

Abstract
The objective of this study was to identify the role of individual amino acid residues in determining the substrate specificity of the yeast mitochondrial citrate transport protein (CTP). Previously, we showed that the CTP contains at least two substrate-binding sites. In this study, utilizing the overexpressed, single-Cys CTP-binding site variants that were functionally reconstituted in liposomes, we examined CTP specificity from both its external and internal surfaces. Upon mutation of residues comprising the more external site, the CTP becomes less selective for citrate with numerous external anions able to effectively inhibit [14C]citrate/citrate exchange. Thus, the site 1 variants assume the binding characteristics of a nonspecific anion carrier. Comparison of [14C]citrate uptake in the presence of various internal anions versus water revealed that, with the exception of the R189C mutant, the other site 1 variants showed substantial uniport activity relative to exchange. Upon mutation of residues comprising site 2, we observed two types of effects. The K37C mutant displayed a markedly enhanced selectivity for external citrate. In contrast, the other site 2 mutants displayed varying degrees of relaxed selectivity for external citrate. Examination of internal substrates revealed that, in contrast to the control transporter, the R181C variant exclusively functioned as a uniporter. This study provides the first functional information on the role of specific binding site residues in determining mitochondrial transporter substrate selectivity. We interpret our findings in the context of our homology-modeled CTP as it cycles between the outward-facing, occluded, and inward-facing states.
Keywords: Anion Transport, Bioenergetics, Membrane, Mitochondria, Mitochondrial Transport
Introduction
Citrate is a prominent intermediate in both carbohydrate and lipid metabolism. Once it is formed within the mitochondrial matrix as part of the tricarboxylic acid cycle, it is then either processed by the cycle enzymes to generate NADH and FADH2 leading ultimately to the formation of ATP, or it can be transported out of the mitochondrial matrix across the inner membrane and into the intermembrane space via the mitochondrial inner membrane citrate transport protein (CTP)2 (1, 2). Citrate then passively diffuses through a voltage-dependent anion-selective channel within the outer membrane, into the cytoplasm, where it is broken down by citrate lyase to acetyl-CoA and oxaloacetate. The resulting acetyl-CoA represents a prime carbon source fueling fatty acid, triacylglycerol, and sterol biosyntheses (3–6). Consequently, the CTP is critical to the energy metabolism of eukaryotic cells.
The mitochondrial CTP catalyzes an obligatory exchange of the dibasic form of tricarboxylates (i.e. citrate and isocitrate) either for each other in yeast (7, 8) or for dicarboxylates or phosphoenolpyruvate in higher eukaryotes (1, 2). Its function is altered in several diseases, including cancer (9) and diabetes (10), and it is thought to figure prominently in the abnormal bioenergetics observed with these pathologies. In terms of its function in the diabetic state, Joseph et al. (11) demonstrated that citrate transport out of mitochondria on the CTP was key in regulating glucose-stimulated insulin secretion from pancreatic islet β-cells. Moreover, suppression or enhancement of CTP function caused a corresponding change in glucose-stimulated insulin secretion leading to the suggestion that the CTP may be of critical importance in regulating insulin secretion from the pancreas (11, 12).
Because of the importance of the CTP in physiology and pathology, our laboratory has carried out extensive investigations to elucidate its structure-based mechanism. Thus, we have purified (13, 14), cloned (15), overexpressed (7, 16), and functionally reconstituted both the rat liver and the yeast mitochondrial CTPs. Based on the x-ray structure of the mitochondrial ADP/ATP carrier (17), we developed a homology-modeled structure of the CTP (18) and validated key features of this model, including localization of the substrate transport pathway (19–22). Importantly, kinetic studies with single-Cys CTP variants in combination with molecular modeling has enabled the identification of two substrate-binding sites that reside at increasing depths within the membrane bilayer (Fig. 1) (23). Furthermore, in silico screening of the ZINC data base coupled with experimental characterization of selected compounds has led to the discovery of a CTP inhibitor (i.e. compound 792949), which likely binds to and spans both substrate-binding sites simultaneously (24). EPR analysis indicates that, as predicted, this inhibitor causes substantial immobilization of multiple CTP domains (25).
FIGURE 1.

Two citrate-binding sites in the cytosol-facing conformation of the CTP. View is from the cytosolic face (see inset on left) looking down into the transport path. Nitrogen atoms are shown in blue, oxygen in red, citrate carbons in orange, site 1 carbons in magenta, and site 2 carbons in cyan. “Site 1” is depicted in a slightly larger and darker font to reflect the fact that it is closer to the membrane surface and thus to the eye of the reader. Site 2 is further away in the middle of the transport pathway. Although two citrate molecules are depicted to indicate substrate interactions at sites 1 and 2, kinetic data indicate that one citrate molecule is bound at a time per CTP monomer during a transport cycle.
Building upon this foundation, this study focused on characterizing the role that CTP-binding site residues play in determining substrate specificity. We examined the specificity of the CTP from both its external and internal surfaces and observed that upon mutation of site 1 residues, one at a time to cysteine, the CTP becomes considerably less selective for citrate and displays the characteristics of a nonspecific anion carrier. Furthermore, certain of these CTP variants functioned primarily as uniporters rather than as strict obligatory exchangers. Upon mutation of site 2 residues, we found a variety of different effects from increased specificity (e.g. K37C) to relaxed specificity (e.g. R181C, K239C, R276C, and R279C), as well as varying levels of change in the transport mode from obligatory exchange to uniport. These findings provide important new insights into the structure-based mechanism of the CTP and are discussed in this context.
EXPERIMENTAL PROCEDURES
Preparation, Expression, and Isolation of Single-Cys CTP Mutants
The nine single-Cys CTP-binding site mutants were constructed using the QuikChange site-directed mutagenesis kit and cloned into storage (NovaBlue) and expression (BL21(DE3)) hosts as described previously (20, 23). Expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. Two hours following induction, cells were harvested and lysed, and the inclusion body fraction was isolated as detailed previously (23). CTP was extracted from inclusion bodies via addition of 6.0 ml of ice-cold 1.2% (w/v) Sarkosyl (23). Following centrifugation at 231,000 × g for 45 min, the resulting supernatant contained the solubilized CTP. The quantity of Sarkosyl-solubilized CTP was determined by the method of Kaplan and Pedersen (26) except the dye was eluted from the Millipore filters with 1.0 ml of elution buffer (25 mm NaOH, 0.05 mm EDTA, 50% (v/v) ethanol).
Incorporation of CTP Mutants into Phospholipid Vesicles and Determination of the Ability of Alternative External Anions to Compete with [14C]Citrate/Citrate Exchange
Solubilized single-Cys CTP mutants were incorporated into phospholipid vesicles in the presence of 0.075 m citrate via the freeze-thaw-sonication procedure as detailed previously (7, 13). The Cys-less CTP was incorporated in the presence of 0.05 m citrate. Immediately prior to the transport assay, a given sample was thawed and sonicated on ice (30 sonic bursts; 0.7 s/bursts; Branson Sonifier 250; small tip), and the extraliposomal citrate was removed on either 1.75- or 3-ml Dowex columns. The sample was immediately assayed for transport.
Sensitivity of the reconstituted citrate/citrate exchange to externally added anions was investigated as follows. Proteoliposomes (45 μl) were preincubated with 5 μl of either buffer (experimental sample) or 1 m BTC (Sigma) (control sample) for 12–13 min at 21 °C. Transport was initiated by the addition of 25 μl of a solution of [1,5-14C]citrate (GE Healthcare; specific radioactivity = 11–65 × 103 cpm/nmol; Cf = 1–2 mm) plus unlabeled alternative anion (Cf = 25–50 mm). In all cases, a 25-fold excess of competing substrate was employed. The unlabeled substrates used were isocitrate, phosphoenolpyruvate, malate, succinate, α-ketoglutarate, malonate, ADP, glutamate, pyruvate, and phosphate. The uptake time ranged from 1 to 165 min depending on the intrinsic activity of a given CTP mutant. Experimental samples were quenched by the addition of 5 μl of 1 m BTC, whereas the control samples received an equal volume of buffer. Following transport, intraliposomal radiolabeled citrate was separated from the external radiolabel via chromatography on 2.6 ml of Dowex resin in Bio-Rad support columns. The eluted intraliposomal radiolabel was quantified via liquid scintillation counting.
The effect of an externally added anionic substrate on the [14C]citrate/citrate exchange of the Cys-less CTP and single-Cys CTP variants is expressed as the percentage of the initial BTC-sensitive [14C]citrate/citrate exchange (measured in the presence of water) that remained in the presence of a given anionic substrate concentration. This was calculated as follows: (i) determining the BTC-sensitive [14C]citrate/citrate exchange via subtraction of the control value from the experimental value, in the absence of the anionic substrate; (ii) subtracting the control value from the experimental value obtained in the presence of 25–50 mm anionic substrate; (iii) determining the ratio of this difference to the uninhibited BTC-sensitive [14C]citrate/citrate exchange; and (iv) application of the formula (1 − ratio) × 100.
Assessment of the Ability of Various Internal Anionic Substrates to Support the Influx of External [14C]Citrate with the Reconstituted Binding Site Mutants
Proteoliposomes, preloaded with 50 mm anionic substrate, were prepared as described above. Following removal of extraliposomal substrate on Dowex columns, transport was conducted as follows. Proteoliposomes (45 μl) were preincubated with 5 μl of either buffer (experimental sample) or 1 m BTC (control sample) for 5–7 min at 21 °C. Transport was initiated by the addition of 25 μl of 6 mm [1,5-14C]citrate (GE Healthcare; specific radioactivity = 18–65 × 103 cpm/nmol). The uptake time ranged from 10 s to 4.5 h depending on the intrinsic activity of a given CTP mutant. Experimental samples were quenched by the addition of 5 μl of 1 m BTC, whereas the control samples received an equal volume of buffer. Following transport, intraliposomal radiolabeled citrate was separated from the external radiolabel via chromatography on 2.6 ml of Dowex resin in Bio-Rad support columns. The eluted intraliposomal radiolabel was quantified via liquid scintillation counting. The BTC-sensitive transport rate was calculated by subtracting the control value from the experimental value.
Calculation of Ki and Selectivity Coefficient Values
For each mutant, each of the anions was modeled as a competitive inhibitor using the following equation: v = Vmax × [S]/(Km × (1 + [I]/Ki) + [S]). Ki, which for a competing substrate is its Km value, was computed from a single inhibitor and single substrate concentration using the following equation: Ki = Km × [I] × (1 − % inhibition)/((Km + [S]) × % inhibition), where % inhibition is calculated from the transport activity measured in the presence and absence of the anionic inhibitor as described above. The relative standard error from the inhibition data is propagated as the error in Ki. The selectivity coefficient is defined as Ki/Km and was calculated for each mutant/anion pair as described in the legend to Table 4. It should be noted that while our use of a competitive model of inhibition may or may not be strictly correct and thus may affect the absolute value of the selectivity coefficient, the trends observed in this parameter should be retained in any model of inhibition that might apply. We note the following: (i) compounds that closely resemble the structure of citrate are in fact predominantly competitive inhibitors (24); and (ii) there exists a close resemblance between citrate and the anions tested in these studies. These points provide strong justification for the premise that the anions tested are likely to behave primarily as competitive inhibitors. To validate the underlying assumptions used in this approach, with two mutant-anion pairs (i.e. R181C/malate and R189C/malate), comprehensive substrate-inhibitor data sets were obtained. As depicted in supplemental Fig. 1, with both mutant-anion pairs a primarily competitive inhibition pattern was observed. Furthermore, the competitive Ki values calculated via a global fit analysis using the above equation revealed Ki values that were within 30% of single point estimates, thus validating the use of this approach.
TABLE 4.
Km and substrate selectivity coefficient values for CTP variants
The substrate selectivity coefficient is defined as Ki/Km. Ki values (mm) were determined as described under “Experimental Procedures” and are presented in Table 3. Standard errors were propagated from the standard errors in Km and Ki. For single-Cys CTP variant-inhibitor combinations where either the percent inhibition was less than 3% or the standard error of the measurement was greater than the calculated Ki value itself, then a minimal estimate for the selectivity coefficient was determined as follows: % inhibition + % S.E. is used to calculate a Ki, then Ki/Km is used to calculate a minimal selectivity coefficient. Thus, poorly inhibiting anions display different thresholds of selectivity based on inherent differences in the Km value of the variant. In these instances, the selectivity coefficient value presented represents a minimal estimate. Those cases where Ki + S.E. ≤ 0 (i.e. no observable inhibition) are denoted as ND as the selectivity coefficient value is not determinable. α-KG is α-ketoglutarate; PEP is phosphoenolpyruvate.
| CTP variant | Km valuesa | Selectivity coefficient values |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Isocitrate | α-KG | Malate | Succinate | Malonate | PEP | ADP | Phosphate | Pyruvate | Glutamate | ||
| Cys-less | 0.62 ± 0.04 | 10.3 ± 0.8 | 61.7 ± 10.2 | 81.0 ± 15.4 | 70.3 ± 8.4 | 61.7 ± 22.5 | 30.0 ± 2.8 | 75.3 ± 27.0 | 156.0 ± 44.7 | >370 | >756 |
| K83C | 28.2 ± 7.2 | 0.9 ± 0.3 | 0.6 ± 0.2 | 0.5 ± 0.2 | 0.7 ± 0.2 | 1.1 ± 0.4 | 0.6 ± 0.2 | 1.0 ± 0.3 | 1.0 ± 0.4 | 27.7 ± 16.3 | 2.6 ± 1.6 |
| R87C | 31.1 ± 4.7 | 0.5 ± 0.1 | 0.5 ± 0.2 | 0.5 ± 0.1 | 0.7 ± 0.2 | 0.9 ± 0.2 | 0.4 ± 0.1 | 1.0 ± 0.3 | 1.4 ± 0.5 | ND | 2.8 ± 1.3 |
| G119C | 25.4 ± 5.1 | 0.7 ± 0.2 | 1.3 ± 0.5 | 2.0 ± 0.8 | 3.2 ± 1.2 | 2.0 ± 0.7 | 0.9 ± 0.2 | 2.0 ± 0.5 | 2.3 ± 0.9 | 14.8 ± 17.8 | 4.0 ± 1.2 |
| R189C | 14.5 ± 1.7 | 0.6 ± 0.1 | 0.5 ± 0.1 | 0.5 ± 0.1 | 0.4 ± 0.1 | 0.8 ± 0.1 | 2.2 ± 0.3 | 3.9 ± 1.0 | 2.3 ± 0.5 | 1.5 ± 0.3 | 4.1 ± 1.2 |
| K37C | 2.3 ± 0.6 | 100.6 ± 40.6 | >750 | 118.7 ± 50.7 | >144 | 100.6 ± 83.8 | 100.6 ± 55.0 | 55.6 ± 37.6 | 76.6 ± 54.0 | >77 | >750 |
| R181C | 25.8 ± 5.2 | 5.3 ± 1.8 | 1.8 ± 0.4 | 1.5 ± 0.5 | 1.6 ± 0.6 | 1.7 ± 0.5 | 2.5 ± 1.0 | 7.5 ± 3.6 | 1.8 ± 0.6 | >6 | ND |
| K239C | 30.9 ± 10.9 | 4.8 ± 2.4 | >7 | 5.2 ± 3.1 | >38 | 6.3 ± 5.1 | 3.3 ± 1.7 | >5 | 3.8 ± 2.5 | 4.8 ± 3.1 | >78 |
| R276C | 16.4 ± 3.3 | 1.4 ± 0.4 | 2.0 ± 0.5 | 1.8 ± 0.4 | 1.8 ± 0.4 | 2.7 ± 0.7 | 0.6 ± 0.2 | 1.2 ± 0.3 | 2.7 ± 0.8 | 27.3 ± 27.3 | 2.2 ± 0.7 |
| R279C | 19.5 ± 5.4 | 1.4 ± 0.5 | 0.7 ± 0.3 | 1.1 ± 0.4 | 1.3 ± 0.4 | 0.9 ± 0.4 | 0.7 ± 0.3 | 1.6 ± 0.7 | 1.0 ± 0.4 | 16.2 ± 11.4 | 2.6 ± 0.9 |
a Km values (mm) were previously published (24) except for the G119C mutant data.
Molecular Modeling of CTP Variants
Molecular modeling was carried out as described previously (18), using Molecular Operating Environment, version 2009.10 (Chemical Computing Group, Montreal, Canada). Substrate conformations were generated and minimized using the MMFF94X force field (27).
RESULTS
We previously demonstrated that, as depicted in Fig. 1, the CTP in its outward-facing conformation contains at least two substrate-binding sites (23). In site 1, the more external site, the residues that primarily interact with citrate are Lys-83, Arg-87, and Arg-189. Furthermore, Gly-119 plays a critical role in both providing the flexibility required for conformational changes that occur during transport and, due to its small size, in enabling the correct packing interactions to occur between residues in TMDs II and IV that are required for formation of this binding site. Site 2, which is located near the midpoint of the membrane bilayer, is also depicted in Fig. 1. The residues that interact with citrate are Lys-37, Arg-181, Lys-239, Arg-276, and Arg-279. This study focused on determining the substrate specificity conferred by mutation of each of the site 1 and 2 residues noted above, one at a time, to cysteine. It should be noted that cysteine was chosen as the replacement residue because it is average in size, relatively hydrophobic (and thus suitable for substitution in transmembrane domains), and highly reactive (28, 29). The latter property enables it to be chemically modified with specific probes that permit a versatile array of approaches to be employed in the study of transporter function. Finally, it has been shown to be minimally disruptive of protein function (30).
The single-Cys CTP mutants were overexpressed and isolated, and their function was reconstituted in liposomal vesicles. We have previously demonstrated the rigor of this approach with regard to CTP functional analysis (8, 19–23). We then assessed the specificity of each CTP variant from both their external and internal surfaces. The latter measurement also enabled a determination of the extent to which substrate exchange versus uniport (i.e. influx in the absence of internal substrate) occurred with a given CTP variant. Importantly, the Cys-less CTP served as the control protein for these studies. We note that both the kinetic properties and the substrate specificity of the Cys-less CTP control are comparable with the values observed with the wild-type transporter (8).
Effect of External Anions on Reconstituted [14C]Citrate/Citrate Exchange Mediated by CTP Single-Cys Substrate-binding Site Mutants
We studied the external specificity of CTP variants by examining the ability of a 25-fold excess of individual external anions to compete with and hence inhibit [14C]citrate uptake into proteoliposomes. The percent inhibition caused by 10 separate anions with each of the nine binding site mutants, as well as with the Cys-less control transporter, was determined. The inhibition data obtained with site 1 and 2 mutants are depicted in Tables 1 and 2, respectively. The inhibition results were then used to calculate a Ki value (i.e. the competitive inhibitor dissociation constant) for each mutant-anion pair. These Ki values are presented in Table 3. A selectivity coefficient, defined as the ratio of the Ki value for a given anion-mutant combination to the Km value of that mutant for citrate transport, was then calculated for each anion-mutant pair. The selectivity coefficient represents a quantitative assessment of the effectiveness of a given mutant in transporting citrate versus an alternative anion. Thus, the higher the selectivity of a given mutant for citrate versus another anion, the higher the value of the selectivity coefficient will be. These coefficient values are presented in Table 4.
TABLE 1.
Effect of externally added anions on the [14C]citrate/citrate exchange catalyzed by reconstituted single-Cys site 1 CTP variants
Reconstituted site 1 single-Cys CTP variants were incubated with 1–2 mm [14C]citrate plus a 25-fold excess of potential competing anion. Controls contained water in place of competing anion. Data are presented as mean percentage inhibition ± S.E. from 3 to 5 experiments. Transport reactions and data calculations were conducted as described under “Experimental Procedures.” Statistical differences among the Cys-less and the site 1 CTP variants with a given anion were evaluated with the one-way analysis of variance test. Significance was then determined with the Bonferroni multiple comparisons post hoc test.
| CTP variant | Isocitrate | α-Ketoglutarate | Malate | Succinate | Malonate | Phosphoenolpyruvate | ADP | Phosphate | Pyruvate | Glutamate |
|---|---|---|---|---|---|---|---|---|---|---|
| Cys-less | 60 ± 1 | 20 ± 2 | 16 ± 2 | 18 ± 1 | 20 ± 6 | 34 ± 1 | 17 ± 5 | 9 ± 2 | 1 ± 3 | −7 ± 9 |
| K83C | 50 ± 7 | 60 ± 1a | 62 ± 6a | 56 ± 3a | 44 ± 3b | 60 ± 5a | 47 ± 1a | 47 ± 7b | 3 ± 1 | 25 ± 9c |
| R87C | 62 ± 5 | 61 ± 10a | 61 ± 4a | 53 ± 7a | 46 ± 4b | 65 ± 3a | 43 ± 5b | 36 ± 7b | −3 ± 2 | 22 ± 7c |
| G119C | 57 ± 3 | 42 ± 8c | 32 ± 6 | 23 ± 4 | 32 ± 4 | 51 ± 1b | 32 ± 2 | 29 ± 6 | 6 ± 6 | 19 ± 2 |
| R189C | 74 ± 2c | 78 ± 1a | 78 ± 4a | 80 ± 2a | 68 ± 4a | 42 ± 1 | 29 ± 4 | 41 ± 4b | 52 ± 4a | 28 ± 5c |
a p < 0.001 versus Cys-less.
b p < 0.01 versus Cys-less.
c p < 0.05 versus Cys-less.
TABLE 2.
Effect of externally added anions on the [14C]citrate/citrate exchange catalyzed by reconstituted single-Cys site 2 CTP variants
Reconstituted site 2 single-Cys CTP variants were incubated with 1–2 mm [14C]citrate plus a 25-fold excess of potential competing anion. Controls contained water in place of competing anion. Data are presented as mean percentage inhibition ± S.E. from 3 to 6 experiments. Transport reactions and data calculations were conducted as described under “Experimental Procedures.” Statistical differences among the Cys-less and the site 2 CTP variants with a given anion were evaluated with the one-way analysis of variance test. Only with pyruvate were no statistical differences found. Significance was then determined with the Bonferroni multiple comparisons post hoc test. α-KG is α-ketoglutarate; PEP is phosphoenolpyruvate.
| CTP variant | Isocitrate | α-KG | Malate | Succinate | Malonate | PEP | ADP | Phosphate | Pyruvate | Glutamate |
|---|---|---|---|---|---|---|---|---|---|---|
| Cys-less | 60 ± 1 | 20 ± 2 | 16 ± 2 | 18 ± 1 | 20 ± 6 | 34 ± 1 | 17 ± 5 | 9 ± 2 | 1 ± 3 | −7 ± 9 |
| K37C | 7 ± 1a | −1 ± 2b | 6 ± 1 | 2 ± 3 | 7 ± 4 | 7 ± 2a | 12 ± 5 | 9 ± 4 | 3 ± 6 | −4 ± 5 |
| R181C | 15 ± 2a | 34 ± 1 | 38 ± 4a | 37 ± 7b | 35 ± 3 | 27 ± 5 | 11 ± 3 | 34 ± 5c | 6 ± 7 | −8 ± 8 |
| K239C | 14 ± 2a | 4 ± 6 | 13 ± 3 | −2 ± 4b | 11 ± 5 | 19 ± 3 | 6 ± 8 | 17 ± 5 | 14 ± 4 | −4 ± 5 |
| R276C | 51 ± 5 | 42 ± 3b | 45 ± 2a | 45 ± 1c | 35 ± 2 | 69 ± 7a | 55 ± 2c | 35 ± 3c | 5 ± 4 | 39 ± 5a |
| R279C | 47 ± 5b | 64 ± 9a | 52 ± 3a | 48 ± 1c | 57 ± 6a | 64 ± 8c | 44 ± 8b | 54 ± 7a | 7 ± 3 | 32 ± 2c |
a p < 0.001 versus Cys-less.
b p < 0.05 versus Cys-less.
c p < 0.01 versus Cys-less.
TABLE 3.
Ki values for single-Cys CTP variants
Ki values (mm) ± S.E. were calculated from the inhibition data depicted in Tables 1 and 2 as described under “Experimental Procedures.” NSI denotes no significant inhibition was observed with a given CTP mutant-anion pair, defined as ≤3% inhibition depicted in Tables 1 and 2. α-KG is α-ketoglutarate; PEP is phosphoenolpyruvate.
| CTP variant | Isocitrate | α-KG | Malate | Succinate | Malonate | PEP | ADP | Phosphate | Pyruvate | Glutamate |
|---|---|---|---|---|---|---|---|---|---|---|
| Cys-less | 6.4 ± 0.1 | 38.3 ± 3.8 | 50.2 ± 6.3 | 43.6 ± 2.4 | 38.3 ± 11.5 | 18.6 ± 0.5 | 46.7 ± 13.7 | 96.7 ± 21.5 | NSI | NSI |
| K83C | 24.1 ± 3.4 | 16.1 ± 0.3 | 14.8 ± 1.4 | 19.0 ± 1.0 | 30.7 ± 2.1 | 16.1 ± 1.3 | 27.2 ± 0.6 | 27.2 ± 4.1 | NSI | 72.4 ± 26.1 |
| R87C | 14.8 ± 1.2 | 15.5 ± 2.5 | 15.5 ± 1.0 | 21.5 ± 2.8 | 28.4 ± 2.5 | 13.0 ± 0.6 | 32.1 ± 3.7 | 43.1 ± 8.4 | NSI | 85.9 ± 27.3 |
| G119C | 18.1 ± 1.0 | 33.2 ± 6.3 | 51.1 ± 9.6 | 80.5 ± 14.0 | 51.1 ± 6.4 | 23.1 ± 0.5 | 51.1 ± 3.2 | 58.9 ± 12.2 | 376.8 ± 376.8 | 102.5 ± 10.8 |
| R189C | 8.2 ± 0.2 | 6.6 ± 0.1 | 6.6 ± 0.3 | 5.8 ± 0.1 | 11.0 ± 0.6 | 32.3 ± 0.8 | 57.3 ± 7.9 | 33.7 ± 3.3 | 21.6 ± 1.7 | 60.1 ± 10.7 |
| K37C | 231.5 ± 33.1 | NSI | 273.0 ± 45.5 | NSI | 231.5 ± 132.3 | 231.5 ± 66.1 | 127.8 ± 53.2 | 176.2 ± 78.3 | NSI | NSI |
| R181C | 136.4 ± 18.2 | 46.7 ± 1.4 | 39.3 ± 4.1 | 41.0 ± 7.8 | 44.7 ± 3.8 | 65.1 ± 12.1 | 194.7 ± 53.1 | 46.7 ± 6.9 | 377.1 ± 439.9 | NSI |
| K239C | 148.8 ± 21.3 | 581.2 ± 871.8 | 162.1 ± 37.4 | NSI | 195.9 ± 89.1 | 103.2 ± 16.3 | 379.4 ± 505.9 | 118.2 ± 34.8 | 148.8 ± 42.5 | NSI |
| R276C | 22.6 ± 2.2 | 32.5 ± 2.4 | 28.8 ± 1.3 | 28.8 ± 0.6 | 43.8 ± 2.5 | 10.6 ± 1.1 | 19.3 ± 0.7 | 43.8 ± 3.8 | 447.7 ± 358.2 | 36.9 ± 4.7 |
| R279C | 26.8 ± 2.9 | 13.4 ± 1.9 | 22.0 ± 1.3 | 25.8 ± 0.5 | 17.9 ± 1.9 | 13.4 ± 1.7 | 30.3 ± 5.5 | 20.3 ± 2.6 | 315.9 ± 135.4 | 50.5 ± 3.2 |
The Cys-less CTP maintains a 10-fold preference for citrate versus isocitrate and a 30-fold preference relative to that for phosphoenolpyruvate (Table 4). The control transporter displays an even lower effectiveness for the other anions tested (e.g. see the elevated Ki values depicted in Table 3). Fig. 2A depicts the interaction of citrate at site 1 in the Cys-less control transporter. Note the ability of citrate to form five ionic hydrogen bonds with residues comprising this site. In sharp contrast, each of the site 1 single-Cys CTP variants lose this unique selectivity for citrate and display sharply reduced selectivity coefficients for all of the dianions tested (see Table 4). Thus, single-Cys replacements for any of the site 1 residues result in CTP variants that display the specificity of a general anion transporter. For instance, the K83C variant caused the Km value for citrate transport to increase 45-fold (Table 4) and the Ki value for malate to decrease over 3-fold (Table 3), resulting in a selectivity coefficient for malate of 0.5. Fig. 2B shows that with the K83C mutant, citrate can now form only four hydrogen bonds, and importantly, one of the carboxylate groups is now no longer tethered to the binding site resulting in a striking elevation in Km value. This elevation can be seen as being due primarily to a loss in binding affinity for citrate. Fig. 2C depicts interactions between malate and site 1 of the K83C variant. Note the formation of three hydrogen bonds in this remnant binding site that results in a Ki value (i.e. 15 mm) that is slightly lower than the Km value for citrate (i.e. 28 mm). Because of its small size and the presence of only two carboxylate groups, malate can only bind two positively charged residues at a time whether in the wild-type (data not shown) or in this CTP mutant. Thus, the loss of a single arginine or lysine does not decrease the affinity for malate (e.g. Ki values of 50, 15, and 16 mm for the Cys-less control, K83C, and R87C variants, respectively). The modest decrease in Ki values of these mutants may be attributed to a decrease in transport rate for reasons discussed below. Finally, it is noteworthy that even though the G119C variant displayed the least significant changes in inhibition values compared with the Cys-less control transporter (Table 1), the fact that its Km value for citrate transport was increased 41-fold resulted in markedly reduced selectivity coefficients with each of the anions tested (see Table 4).
FIGURE 2.

Citrate and selected alternate substrates in binding site 1. Substrate is shown as a stick figure with carbon atoms in magenta and oxygen atoms red. Binding site residues are shown with carbon atoms in green, oxygen atoms red, nitrogen atoms blue, and sulfur atoms yellow. Ionic hydrogen bonds are shown as dotted orange lines. A, citrate in site 1 of the Cys-less control CTP binds to three positively charged side chains. B, citrate in the K83C mutant binds to two positively charged side chains. C, malate in the K83C mutant can bind to two positively charged side chains, as could citrate. D, citrate in the R189C mutant binds to two positively charged side chains. E, malate in the R189C mutant can bind to two positively charged side chains, as could citrate. F, pyruvate in the R189C mutant can bind to two positively charged side chains, as could citrate.
The R189C mutant represents the most extreme example of this trend of an increased affinity for dicarboxylates versus citrate in site 1. We note that this single replacement caused a 23-fold increase in the Km value for citrate and an 8-fold decrease in the Ki value for malate. Fig. 2D depicts the interaction of citrate with the R189C mutant. Again, we note a decrease in the number of hydrogen bonding interactions and an untethered carboxylate group, causing a substantial elevation in the Km value for citrate. Fig. 2E shows the likely interaction of malate with this mutated site. Note the ability of the dicarboxylate to adopt a different, higher affinity conformation in this site compared with the K83C mutant (i.e. the Ki value decreases 2-fold), as it now is able to form three hydrogen bonds with Arg-87 and Lys-83. Importantly, the overall decrease in the Ki values of the mutants for malate compared with the Cys-less transporter likely reflects both binding and velocity components. Fig. 2F depicts the ability of mono-anionic pyruvate to form three hydrogen bonds with this remnant site and to effectively inhibit citrate transport (see Tables 1 and 3). Finally, it should be noted that although we have chosen to focus primarily on malate-mutant interactions, these general trends are seen throughout site 1 mutant-anion pairs (Tables 3 and 4).
Mutation of site 2 residues resulted in two strikingly different findings. First, as depicted in Tables 2 and 3, the K37C mutant displayed a dramatically increased selectivity for citrate relative to the control transporter. Thus, at a 25-fold excess concentration (over the citrate concentration), none of the anions tested displayed substantial inhibition (Table 2). Even for isocitrate, the substrate most closely resembling citrate in structure, the selectivity coefficient increased 10-fold compared with the Cys-less control (i.e. from 10 to 101; Table 4). With the other anions tested, the selectivity coefficient values ranged from 77 to >750. The second major finding was that upon mutation of all other site 2 residues, we observed varying degrees of relaxation of CTP substrate selectivity, which in some cases approached the levels observed with the site 1 mutants. For example, the K239C mutant displayed a moderate decrease in selectivity for citrate (see Table 4). In contrast the R181C, R276C, and R279C variants showed a substantial reduction in citrate selectivity with nearly all of the anions tested, relative to the Cys-less control. In many cases coefficient values of 1–2 were obtained indicating a similar effectiveness for citrate and the tested anion. Thus, many of the site 2 mutants, like the site 1 mutants, converted CTP binding specificity to that of a general anion carrier. It should be noted that one exception was the interaction of the R181C mutant with isocitrate, with which a selectivity coefficient of 5 was observed, about 50% of the value obtained with the control transporter.
Fig. 3 portrays a typical example of the proposed substrate interactions that occur in site 2 of the control and the mutated transporters. Fig. 3A shows citrate interacting with site 2 of the Cys-less control transporter and indicates that it can interact with five positively charged residues and form six ionic hydrogen bonds with these residues. Fig. 3B shows citrate binding to site 2 in the R181C mutant. With this mutant, citrate can only interact with four positively charged residues and form five ionic hydrogen bonds. Note that the inability of the C1-carboxylate group of citrate to form an ionic hydrogen bond (with residue 181) in the R181C mutant likely gives rise to the observed elevation in Km value for citrate transport (i.e. 26 versus 0.6 mm and R181C versus control values, respectively). Fig. 3C depicts malate in site 2 of the Cys-less control transporter. Its two carboxyl groups can form three ionic hydrogen bonds with residues Arg-279 and Lys-239. Notice that Arg-276 from TMD VI and Arg-181 from TMD IV are not thought to participate in malate binding. Presumably, the lack of these interactions gives rise to the relatively high Ki value of the control transporter for malate versus its Km value for citrate (i.e. 50 versus 0.6 mm, respectively). Fig. 3D depicts malate binding to site 2 in the R181C mutant. Notice its binding interactions are not significantly changed compared with the control protein, and this is reflected by the very similar Ki values for malate (i.e. 39 versus 50 mm, R181C versus Cys-less, respectively). Thus, in this example it is the decrease in the affinity for citrate and the constancy in the affinity for malate that give rise to the observed reduction in the selectivity coefficient from 81 (Cys-less) to 1.5 (R181C mutant). This general pattern is seen with many of the site 2 mutant-anion pairs except with the K37C mutant as described above.
FIGURE 3.

Citrate and malate in binding site 2. Substrate is shown as a stick figure with carbon atoms in magenta and oxygen atoms red. Binding site residues are shown with carbon atoms in green, nitrogen atoms blue, and sulfur atoms yellow. Ionic hydrogen bonds are shown as dotted orange lines. A, citrate in site 2 of the Cys-less control CTP binds to five positively charged side chains. B, citrate in the R181C mutant can bind to four positively charged side chains. C, malate in site 2 of the Cys-less control transporter can bind to two positively charged side chains. D, malate in R181C mutant can bind to two charged side chains.
Examination of the Internal Substrate Specificity of the Single-Cys CTP-binding Site Variants
We studied the ability of different intraliposomal anions to support [14C]citrate uptake into proteoliposomes. As depicted in Fig. 4, the Cys-less control CTP maintains a clear selectivity for internal citrate. Isocitrate can partially support citrate uptake such that in the presence of this anion the uptake rate was 18% of the value observed in the presence of internal citrate. (Note: in all cases the reported percentage values represent means of the samples taken throughout the time course.) None of the other eight anions tested were able to support citrate uptake to a significantly greater extent than that observed in the absence of internal anion. It should be noted that the condition of citrate uptake in the absence of internal counteranion constitutes a uniport reaction in contrast to obligatory substrate exchange, which is the transport mode primarily observed with the control transporter. Thus, with the Cys-less CTP, the level of uniport is 9% of the level of the exchange reaction when citrate is the intraliposomal substrate.
FIGURE 4.

Ability of various internal anions to support the influx of external [14C]citrate with the Cys-less control CTP and binding site 1 CTP variants. Time courses of [14C]citrate uptake into liposomes reconstituted with CTP variants are presented. The proteoliposomes were reconstituted with 50 mm anionic substrate and the exchange was triggered by addition of 2 mm [14C]citrate. The internal anions included are defined in the inset within a given panel. Data points are presented as means ± S.E. Transport reactions were conducted as described under “Experimental Procedures.”
Fig. 4 further depicts the data obtained with four site 1 mutants. With the K83C mutant, the transport activity observed in the presence of internal substrate is about double the level observed in the absence of internal anion. Moreover, there is some relaxation in specificity as citrate and isocitrate support citrate uptake to similar extents. Malate also supports uptake, albeit to a moderately lesser degree. A similar specificity was observed with the R87C mutant, except in this case the transport activity observed in the presence of internal anion is only somewhat greater than in the absence of internal counteranion (i.e. the magnitude of the uniport activity was 68% of the maximum observed exchange activity). The G119C mutant primarily functions as a uniporter. Thus, internal citrate, isocitrate, or malate caused little discernable change in the rate of citrate uptake relative to that observed in the absence of internal anion. In sharp contrast, the R189C variant primarily catalyzes an exchange reaction with the level of uniport being only 26% the level of exchange observed with isocitrate as the internal substrate. Most interestingly, with this mutant the internal specificity is altered. Thus citrate, isocitrate, malate, succinate, and α-ketoglutarate all support [14C]citrate uptake to generally similar extents. Other anions tested supported uptake only marginally better than did water alone.
Fig. 5 depicts the data obtained with the five site 2 mutants. The K37C mutant resembles the Cys-less control transporter in that it maintains a strict requirement for internal anion and displays a high selectivity for internal citrate. Thus, the level of uniport activity is only 11% the level of the exchange activity. Moreover, similar to the control transporter, this CTP variant displays a strong preference for internal citrate. Other anions such as isocitrate, malate, and phosphoenolpyruvate supported citrate uptake at 16–19% of the level observed with internal citrate. Similar to the external substrate specificity studies, the K239C mutant displays an intermediate level of selectivity. With this mutant citrate uptake is highly dependent on internal anions with the uniport activity being only 25% of the exchange activity. Furthermore, isocitrate and malate are able to support citrate uptake to moderate extents (i.e. 38–39%) relative to the activity observed in the presence of internal citrate. The R181C mutant represents an example of the opposite extreme (i.e. compared with the K37C variant), where [14C]citrate uptake is independent of internal substrate. Thus, the levels of uptake observed in the presence of internal citrate, isocitrate, or malate are similar to the uptake level observed in the absence of any internal counteranion. This mutant appears to operate exclusively in a uniporter mode. Finally, the R276C and R279C mutants displayed intermediate properties. With these mutants the level of citrate uptake was ∼2-fold higher in the presence of internal citrate versus in the absence of any internal anion. Furthermore, isocitrate supported exchange at 79% of the level observed with internal citrate. In contrast, malate was ineffective at supporting substrate exchange.
FIGURE 5.

Ability of various internal anions to support the influx of external [14C]citrate with binding site 2 CTP variants. Time courses of [14C]citrate uptake into liposomes reconstituted with various site 2 CTP variants are presented. The proteoliposomes were reconstituted with 50 mm anionic substrate, and the exchange was triggered via addition of 2 mm [14C]citrate. The internal anions present are defined in the inset within a given panel. Data points are presented as means ± S.E. Transport reactions were conducted as described under “Experimental Procedures.”
DISCUSSION
This study examined the substrate specificity of the CTP from its external and internal surfaces. Two types of studies were conducted. To examine the role of a residue in transport specificity from the external surface, we measured the ability of 10 alternative anions to inhibit [14C]citrate influx with each of nine single-Cys replacements for residues known to comprise the two substrate-binding sites of CTP. Internal specificity was examined by preloading liposomes with various internal anions and measuring the ability of these anions to support [14C]citrate influx. The results from our studies shed light not only on the role of individual binding site residues on substrate specificity but also enhance our understanding of the mechanism by which the CTP transports citrate across the membrane bilayer. Specifically, previously we (25) and others (31–33) have conjectured that mitochondrial carriers exist in an outward-facing and an inward-facing conformation. We have previously modeled the outward-facing conformation (18) based on the crystal structure of the ATP carrier (17) and have experimentally confirmed the validity of this model (19–23). A key conclusion from this study, which builds on this model, is that residues within site 1, the more external of the two sites, are accessible and play a role in determining specificity with regard to internal substrate as it moves through the CTP. This result leads us to postulate the existence of an intermediate occluded conformation that CTP assumes during the transport cycle. There is precedence for this with the amino acid antiporter AdiC (34).
Relationship of Km to Kd
Below, we depict a model for citrate transport that includes one or more intermediate states and Equations 1 and 2 defining Km for such a model (35). We define k1 and k−1 as the on- and off-rates for the outward-facing CTP conformation. We condense the intermediate steps to a single conformational change depicted by the rate constants k2 and k−2. Finally, we define k3 as representing the final conformational change and the release of substrate to the opposite side of the membrane. If we assume that the rate constants k2 and k−2 are within a factor of 10 of each other, then for reasonable values of k1 and k−1 (given the observed Km value for citrate with the Cys-less CTP), we conclude that if the final step in the transport mechanism (k3) is the rate-determining step, then a reduction in that rate would tend to show a decrease in the Km value. For example, if k2 is equal to k−2 and k3 is much less than either of these rates, then Km will be equal to 0.5 Kd. Furthermore, if the slow step for any particular mutant is the final conformational change that allows citrate to be transported and released on the opposite side of the membrane (i.e. k3), then changes in the citrate binding (Kd) would once again be required to explain the changes in the Km value (and the change in Km would underestimate the change in Kd). Therefore, the ratio of Km for citrate transport in the mutant to the Km value for citrate transport in the Cys-less control would largely reflect the underlying Kd values. Finally, if one were to assume that an intermediate step in the transport mechanism is rate-limiting (e.g. k2), once again the change in Km would be expected to largely reflect the change in Kd (Equations 1 and 2).
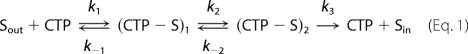 |
Relationship of Ki to Kd
One can classify the alternative anions tested in this study either as classical inhibitors (i.e. molecules that compete for the CTP-binding sites and prevent citrate from binding but are not themselves actually transported) or as poor substrates (i.e. molecules that are actually transported across the membrane bilayer). If they act as classical inhibitors, then the competitive Ki can be interpreted to be equal to the Kd value. This must be the case for the strong, predominantly competitive inhibitor BTC, which has been shown to not be transported by CTP (36). Relatedly, we previously noted that each of nine single-Cys substrate-binding site mutants binds citrate less effectively compared with the Cys-less control transporter, as evidenced by both an increase in mutant Km values for citrate transport (24) as well as a proportional increase in mutant Ki values for BTC (24). Consequently, there was little change in the ratio of the Ki value for BTC to the Km value for citrate transport, thus suggesting that the changes in the Km value are largely due to changes in the Kd value for citrate.
If instead one assumes that the alternative anions tested exhibit substrate behavior, then the logic applied above to citrate should also hold for these alternative substrates. The Ki value measured with these anions is the Km value and should largely reflect the Kd value for the reasons described above. Thus, the ratio of the measured Ki value for an alternative anion to the Km value for citrate in most cases represents the ratio of their underlying Kd values for the anion pair. We recognize that any mutation is likely to perturb a number of the rate constants in the transport mechanism, only some of which will be reflected in Kd values, although others may change the overall rate of the reaction and thus may also change Km values. However, given the observed changes in Vmax with each of these mutants for citrate, it is likely that similar changes are seen for the alternative substrates indicating the changes in Km values are largely reflecting changes in the underlying Kd values.
Assessment of Mutant Selectivity for External Anions
This study sought to quantify the selectivity of single-Cys substrate-binding site CTP mutants for citrate versus other physiologically relevant substrate-like anions. We have quantitatively defined the substrate selectivity of a given CTP variant as the ratio of its Ki value for alternative anion to the Km value for citrate (i.e. the affinity for citrate; note: differences in Km values are likely to reflect differences in Kd values with the mutants for the reasons outlined above). We realize that this analysis is limited to an understanding of binding selectivity and thus is unlikely to fully capture the preference by CTP for citrate over alternative anions. However, we feel that it is quite unlikely that any of these alternative anions will ever have Vmax values that are substantially greater than that with citrate, and thus the selectivity coefficients that we display in Table 4 represent a minimal estimate of how a particular CTP variant discriminates for each of the anions tested (i.e. if the Vmax,alt. ≤ Vmax,citrate then the preference for citrate over the alternative anion (Vmax/Km)citrate/(Vmax/Km)alt. will be greater than or equal to the calculated selectivity coefficient). Lower selectivity coefficient values, as observed with most cases of single-Cys mutations compared with the Cys-less control, reflect a smaller increase in the Ki value for an alternative anion compared with the previously documented increase in the Km value for citrate transport (24) and therefore indicate a reduced selectivity for citrate. The extent to which one anion is discriminated against versus another anion for a particular binding site mutant helps us understand how that residue normally binds citrate.
Site 1 Mutants: External Anion-mediated Inhibition of [14C]Citrate Influx
We note that each site 1 mutant is characterized by much lower selectivity coefficient values compared with the Cys-less control transporter, indicating the ability of a wide spectrum of anions to bind to a given CTP variant and inhibit citrate transport. We further note that each mutation accommodates a different spectrum of anions more or less effectively, presumably by taking advantage of the altered fit between the remnant binding site and each of the anions tested. The R189C mutant is the most extreme example of the increased promiscuity of this site. Upon creation of this mutant, the remnant of binding site 1 consists of Arg-87 and Lys-83, and although it does not bind citrate very well, we observe that almost any dianion can effectively compete with citrate, as can pyruvate, a small monoanion. In contrast, mutation of either Lys-83 or Arg-87 does not allow pyruvate to compete with citrate. However, the dianion malate appears to bind the remnant sites of the K83C, R87C, and the R189C mutations to similar extents due to its slightly larger size and additional charge. As depicted in Fig. 2, malate can bind either to Arg-87 and Arg-189 in the K83C mutant or, alternatively, to Arg-87 and Lys-83 in the R189C mutant. In both instances, malate binding effectively inhibits citrate transport. Finally, with the G119C mutant, the addition of the cysteine side chain at location 119 apparently sufficiently distorts site 1 such that productive citrate binding affinity is substantially reduced (i.e. a dramatically elevated Km value is observed). Smaller and/or more flexible dianions apparently are less affected by the distortion of the binding site caused by this mutation.
Site 1 Mutants: Internal Anion-supported [14C]Citrate Influx
We next examined the ability of internal anions to trigger citrate uptake. We note that the Cys-less CTP is highly specific for internal citrate. Thus, whereas intra-liposomal isocitrate supports an exchange reaction that is 18% of the level observed with internal citrate, other anions do not support substrate exchange. In contrast, with the R189C mutant, internal isocitrate, malate, succinate, and α-ketoglutarate all support citrate uptake to similar or slightly greater extents than does internal citrate. We interpret the broader substrate specificity of the R189C mutant as arising from the ability of the Lys-83/Arg-87/Gly-119 remnant site 1 to bind a variety of internal anions that have moved in an outward direction through the CTP and approach this site as they enter the external leaflet of the membrane bilayer. Upon reaching this remnant site 1, these anions then trigger the obligatory conformational change(s), and hence this mutant catalyzes an exchange activity with a broad substrate specificity. We posit that anion binding to residues Lys-83 and Arg-87 (see Fig. 2E), both of which reside on TMD helix II, is necessary for triggering the required conformational change(s) associated with exchange. Note this explanation is in complete agreement with the increased promiscuity of this mutant for external anions (Table 4). Importantly, the observation that upon mutation of either Lys-83 or Arg-87, internal ligand loses much of its effectiveness at triggering citrate uptake such that the transport rate observed with internal anion is much closer to the low level observed with water alone (Fig. 4), reinforces the notion that these residues are required for the exchange reaction. With regard to the G119C variant, we posit that the reason it is no longer stimulated by internal citrate or other anions (i.e. functions as a uniporter) is that the flexibility of glycine is required for the substrate-triggered conformational change(s) that allow the conversion of the CTP inward-facing conformation to the outward-facing conformation.
Site 1 Mutants: Effect on Exchange Versus Uniport Activities
As stated above, a key conclusion emerging from our studies is that site 1 residues, located near the external portion of the bilayer, play a key role in determining the selectivity of the efflux reaction. Our data suggest that internal substrates are able to and in fact are required to bind to site 1 during their efflux, possibly triggering a final conformational change(s) leading to their release. An additional mechanistic consequence of the posited role for site 1 residues is that they are key to facilitating substrate exchange but may also be involved to a lesser extent in the facilitation of substrate uniport (i.e. one-way uptake). Consistent with this idea, our data indicate that mutation of site 1 residues affects the exchange reaction to a greater extent than the uniport reaction. For example, with the Cys-less CTP, the uniport activity is 9% of the exchange activity. In contrast, with the G119C, R87C, K83C, and R189C mutants, the uniport activities are 88, 68, 51, and 26% of the maximum observed anion-supported exchange activities. One interpretation for the previously reported (23, 24) dramatic reduction in the Vmax value for each of the site 1 mutants is that the removal of a positive charge from site 1 stabilizes the inward-facing conformation, thereby reducing the rate at which the outward-facing conformation can be adopted, thus slowing down both the rates of exchange and uniport, but perhaps to different extents.
Site 2 Mutant, K37C
The K37C mutant is unique among the CTP mutants in that it displays a remarkably enhanced anion selectivity, such that none of the alternative anions tested substantially inhibited [14C]citrate uptake (Table 2). Thus, this mutant displayed substantially elevated selectivity coefficient values (Table 4) for each of the tri- and dicarboxylate anions tested compared with the Cys-less control transporter. The following question arises. What accounts for the high selectivity of this mutant? We posit that the K37C mutation has two effects on CTP function. One effect is on anion binding where the alternative substrates rely on interaction with this lysine to productively bind to site 2 prior to transport in a way that citrate does not giving rise to the enhanced selectivity that is observed with this mutant.
Alternatively, anions may be unable to trigger the conformational changes required for transport in the absence of Lys-37. Consistent with this interpretation, we note that with the K37C mutant the Km value for citrate is increased 4-fold, and the Vmax value is decreased by ∼600-fold (24), suggesting that in the wild-type transporter Lys-37 plays a more prominent role in the substrate-dependent conformational change(s) required for transport versus its role in substrate binding per se. In support of this concept is our earlier finding that chemical modification of this mutant with a positively charged methanethiosulfonate reagent restores Km to the value observed with the Cys-less control but does not restore Vmax suggesting that the positive charge of lysine in the control transporter facilitates substrate binding, whereas the high degree of lysine flexibility is required for the conformational changes needed for transport (23). We note that Lys-37 is highly conserved (32), and it represents one of the mitochondrial transporter family signature sequence residues that appear to participate in a salt bridge network thought to be key to conformational changes in mitochondrial carriers (37).
Site 2 Mutant, K239C
The K239C site 2 mutant displays a moderate decrease in substrate selectivity relative to the Cys-less control protein. Modeling suggests that Lys-239 likely forms a salt bridge to a citrate carboxylate and an ionic hydrogen bond to the citrate hydroxyl group (see Figs. 1 and 3). We posit that the decrease in specificity likely arises from the loss of these direct interactions with the substrate. This interpretation is supported by the substantially elevated Km value for citrate transport observed with the K239C mutant relative to the Cys-less control protein (i.e. 31 versus 0.6 mm, respectively). The Ki values for the alternative anions are increased, albeit to a lesser extent, resulting in a mutant that retains a reasonable degree of selectivity. Finally, although Lys-239 is part of the same salt bridge network as Lys-37 (37), both the Km data and modeling suggest that this residue assumes a more prominent role in substrate binding than in catalyzing conformational change.
Site 2 Mutant, R181C
With the R181C mutant, we note that citrate as well as isocitrate lose a salt bridge between the C1-carboxylate group and the Arg-181 side chain that exists in the Cys-less control transporter. With citrate, this loss causes a dramatic increase in Km (0.6 versus 25.8 mm; Cys-less versus R181C, respectively). In terms of the substrate specificity of this mutant, we note that although the Ki value for external isocitrate has increased 21-fold (Table 3), the Km value for citrate has increased to a greater extent (42-fold) resulting in a selectivity coefficient that is only moderately reduced (i.e. 10.3 versus 5.3, Cys-less versus R181C mutant, respectively; see Table 4). We also observed that the dicarboxylates malate and succinate were moderately effective inhibitors of this mutant. Each of these anions can be modeled in the CTP-binding site such that the main interactions that occur with the control transporter also occur with the residues of remnant site 2 (see Fig. 3, C and D). Thus, removal of the Arg-181 side chain more drastically decreases the affinity of CTP for citrate versus its affinity for these dicarboxylates, thereby resulting in a reduced selectivity coefficient.
Additionally, we note that this mutant does not show a dependence upon intra-liposomal substrate (Fig. 5). Thus, R181C behaves as a uniporter such that internal substrate does not facilitate reorientation of the transporter from the inward-facing to the outward-facing conformation. Based on these findings, we conclude that Arg-181 is critical in positioning both extra- and intra-liposomal citrate in a proper orientation to trigger the conformational change(s) that occur during transport. We further posit that Arg-181, which resides in TMD IV, is critical for the citrate-dependent rearrangement of this TMD, possibly bringing it in closer proximity to TMD VI. In the absence of Arg-181, internal substrates are not able to induce this conformational change and hence the exchange function of this mutant is lost.
Site 2 Mutants, R276C and R279C
With the R276C and R279C mutants, we note the ability of a wide spectrum of anions to effectively compete with citrate for transport. Furthermore, we have previously reported that both of these mutants display large increases in their Km values for citrate and decreases in Vmax (see Table 4) (24). We note that Arg-276 is highly conserved among mitochondrial CTPs, but not as conserved among other mitochondrial anion carriers, whereas the Arg-279 is conserved among most mitochondrial transporter types (7, 32).
Our modeling studies with the control transporter indicated that, upon binding to site 2 residues, the C1-COO− of citrate forms an ionic hydrogen bond with Arg-276 and the C5-COO− bonds with Arg-279 (see Figs. 1 and 3). These two residues are separated by one turn of TMD helix VI. Moreover, modeling with the control protein suggests that dicarboxylate substrates such as malate interact only with Arg-279 and Lys-239 but not Arg-276 (see Fig. 3C). Importantly, mutation of either Arg-276 or Arg-279 impairs the ability of the CTP to bind and transport citrate (i.e. the Km is increased 26–31-fold in these mutants) to a greater extent than it affects the binding of the smaller dicarboxylates at this site (i.e. the Ki values are actually decreased an average of ∼2-fold). Thus, these mutant CTPs display a decrease in the selectivity coefficient for all the dianions studied. We postulate that with the R276C mutant, the malate carboxyl group, which normally binds to this location, can now bind instead to another nearby positive charge (e.g. possibly Lys-37), thereby enabling the mutated transporter to maintain a reasonable Ki value with malate and other dicarboxylates.
It is noteworthy that upon mutation of either Arg-276 or Arg-279, there is no longer a pronounced preference for citrate relative to isocitrate (i.e. the selectivity coefficient is 1.4). Thus, the absence of either of these positive charges increases the Km value for citrate to a greater extent (i.e. 26–31-fold) than the Ki value for isocitrate (i.e. 4-fold), both of which now become of similar magnitude to the values observed with the dicarboxylates studied (see Table 3). Furthermore, these mutants display a significant decrease in transport stimulated by internal anions, such that only citrate and isocitrate are able to confer a modest increase in the level of citrate uptake compared with the level of the uniport activity observed in the absence of internal counteranion. We posit that these residues are key to enabling internal citrate to promote the correct spatial relationships between TMDs IV and VI in site 2 that are required to promote a switch from the inward-facing conformation to the outward-facing conformation.
CTP Transport Mechanism
Previously, we (25) and others (31–33) have discussed mitochondrial transporter mechanisms in terms of inward-facing and outward-facing transporter conformations. Our current specificity data suggest that the CTP adapts a complex set of conformational states similar to those hypothesized for the AdiC transporter (34), which would include at least one occluded intermediate conformation in addition to the open conformations. With regard to the CTP, a surprising and key finding with important mechanistic implications that may extend to the entire mitochondrial carrier family is the observation that binding site 1 is kinetically accessible to anions from the inner surface as demonstrated by the altered specificity of certain of the site 1 mutants to internal substrate. We define kinetically accessible to mean that either site 1 is able to directly contact internal anions when CTP is in the inward-facing conformation or site 1 is in direct contact with the internal anions after the CTP has undergone a conformational change to an occluded state. Because we postulate that conformational change is triggered prior to internal substrate actually reaching site 1, we favor the latter interpretation. Furthermore, based on crystallographic studies (34), it is interesting to note that with AdiC it is thought that a rearrangement of several transmembrane-spanning α-helices results in the conversion of its outward-facing conformation to its occluded conformation. With regard to the CTP, the fact that site 1 residues Lys-83 and Arg-87 are found to be on the same TMD II and are critical to the selectivity for internal anions suggests that the conformational change that enables the conversion of the occluded to the outward-facing conformation may involve winding/unwinding of this helix. We have previously hypothesized that the inward-facing conformation of the CTP forms a third substrate-binding site near the internal surface of the bilayer (25). Whether this proposed site is kinetically accessible to external anions via the existence of an occluded state(s) will be the subject of future investigations.
Several comments are in order regarding comparison of our findings with the important work of Kunji and co-workers (32, 37). Their approach to understanding substrate specificity is based on analysis of amino acid conservation, symmetry, and distance and chemical constraints across the mitochondrial transporter family. This has enabled prediction of residues of mechanistic importance with individual mitochondrial transporters. In contrast, our work has employed the methods of mechanistic enzymology with site-specific CTP mutants to characterize in detail the role of individual CTP residues in substrate binding, specificity, and the translocation mechanism. Recently, they suggested that several of the residues, which we indicated participate in substrate binding (23), may be involved instead in other aspects of the translocation mechanism (37). We think this unlikely because of the following: (i) upon mutation of these residues we observe prominent increases in both the Km value for citrate transport and the Ki value for the binding of BTC (24), a primarily competitive inhibitor of the CTP (1, 24, 38); and (ii) modeling studies show the ability of these residues to form several ionic hydrogen bonds with citrate (23). Although our findings do not preclude involvement in other mechanistic steps as well, and in fact as mentioned above our data suggest such a scenario with Lys-37, the totality of our evidence strongly supports the direct participation of eight of the nine residues depicted in Fig. 1 in substrate binding. Gly-119 is the sole exception as its role in substrate binding is indirect; its lack of a side chain permits the proper packing of TMDs II and III in the formation of site 1, thereby allowing space for the Arg-87 side chain to interact with citrate. Furthermore, we believe that the assertion by Robinson and Kunji (32, 37) that Gln-182 is a key residue in substrate binding is likely to be incorrect based on our earlier findings that the Q182C mutant displays reduced Km and Vmax values, with the reduction in the latter parameter being considerably more pronounced (22, 23). Our findings suggest that Gln-182 is primarily involved in the conformational change-triggering mechanism that allows citrate to be transported, rather than in substrate binding. Moreover, if the wild-type residue formed important interactions with citrate, its mutation would be expected to cause a significant elevation in the Km parameter. It should be noted that our modeling of site 1 does indicate that Gln-182 is one of several residues that helps to shape the citrate binding pocket (23), and thus it may play a minor role in the citrate-site 1 interaction. Finally, we note that this study with the Cys-less CTP (see Fig. 4, Cys-less panel) confirms our earlier findings (8) that this transporter maintains a strong requirement for intra-liposomal substrate. Thus, in the absence of internal substrate, the residual uniport reaction is ≤10% of the exchange reaction. We have observed this requirement for internal substrate not only with the Cys-less yeast CTP but also with the wild-type yeast transporter (7, 8) and the rat liver CTP (39) as well, where the residual uniport reaction occurs to an even lesser extent. Thus, we take issue with the argument of Robinson et al. (37) that a weak cytoplasmic charge network within the CTP favors the catalysis of a substantive uniport reaction. Because we do not observe a substantial uniport reaction with CTPs from multiple sources, our data do not support their interpretation.
In conclusion, the investigations described above have provided the first function-based information with a mitochondrial transporter on the role that specific substrate-binding site residues play in determining transporter specificity. Our future experiments will be aimed at determining whether a proposed third substrate-binding site exists near the inner surface of the CTP (25), and if so how its component residues influence CTP specificity.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant GM-054642 (to R. S. K.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
- CTP
- citrate transport protein
- BTC
- 1,2,3-benzenetricarboxylate
- TMD
- transmembrane domain.
REFERENCES
- 1.Palmieri F., Stipani I., Quagliariello E., Klingenberg M. (1972) Eur. J. Biochem. 26, 587–5945025933 [Google Scholar]
- 2.Robinson B. H., Williams G. R., Halperin M. L., Leznoff C. C. (1971) J. Biol. Chem. 246, 5280–5286 [PubMed] [Google Scholar]
- 3.Watson J. A., Lowenstein J. M. (1970) J. Biol. Chem. 245, 5993–6002 [PubMed] [Google Scholar]
- 4.Endemann G., Goetz P. G., Edmond J., Brunengraber H. (1982) J. Biol. Chem. 257, 3434–3440 [PubMed] [Google Scholar]
- 5.Brunengraber H., Lowenstein J. M. (1973) FEBS Lett. 36, 130–132 [DOI] [PubMed] [Google Scholar]
- 6.Conover T. E. (1987) Trends Biochem. Sci. 12, 88–89 [Google Scholar]
- 7.Kaplan R. S., Mayor J. A., Gremse D. A., Wood D. O. (1995) J. Biol. Chem. 270, 4108–4114 [DOI] [PubMed] [Google Scholar]
- 8.Xu Y., Kakhniashvili D. A., Gremse D. A., Wood D. O., Mayor J. A., Walters D. E., Kaplan R. S. (2000) J. Biol. Chem. 275, 7117–7124 [DOI] [PubMed] [Google Scholar]
- 9.Kaplan R. S., Morris H. P., Coleman P. S. (1982) Cancer Res. 42, 4399–4407 [PubMed] [Google Scholar]
- 10.Kaplan R. S., Oliveira D. L., Wilson G. L. (1990) Arch. Biochem. Biophys. 280, 181–191 [DOI] [PubMed] [Google Scholar]
- 11.Joseph J. W., Jensen M. V., Ilkayeva O., Palmieri F., Alárcon C., Rhodes C. J., Newgard C. B. (2006) J. Biol. Chem. 281, 35624–35632 [DOI] [PubMed] [Google Scholar]
- 12.Jensen M. V., Joseph J. W., Ronnebaum S. M., Burgess S. C., Sherry A. D., Newgard C. B. (2008) Am. J. Physiol. Endocrinol. Metab. 295, E1287–E1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaplan R. S., Mayor J. A., Johnston N., Oliveira D. L. (1990) J. Biol. Chem. 265, 13379–13385 [PubMed] [Google Scholar]
- 14.Eriks L. R., Mayor J. A., Kaplan R. S. (2003) Anal. Biochem. 323, 234–241 [DOI] [PubMed] [Google Scholar]
- 15.Kaplan R. S., Mayor J. A., Wood D. O. (1993) J. Biol. Chem. 268, 13682–13690 [PubMed] [Google Scholar]
- 16.Xu Y., Mayor J. A., Gremse D., Wood D. O., Kaplan R. S. (1995) Biochem. Biophys. Res. Commun. 207, 783–789 [DOI] [PubMed] [Google Scholar]
- 17.Pebay-Peyroula E., Dahout-Gonzalez C., Kahn R., Trézéguet V., Lauquin G. J., Brandolin G. (2003) Nature 426, 39–44 [DOI] [PubMed] [Google Scholar]
- 18.Walters D. E., Kaplan R. S. (2004) Biophys. J. 87, 907–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaplan R. S., Mayor J. A., Brauer D., Kotaria R., Walters D. E., Dean A. M. (2000) J. Biol. Chem. 275, 12009–12016 [DOI] [PubMed] [Google Scholar]
- 20.Ma C., Kotaria R., Mayor J. A., Eriks L. R., Dean A. M., Walters D. E., Kaplan R. S. (2004) J. Biol. Chem. 279, 1533–1540 [DOI] [PubMed] [Google Scholar]
- 21.Ma C., Kotaria R., Mayor J. A., Remani S., Walters D. E., Kaplan R. S. (2005) J. Biol. Chem. 280, 2331–2340 [DOI] [PubMed] [Google Scholar]
- 22.Ma C., Remani S., Kotaria R., Mayor J. A., Walters D. E., Kaplan R. S. (2006) Biochim. Biophys. Acta 1757, 1271–1276 [DOI] [PubMed] [Google Scholar]
- 23.Ma C., Remani S., Sun J., Kotaria R., Mayor J. A., Walters D. E., Kaplan R. S. (2007) J. Biol. Chem. 282, 17210–17220 [DOI] [PubMed] [Google Scholar]
- 24.Aluvila S., Sun J., Harrison D. H., Walters D. E., Kaplan R. S. (2010) Mol. Pharmacol. 77, 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayor J. A., Sun J., Kotaria R., Walters D. E., Oh K. J., Kaplan R. S. (2010) J. Bioenerg. Biomembr. 42, 99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaplan R. S., Pedersen P. L. (1985) Anal. Biochem. 150, 97–104 [DOI] [PubMed] [Google Scholar]
- 27.Halgren T. A. (1996) J. Comput. Chem. 17, 490–519 [Google Scholar]
- 28.Guan L., Kaback H. R. (2007) Nat. Protoc. 2, 2012–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rudnick G. (2002) in Transmembrane Transporters (Quick M. W. ed) pp. 125–141, Wiley-Liss, Hoboken, NJ [Google Scholar]
- 30.Kaback H. R., Sahin-Toth M., Weinglass A. B. (2001) Nat. Rev. Mol. Cell Biol. 2, 610–620 [DOI] [PubMed] [Google Scholar]
- 31.Buchanan B. B., Eiermann W., Riccio P., Aquila H., Klingenberg M. (1976) Proc. Natl. Acad. Sci. U.S.A. 73, 2280–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinson A. J., Kunji E. R. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 2617–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pebay-Peyroula E., Brandolin G. (2004) Curr. Opin. Struct. Biol. 14, 420–425 [DOI] [PubMed] [Google Scholar]
- 34.Gao X., Zhou L., Jiao X., Lu F., Yan C., Zeng X., Wang J., Shi Y. (2010) Nature 463, 828–832 [DOI] [PubMed] [Google Scholar]
- 35.Van Winkle L. J. (1999) Biomembrane Transport, pp. 90–91, Academic Press, NY [Google Scholar]
- 36.Palmieri F., Genchi G., Stipani I., Quagliariello E. (1977) in Structure and Function of Energy Transducing Membranes (van Dam K., van Gelder B. F. eds) pp. 251–260, Elsevier Science Publishing Co., Inc., New York [Google Scholar]
- 37.Robinson A. J., Overy C., Kunji E. R. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 17766–17771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson B. H., Williams G. R., Halperin M. L., Leznoff C. C. (1971) Eur. J. Biochem. 20, 65–71 [DOI] [PubMed] [Google Scholar]
- 39.Kaplan R. S., Mayor J. A., Oliveira D. L., Johnston N. (1989) in Anion Carriers of Mitochondrial Membranes (Azzi A., Nalecz K. A., Nalecz M. J., Wojtczak L. eds) pp. 59–69, Springer-Verlag Inc., New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.