J. C. (2004) J. Biol. Chem. 279, 18759–18766 [DOI] [PubMed] [Google Scholar]
- 39.Presta A., Weber-Main A. M., Stankovich M. T., Stuehr D. (1998) J. Am. Chem. Soc. 120, 9460–9465 [Google Scholar]
- 40.Ost T. W., Daff S. (2005) J. Biol. Chem. 280, 965–973 [DOI] [PubMed] [Google Scholar]
- 41.Adak S., Wang Q., Stuehr D. J. (2000) J. Biol. Chem. 275, 17434–17439 [DOI] [PubMed] [Google Scholar]
- 42.Haque M. M., Panda K., Tejero J., Aulak K. S., Fadlalla M. A., Mustovich A. T., Stuehr D. J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 9254–9259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salerno J. C. (2008) FEBS Lett. 582, 1395–1399 [DOI] [PubMed] [Google Scholar]
- 44.Feng C., Roman L. J., Hazzard J. T., Ghosh D. K., Tollin G., Masters B. S. (2008) FEBS Lett. 582, 2768–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tetreault M., Cusanovich M., Meyer T., Axelrod H., Okamura M. Y. (2002) Biochemistry 41, 5807–5815 [DOI] [PubMed] [Google Scholar]
- 46.Hall D. A., Vander Kooi C. W., Stasik C. N., Stevens S. Y., Zuiderweg E. R., Matthews R. G. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 9521–9526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Medina M., Abagyan R., Gomez-Moreno C., Fernandez-Recio J. (2008) Proteins 72, 848–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheinerman F. B., Norel R., Honig B. (2000) Curr. Opin. Struct. Biol. 10, 153–159 [DOI] [PubMed] [Google Scholar]
- 49.Wang M., Roberts D. L., Paschke R., Shea T. M., Masters B. S., Kim J. J. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 8411–8416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santolini J., Adak S., Curran C. M., Stuehr D. J. (2001) J. Biol. Chem. 276, 1233–1243 [DOI] [PubMed] [Google Scholar]
- 51.Haque M. M., Fadlalla M., Wang Z. Q., Ray S. S., Panda K., Stuehr D. J. (2009) J. Biol. Chem. 284, 19237–19247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei C. C., Wang Z. Q., Tejero J., Yang Y. P., Hemann C., Hille R., Stuehr D. J. (2008) J. Biol. Chem. 283, 11734–11742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ilagan R. P., Tejero J., Aulak K. S., Ray S. S., Hemann C., Wang Z. Q., Gangoda M., Zweier J. L., Stuehr D. J. (2009) Biochemistry 48, 3864–3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tiso M., Tejero J., Panda K., Aulak K. S., Stuehr D. J. (2007) Biochemistry 46, 14418–14428 [DOI] [PubMed] [Google Scholar]
- 55.Roman L. J., Masters B. S. (2006) J. Biol. Chem. 281, 23111–23118 [DOI] [PubMed] [Google Scholar]
- 56.Stuehr D. J., Tejero J., Haque M. M. (2009) FEBS J. 276, 3959–3974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghosh D. K., Salerno J. C. (2003) Front Biosci. 8, d193–d209 [DOI] [PubMed] [Google Scholar]
- 58.Toogood H. S., van Thiel A., Scrutton N. S., Leys D. (2005) J. Biol. Chem. 280, 30361–30366 [DOI] [PubMed] [Google Scholar]
- 59.Leys D., Basran J., Talfournier F., Sutcliffe M. J., Scrutton N. S. (2003) Nat. Struct. Biol. 10, 219–225 [DOI] [PubMed] [Google Scholar]
- 60.Kang S. A., Crane B. R. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 15465–15470 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.